Mutation-Associated Phenotypic Heterogeneity in Novel and Canonical PIK3CA Helical and Kinase Domain Mutants


Abstract
:1. Introduction
2. Materials and Methods
2.1. Cloning and Site-Directed Mutagenesis of Wild Type and PIK3CA Mutants
2.2. Cell Culture and Transfection
2.3. Morphological Characterization
2.4. Actin Cytoskeletal Staining and Analysis
2.5. Scratch Wound Healing Assay
2.6. Cell Proliferation Assay
2.7. Apoptosis (Caspase 3/7) Assay
2.8. Western Blot Analyses
2.9. Spheroid Formation Assay
2.10. Bioinformatics-Based Prediction of the Functional Impact of PIK3CA Mutations
2.11. Statistical Analyses
3. Results
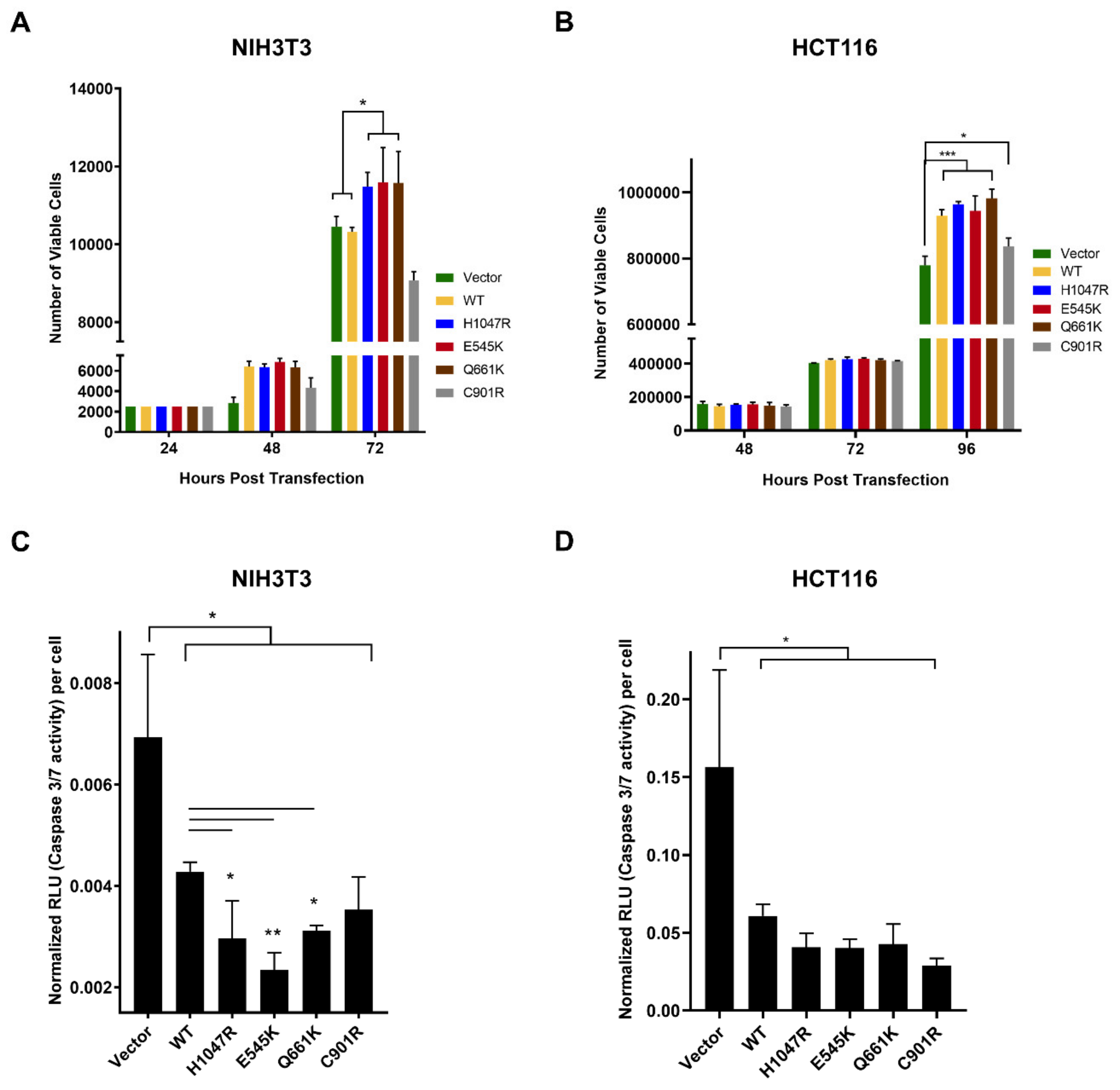
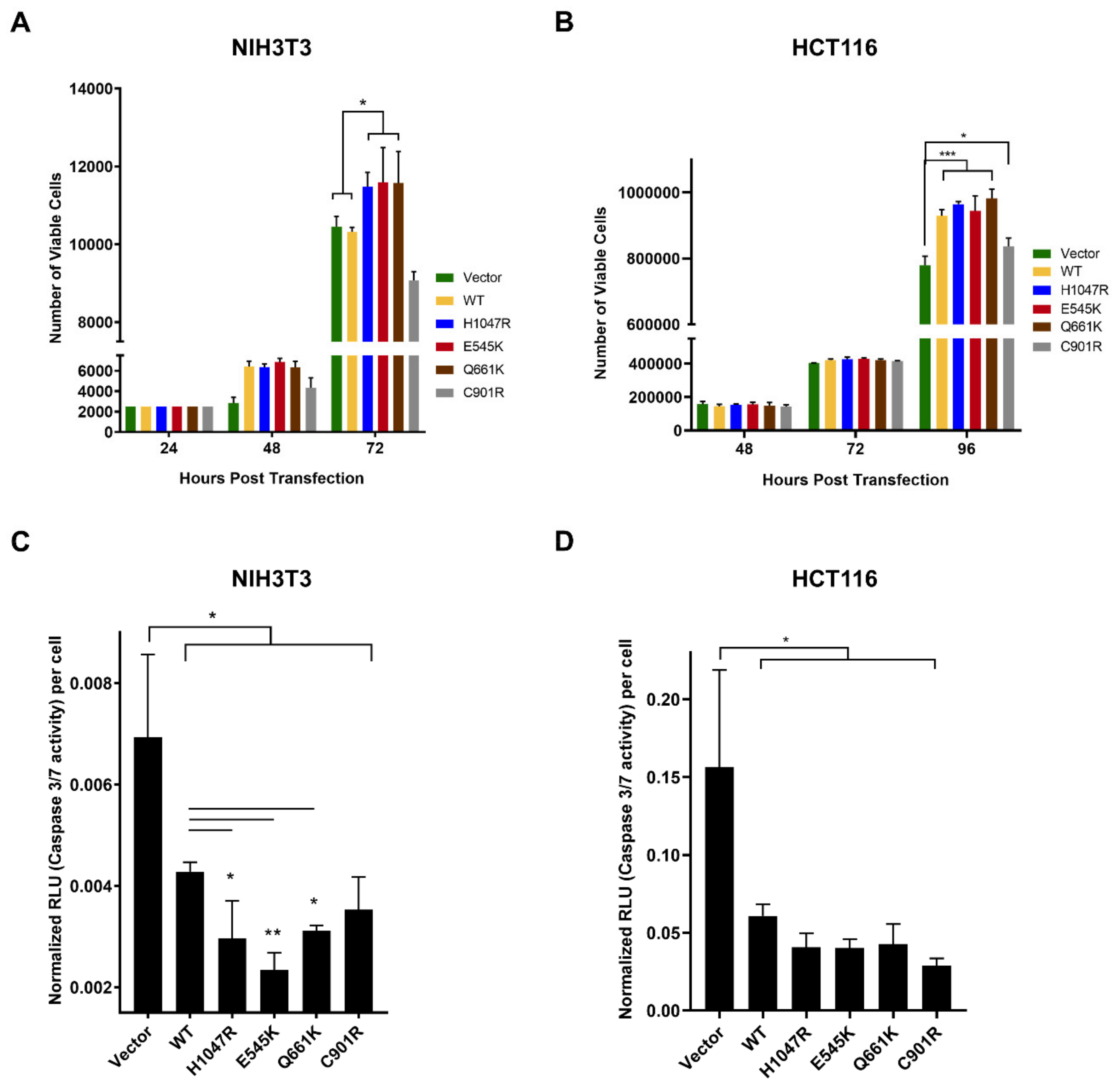
3.1. The PIK3CA Mutations Had Variable Effects on Proliferative Rates of NIH3T3 and HCT116 Cells
3.2. Variable Effects of the Canonical Mutants E545K and H1047R, and the Novel Mutants Q661K and C901R on Apoptosis Resistance in NIH3T3 and HCT116 Cells
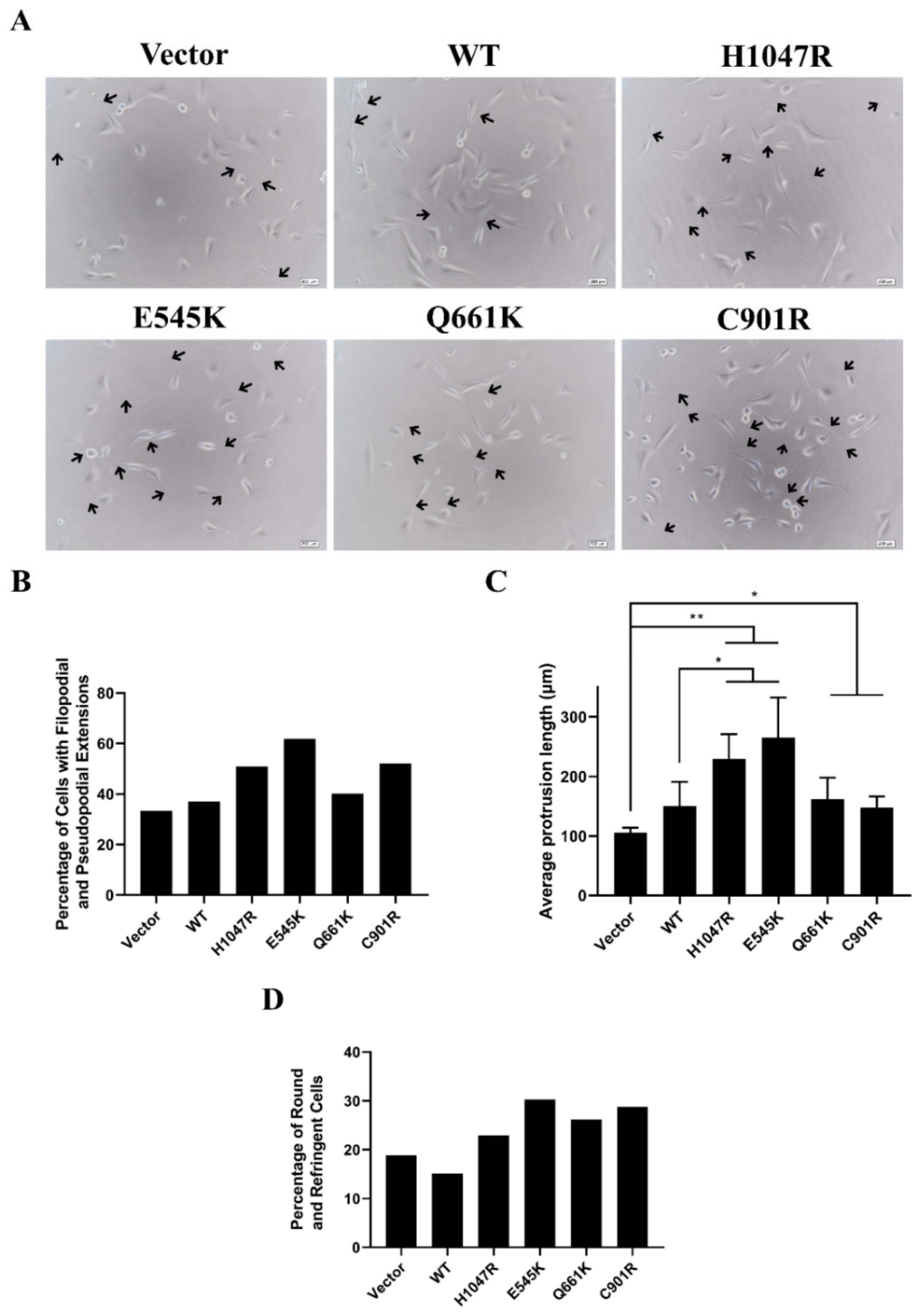
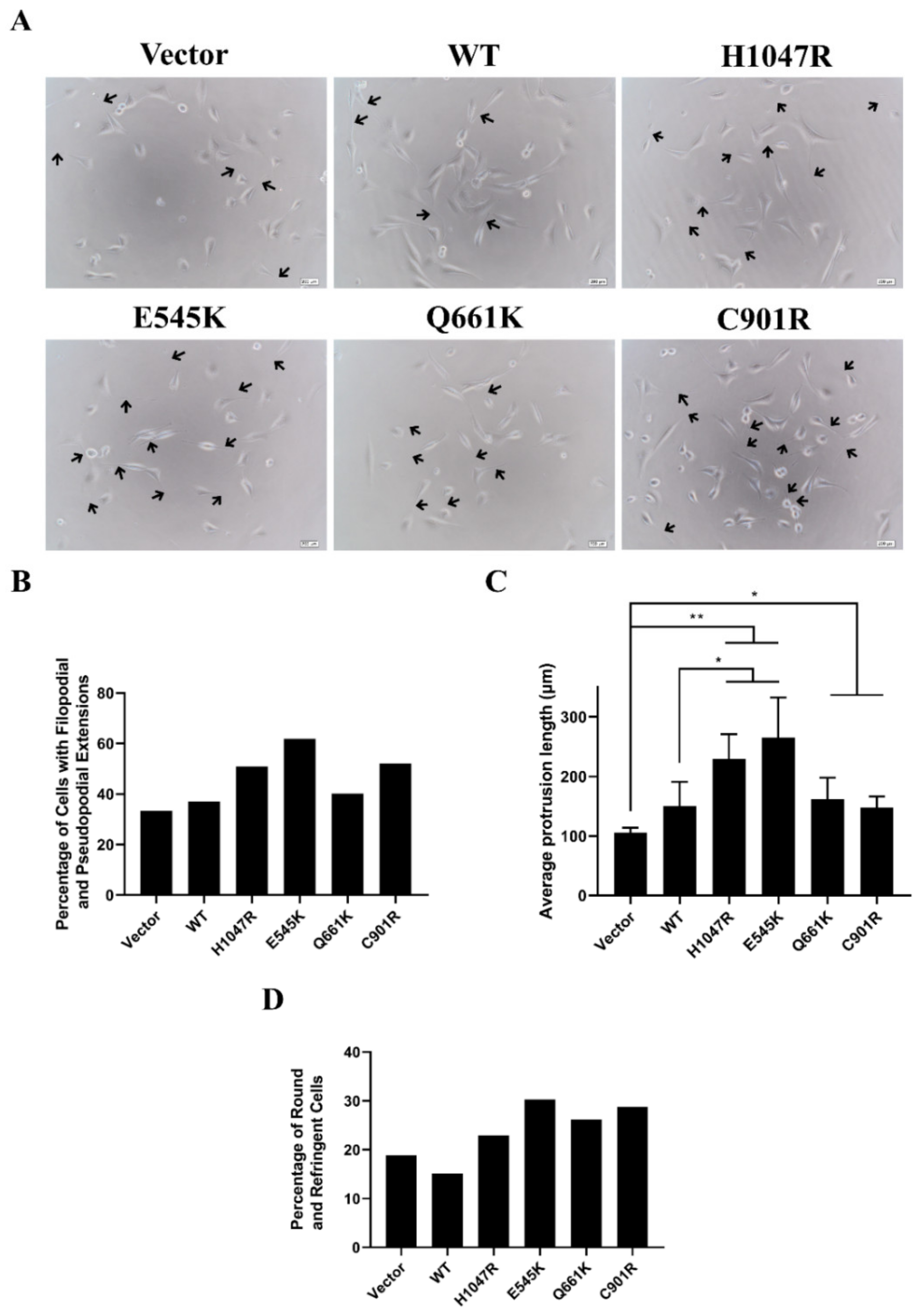
3.3. Novel and Canonical PIK3CA Mutants Induced Gross Morphological Alterations and Enhanced Formation of Pseudopodial Extensions
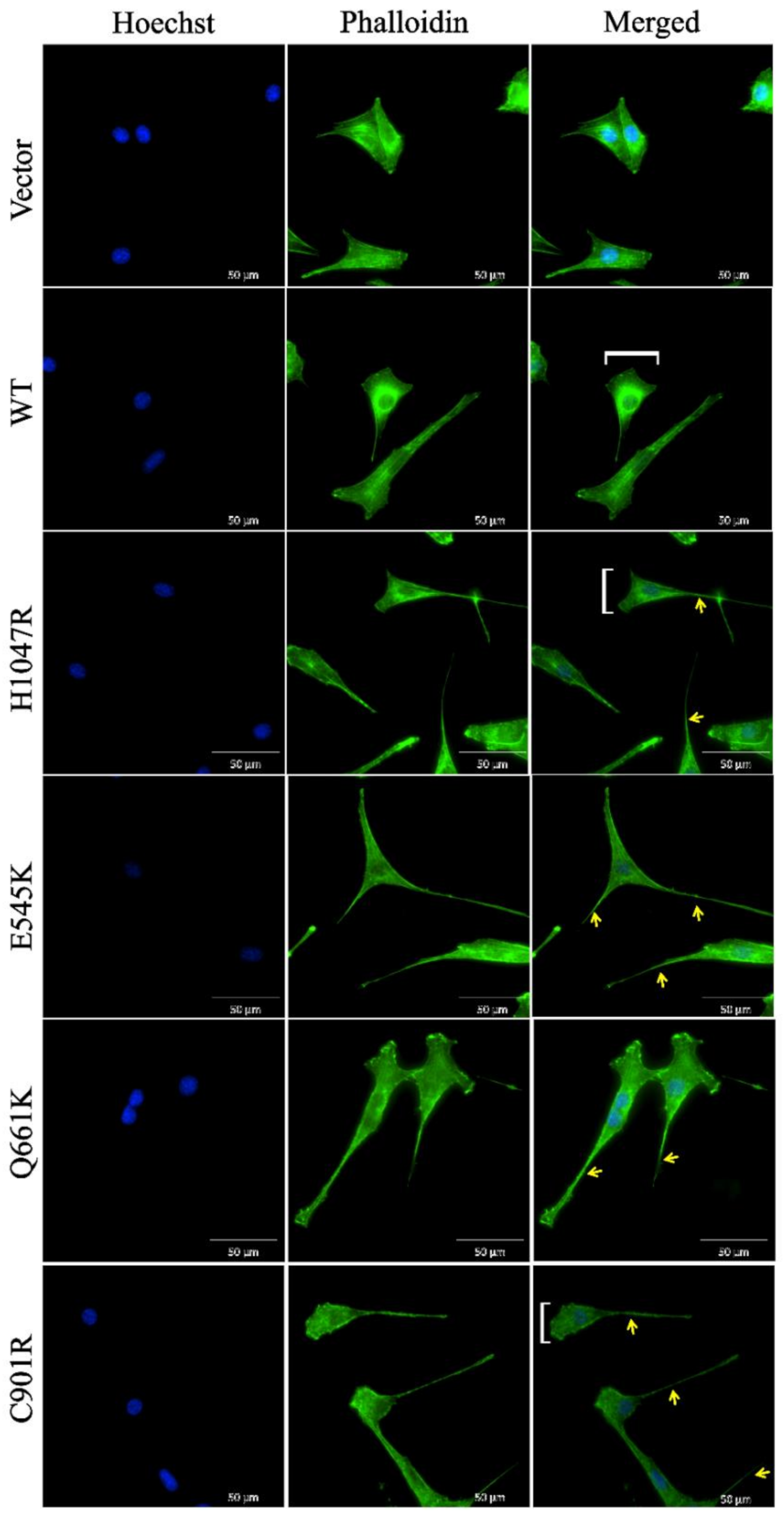
3.4. Cells Overexpressing Wild Type PIK3CA as well as Novel and Canonical PIK3CA Mutants Were Highly Depolarized with Long Cellular Protrusions
3.5. E545K, but not the Other PIK3CA Mutants, Enhanced Migration of NIH3T3 Cells
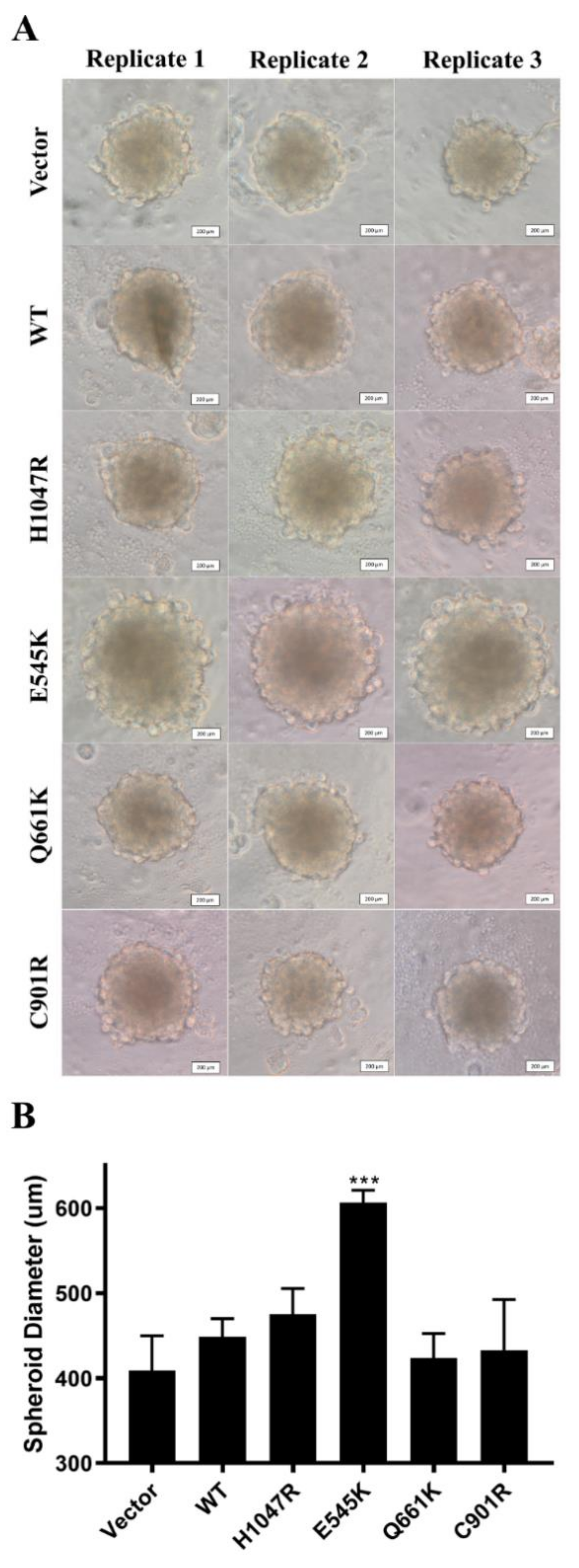
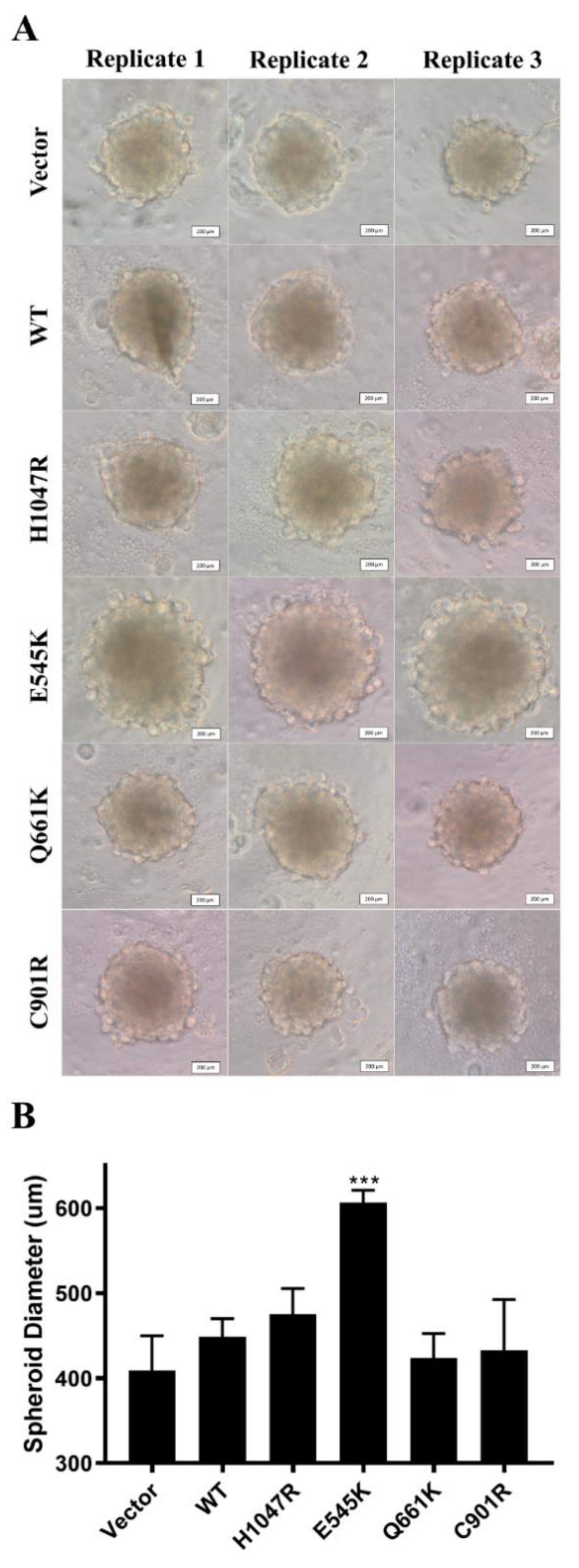
3.6. E545K Canonical Mutation Enhanced Spheroid Formation in NIH3T3 Cells
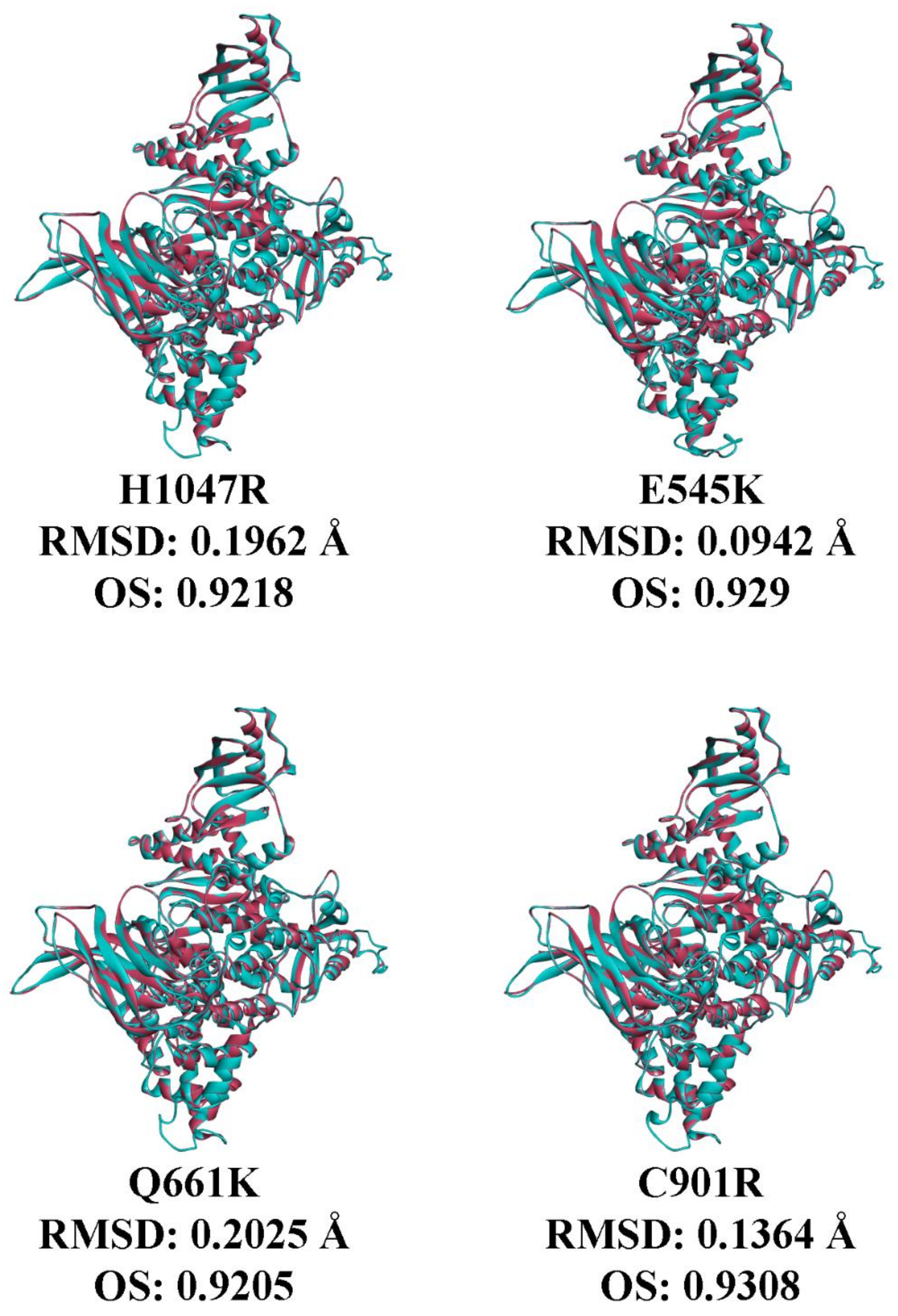
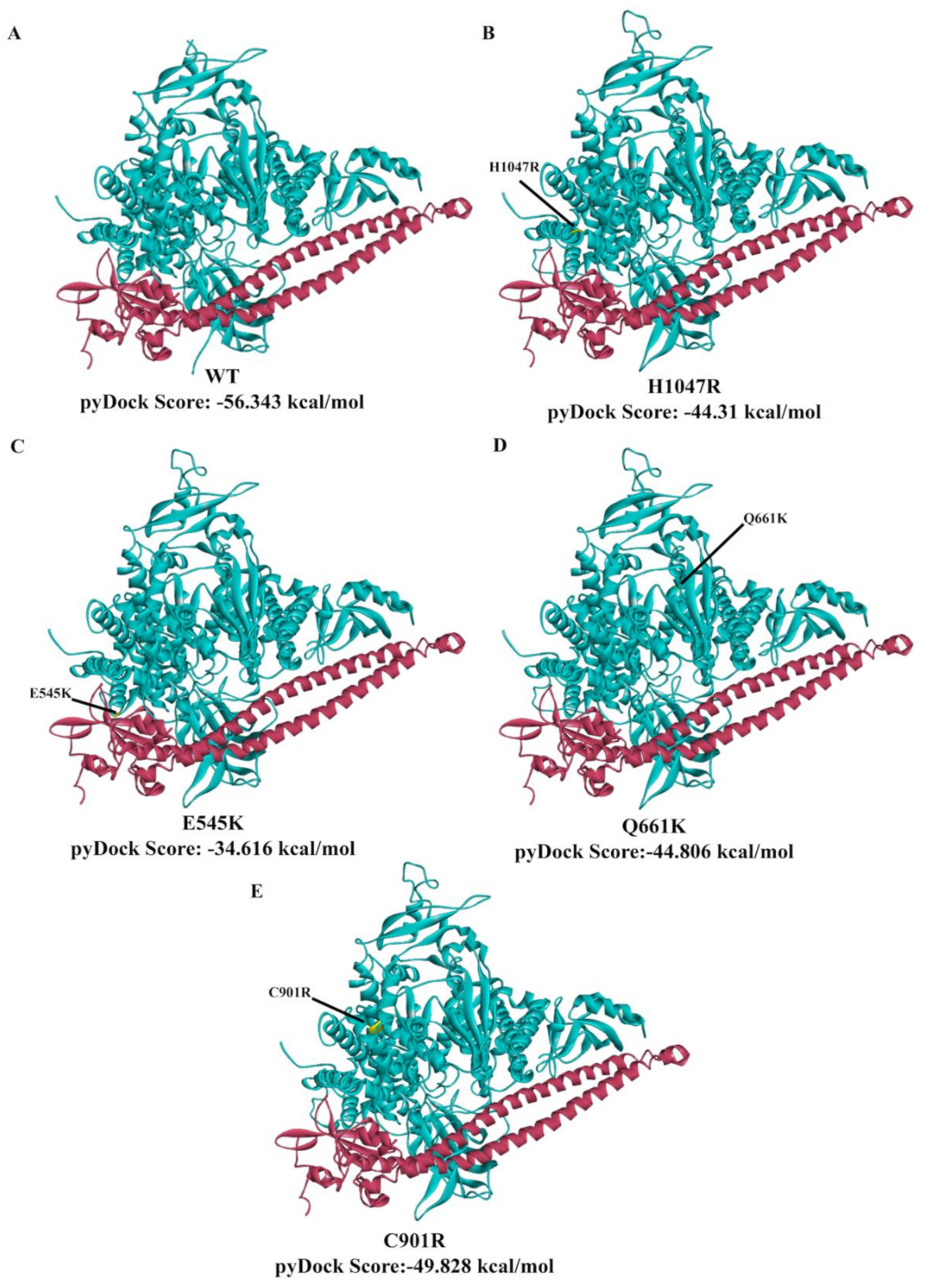
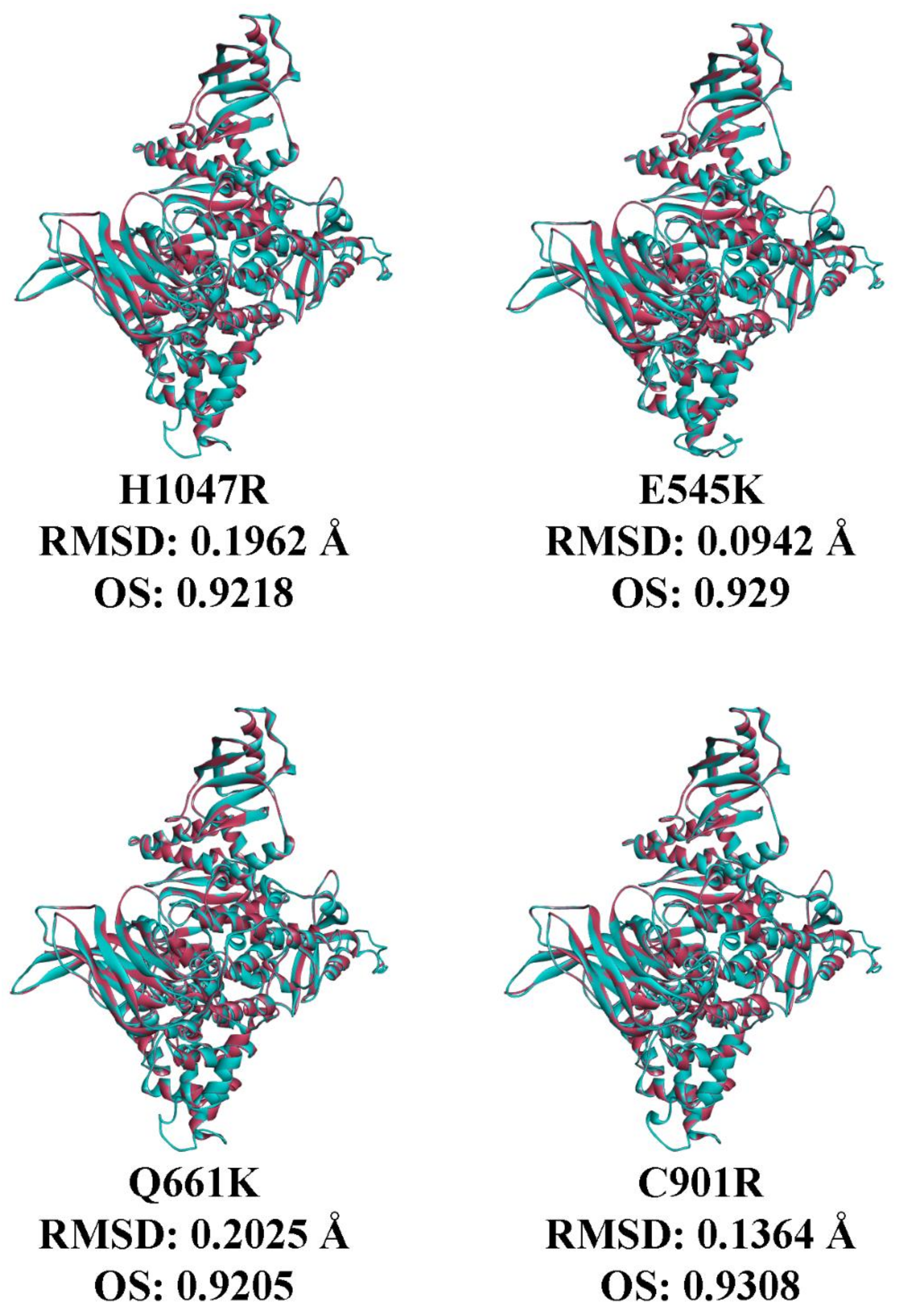
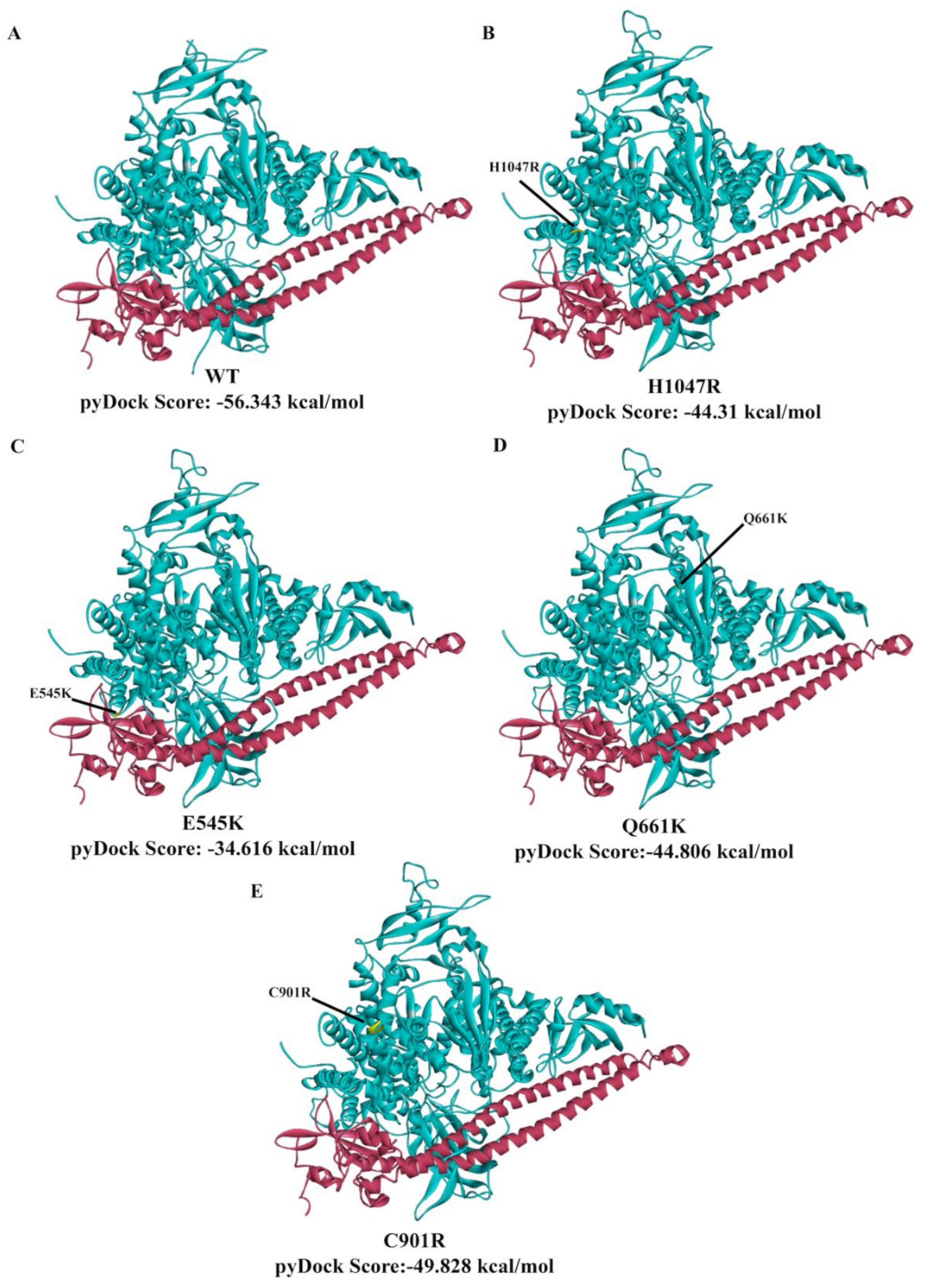
3.7. In Silico Prediction of the Impact of PIK3CA Mutations on Protein Structure and p85 Alpha-niSH2 Binding
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Herreros-Villanueva, M.; Gomez-Manero, N.; Muñiz, P.; García-Girón, C.; Coma del Corral, M.J. PIK3CA mutations in KRAS and BRAF wild type colorectal cancer patients. A study of Spanish population. Mol. Biol. Rep. 2011, 38, 1347–1351. [Google Scholar] [CrossRef] [PubMed]
- Perrone, F.; Lampis, A.; Orsenigo, M.; Di Bartolomeo, M.; Gevorgyan, A.; Losa, M.; Frattini, M.; Riva, C.; Andreola, S.; Bajetta, E.; et al. PI3KCA/PTEN deregulation contributes to impaired responses to cetuximab in metastatic colorectal cancer patients. Ann. Oncol. 2009, 20, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Nosho, K.; Kawasaki, T.; Longtine, J.A.; Fuchs, C.S.; Ohnishi, M.; Suemoto, Y.; Kirkner, G.J.; Zepf, D.; Yan, L.; Ogino, S. PIK3CA Mutation in Colorectal Cancer: Relationship with Genetic and Epigenetic Alterations. Neoplasia 2008, 10, 534–541. [Google Scholar] [CrossRef] [Green Version]
- Prenen, H.; De Schutter, J.; Jacobs, B.; De Roock, W.; Biesmans, B.; Claes, B.; Lambrechts, D.; Van Cutsem, E.; Tejpar, S. PIK3CA Mutations Are Not a Major Determinant of Resistance to the Epidermal Growth Factor Receptor Inhibitor Cetuximab in Metastatic Colorectal Cancer. Clin. Cancer Res. 2009, 15, 3184–3188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Roock, W.; Claes, B.; Bernasconi, D.; De Schutter, J.; Biesmans, B.; Fountzilas, G.; Kalogeras, K.T.; Kotoula, V.; Papamichael, D.; Laurent-Puig, P.; et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010, 11, 753–762. [Google Scholar] [CrossRef]
- Zhao, L.; Vogt, P.K. Helical domain and kinase domain mutations in p110 of phosphatidylinositol 3-kinase induce gain of function by different mechanisms. Proc. Natl. Acad. Sci. USA 2008, 105, 2652–2657. [Google Scholar] [CrossRef] [Green Version]
- Sartore-Bianchi, A.; Martini, M.; Molinari, F.; Veronese, S.; Nichelatti, M.; Artale, S.; Di Nicolantonio, F.; Saletti, P.; De Dosso, S.; Mazzucchelli, L.; et al. PIK3CA Mutations in Colorectal Cancer Are Associated with Clinical Resistance to EGFR-Targeted Monoclonal Antibodies. Cancer Res. 2009, 69, 1851–1857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.-Y.; Wu, X.-Y.; Huang, Y.-F.; Di, M.-Y.; Zheng, D.-Y.; Chen, J.-Z.; Ding, H.; Mao, C.; Tang, J.-L. Promising biomarkers for predicting the outcomes of patients with KRAS wild-type metastatic colorectal cancer treated with anti-epidermal growth factor receptor monoclonal antibodies: A systematic review with meta-analysis. Int. J. Cancer 2013, 133, 1914–1925. [Google Scholar] [CrossRef]
- Catasus, L.; Gallardo, A.; Cuatrecasas, M.; Prat, J. PIK3CA mutations in the kinase domain (exon 20) of uterine endometrial adenocarcinomas are associated with adverse prognostic parameters. Mod. Pathol. 2008, 21, 131–139. [Google Scholar] [CrossRef]
- Pang, H.; Flinn, R.; Patsialou, A.; Wyckoff, J.; Roussos, E.T.; Wu, H.; Pozzuto, M.; Goswami, S.; Condeelis, J.S.; Bresnick, A.R.; et al. Differential Enhancement of Breast Cancer Cell Motility and Metastasis by Helical and Kinase Domain Mutations of Class IA Phosphoinositide 3-Kinase. Cancer Res. 2009, 69, 8868–8876. [Google Scholar] [CrossRef] [Green Version]
- Ross, R.L.; Askham, J.M.; Knowles, M.A. PIK3CA mutation spectrum in urothelial carcinoma reflects cell context-dependent signaling and phenotypic outputs. Oncogene 2013, 32, 768–776. [Google Scholar] [CrossRef] [Green Version]
- Meyer, D.S.; Koren, S.; Leroy, C.; Brinkhaus, H.; Müller, U.; Klebba, I.; Müller, M.; Cardiff, R.D.; Bentires-Alj, M. Expression of PIK3CA mutant E545K in the mammary gland induces heterogeneous tumors but is less potent than mutant H1047R. Oncogenesis 2013, 2, e74. [Google Scholar] [CrossRef]
- Hanna, M.C.; Go, C.; Roden, C.; Jones, R.T.; Pochanard, P.; Javed, A.Y.; Javed, A.; Mondal, C.; Palescandolo, E.; Van Hummelen, P.; et al. Colorectal Cancers from Distinct Ancestral Populations Show Variations in BRAF Mutation Frequency. PLoS ONE 2013, 8, e74950. [Google Scholar] [CrossRef]
- Zulhabri, O.; Rahman, J.; Ismail, S.; Isa, M.R.; Wan Zurinah, W.N. Predominance of G to A codon 12 mutation K-ras gene in Dukes’ B colorectal cancer. Singapore Med. J. 2012, 53, 26–31. [Google Scholar] [PubMed]
- Tong, J.H.; Lung, R.W.; Sin, F.M.; Law, P.P.; Kang, W.; Chan, A.W.; Ma, B.B.; Mak, T.W.; Ng, S.S.; To, K.F. Characterization of rare transforming KRAS mutations in sporadic colorectal cancer. Cancer Biol. Ther. 2014, 15, 768–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudd, M.L.; Price, J.C.; Fogoros, S.; Godwin, A.K.; Sgroi, D.C.; Merino, M.J.; Bell, D.W. A Unique Spectrum of Somatic PIK3CA (p110α) Mutations Within Primary Endometrial Carcinomas. Clin. Cancer Res. 2011, 17, 1331–1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Yang, L.; Yao, L.; Kuang, X.-Y.; Zuo, W.-J.; Li, S.; Qiao, F.; Liu, Y.-R.; Cao, Z.-G.; Zhou, S.-L.; et al. Characterization of PIK3CA and PIK3R1 somatic mutations in Chinese breast cancer patients. Nat. Commun. 2018, 9, 1357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikenoue, T.; Kanai, F.; Hikiba, Y.; Obata, T.; Tanaka, Y.; Imamura, J.; Ohta, M.; Jazag, A.; Guleng, B.; Tateishi, K.; et al. Functional Analysis of PIK3CA Gene Mutations in Human Colorectal Cancer. Cancer Res. 2005, 65, 4562–4567. [Google Scholar] [CrossRef] [Green Version]
- Gymnopoulos, M.; Elsliger, M.-A.; Vogt, P.K. Rare cancer-specific mutations in PIK3CA show gain of function. Proc. Natl. Acad. Sci USA. 2007, 104, 5569–5574. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Liu, G.; Dziubinski, M.; Yang, Z.; Ethier, S.P.; Wu, G. Comprehensive analysis of oncogenic effects of PIK3CA mutations in human mammary epithelial cells. Breast Cancer Res. Treat. 2008, 112, 217–227. [Google Scholar] [CrossRef]
- Croessmann, S.; Sheehan, J.H.; Lee, K.; Sliwoski, G.; He, J.; Nagy, R.; Riddle, D.; Mayer, I.A.; Balko, J.M.; Lanman, R.; et al. PIK3CA C2 Domain Deletions Hyperactivate Phosphoinositide 3-kinase (PI3K), Generate Oncogene Dependence, and Are Exquisitely Sensitive to PI3K α Inhibitors. Clin. Cancer Res. 2018, 24, 1426–1435. [Google Scholar] [CrossRef] [Green Version]
- Vasudevan, K.M.; Barbie, D.A.; Davies, M.A.; Rabinovsky, R.; McNear, C.J.; Kim, J.J.; Hennessy, B.T.; Tseng, H.; Pochanard, P.; Kim, S.Y.; et al. AKT-Independent Signaling Downstream of Oncogenic PIK3CA Mutations in Human Cancer. Cancer Cell 2009, 16, 21–32. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Juvekar, A.; Lyssiotis, C.A.; Lien, E.C.; Albeck, J.G.; Oh, D.; Varma, G.; Hung, Y.P.; Ullas, S.; Lauring, J.; et al. Phosphoinositide 3-Kinase Regulates Glycolysis through Mobilization of Aldolase from the Actin Cytoskeleton. Cell 2016, 164, 433–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juvekar, A.; Hu, H.; Yadegarynia, S.; Lyssiotis, C.A.; Ullas, S.; Lien, E.C.; Bellinger, G.; Son, J.; Hok, R.C.; Seth, P.; et al. Phosphoinositide 3-kinase inhibitors induce DNA damage through nucleoside depletion. Proc. Natl. Acad. Sci. USA 2016, 113, E4338–E4347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lien, E.C.; Dibble, C.C.; Toker, A. PI3K signaling in cancer: Beyond AKT. Curr. Opin. Cell Biol. 2017, 45, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Samuels, Y.; Wang, Z.; Bardelli, A.; Silliman, N.; Ptak, J.; Szabo, S.; Yan, H.; Gazdar, A.; Powell, S.M.; Riggins, G.J.; et al. High Frequency of Mutations of the PIK3CA Gene in Human Cancers. Science 2004, 304, 554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [Green Version]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Wang, B.; Xu, J.; Wang, X.; Xie, L.; Zhang, B.; Li, Y.; Li, J. CanProVar 2.0: An Updated Database of Human Cancer Proteome Variation. J. Proteome Res. 2017, 16, 421–432. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Kong, L.; Liu, Y.; Qu, H. dbEMT: An epithelial-mesenchymal transition associated gene resource. Sci. Rep. 2015, 5, 11459. [Google Scholar] [CrossRef]
- Sacdalan, D.L.; Uy, C.J.; Cutiongco-de la Paz, E.M.; Garcia, R.L. Next-Generation Sequencing reveals putative novel non-hotspot mutations in EGFR pathway genes in Filipino young-onset sporadic colorectal cancer patients. unpublished.
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open source platform for biological image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 2011, 39, e118. [Google Scholar] [CrossRef] [Green Version]
- Shihab, H.A.; Gough, J.; Cooper, D.N.; Stenson, P.D.; Barker, G.L.A.; Edwards, K.J.; Day, I.N.M.; Gaunt, T.R. Predicting the Functional, Molecular, and Phenotypic Consequences of Amino Acid Substitutions using Hidden Markov Models. Hum. Mutat. 2013, 34, 57–65. [Google Scholar] [CrossRef]
- Sim, N.-L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Zhang, X.; Chen, Y.; Lu, S.; Peng, Y.; Wang, X.; Guo, C.; Zhou, A.; Zhang, J.; Luo, Y.; et al. Crystal Structures of PI3Kα Complexed with PI103 and Its Derivatives: New Directions for Inhibitors Design. ACS Med. Chem. Lett. 2014, 5, 138–142. [Google Scholar] [CrossRef] [Green Version]
- Jiménez-García, B.; Pons, C.; Fernández-Recio, J. pyDockWEB: A web server for rigid-body protein–protein docking using electrostatics and desolvation scoring. Bioinformatics 2013, 29, 1698–1699. [Google Scholar] [CrossRef] [Green Version]
- Cheng, T.M.-K.; Blundell, T.L.; Fernandez-Recio, J. pyDock: Electrostatics and desolvation for effective scoring of rigid-body protein-protein docking. Proteins Struct. Funct. Bioinforma. 2007, 68, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Krissinel, E.; Henrick, K. Inference of Macromolecular Assemblies from Crystalline State. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef] [PubMed]
- Chalhoub, N.; Baker, S.J. PTEN and the PI3-Kinase Pathway in Cancer. Annu. Rev. Pathol. Mech. Dis. 2009, 4, 127–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelman, J.A.; Luo, J.; Cantley, L.C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 2006, 7, 606–619. [Google Scholar] [CrossRef]
- Petty, E.M. Cellular cancer markers. Am. J. Med. Genet. 1997, 68, 492–493. [Google Scholar] [CrossRef]
- Guerrero, S.; Casanova, I.; Farré, L.; Mazo, A.; Capellà, G.; Mangues, R. K-ras Codon 12 Mutation Induces Higher Level of Resistance to Apoptosis and Predisposition to Anchorage-independent Growth Than Codon 13 Mutation or Proto-Oncogene Overexpression. Cancer Res. 2000, 60, 6750–6756. [Google Scholar]
- Pawlak, G.; Helfman, D.M. Cytoskeletal changes in cell transformation and tumorigenesis. Curr. Opin. Genet. Dev. 2001, 11, 41–47. [Google Scholar] [CrossRef]
- Alcantara, K.M.; Garcia, R. MicroRNA-92a promotes cell proliferation, migration and survival by directly targeting the tumor suppressor gene NF2 in colorectal and lung cancer cells. Oncol. Rep. 2019, 41, 2103–2116. [Google Scholar] [CrossRef] [Green Version]
- Alcantara, K.M.M.; Malapit, J.R.P.; Yu, R.T.D.; Garrido, J.A.M.G.; Rigor, J.P.T.; Angeles, A.K.J.; Cutiongco-de la Paz, E.M.; Garcia, R.L. Non-Redundant and Overlapping Oncogenic Readouts of Non-Canonical and Novel Colorectal Cancer KRAS and NRAS Mutants. Cells 2019, 8, 1557. [Google Scholar] [CrossRef] [Green Version]
- Nakhaeizadeh, H.; Amin, E.; Nakhaei-Rad, S.; Dvorsky, R.; Ahmadian, M.R. The RAS-Effector Interface: Isoform-Specific Differences in the Effector Binding Regions. PLoS ONE 2016, 11, e0167145. [Google Scholar] [CrossRef]
- Lauffenburger, D.A.; Horwitz, A.F. Cell migration: A physically integrated molecular process. Cell 1996, 84, 359–369. [Google Scholar] [CrossRef] [Green Version]
- Thiery, J.P. Epithelial–mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Pećina-Šlaus, N. Tumor suppressor gene E-cadherin and its role in normal and malignant cells. Cancer Cell Int. 2003, 3, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Devreotes, P.; Horwitz, A.R. Signaling Networks that Regulate Cell Migration. Cold Spring Harb. Perspect. Biol. 2015, 7, a005959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7.20.1–7.20.41. [Google Scholar] [CrossRef] [Green Version]
- Angeles, A.; Yu, R.; Cutiongco-De la Paz, E.; Garcia, R. Phenotypic characterization of the novel, non-hotspot oncogenic KRAS mutants E31D and E63K. Oncol. Lett. 2019. [Google Scholar] [CrossRef] [Green Version]
- Yap, T.A.; Gerlinger, M.; Futreal, P.A.; Pusztai, L.; Swanton, C. Intratumor Heterogeneity: Seeing the Wood for the Trees. Sci. Transl. Med. 2012, 4, 127ps10. [Google Scholar] [CrossRef] [Green Version]
- McGranahan, N.; Swanton, C. Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell 2015, 27, 15–26. [Google Scholar] [CrossRef] [Green Version]
- Nisa, L.; Häfliger, P.; Poliaková, M.; Giger, R.; Francica, P.; Aebersold, D.M.; Charles, R.-P.; Zimmer, Y.; Medová, M. PIK3CA hotspot mutations differentially impact responses to MET targeting in MET-driven and non-driven preclinical cancer models. Mol. Cancer 2017, 16, 93. [Google Scholar] [CrossRef]
- Miled, N.; Yan, Y.; Hon, W.-C.; Perisic, O.; Zvelebil, M.; Inbar, Y.; Schneidman-Duhovny, D.; Wolfson, H.J.; Backer, J.M.; Williams, R.L. Mechanism of Two Classes of Cancer Mutations in the Phosphoinositide 3-Kinase Catalytic Subunit. Science (80) 2007, 317, 239–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bader, A.G.; Kang, S.; Zhao, L.; Vogt, P.K. Oncogenic PI3K deregulates transcription and translation. Nat. Rev. Cancer 2005, 5, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Campbell, I.G.; Russell, S.E.; Choong, D.Y.H.; Montgomery, K.G.; Ciavarella, M.L.; Hooi, C.S.F.; Cristiano, B.E.; Pearson, R.B.; Phillips, W.A. Mutation of the PIK3CA Gene in Ovarian and Breast Cancer. Cancer Res. 2004, 64, 7678–7681. [Google Scholar] [CrossRef] [Green Version]
- Bachman, K.E.; Argani, P.; Samuels, Y.; Silliman, N.; Ptak, J.; Szabo, S.; Konishi, H.; Karakas, B.; Blair, B.G.; Lin, C.; et al. The PIK3CA gene is mutated with high frequency in human breast cancers. Cancer Biol. Ther. 2004, 3, 772–775. [Google Scholar] [CrossRef] [Green Version]
- Levine, D.A. Frequent Mutation of the PIK3CA Gene in Ovarian and Breast Cancers. Clin. Cancer Res. 2005, 11, 2875–2878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saal, L.H.; Holm, K.; Maurer, M.; Memeo, L.; Su, T.; Wang, X.; Yu, J.S.; Malmström, P.-O.; Mansukhani, M.; Enoksson, J.; et al. PIK3CA Mutations Correlate with Hormone Receptors, Node Metastasis, and ERBB2, and Are Mutually Exclusive with PTEN Loss in Human Breast Carcinoma. Cancer Res. 2005, 65, 2554–2559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broderick, D.K.; Di, C.; Parrett, T.J.; Samuels, Y.R.; Cummins, J.M.; McLendon, R.E.; Fults, D.W.; Velculescu, V.E.; Bigner, D.D.; Yan, H. Mutations of PIK3CA in Anaplastic Oligodendrogliomas, High-Grade Astrocytomas, and Medulloblastomas. Cancer Res. 2004, 64, 5048–5050. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, C.; Bartels, G.; Gehlhaar, C.; Holtkamp, N.; von Deimling, A. PIK3CA mutations in glioblastoma multiforme. Acta Neuropathol. 2005, 109, 639–642. [Google Scholar] [CrossRef]
- Lee, J.W.; Soung, Y.H.; Kim, S.Y.; Lee, H.W.; Park, W.S.; Nam, S.W.; Kim, S.H.; Lee, J.Y.; Yoo, N.J.; Lee, S.H. PIK3CA gene is frequently mutated in breast carcinomas and hepatocellular carcinomas. Oncogene 2005, 24, 1477–1480. [Google Scholar] [CrossRef] [Green Version]
- Li, V.S.W.; Wong, C.W.; Chan, T.L.; Chan, A.S.W.; Zhao, W.; Chu, K.-M.; So, S.; Chen, X.; Yuen, S.T.; Leung, S.Y. Mutations of PIK3CAin gastric adenocarcinoma. BMC Cancer 2005, 5, 29. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Helland, Å.; Holm, R.; Kristensen, G.B.; Børresen-Dale, A.-L. PIK3CA mutations in advanced ovarian carcinomas. Hum. Mutat. 2005, 25, 322. [Google Scholar] [CrossRef] [PubMed]
- Stephens, B. The Mutational Spectra of Cancer Genes in TCGA Data. Available online: https://www.cancer.gov/research/key-initiatives/ras/ras-central/blog/2017/cancer-mutation-spectra?fbclid=IwAR3MFtXSJ9BlFJtCtEr01p0-CGhaaKKeK8wzC6ZXkTMiyxCNnpkDaQQoBXs (accessed on 21 April 2020).
- Parsons, J.T.; Horwitz, A.R.; Schwartz, M.A. Cell adhesion: Integrating cytoskeletal dynamics and cellular tension. Nat. Rev. Mol. Cell Biol. 2010, 11, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Mandelker, D.; Gabelli, S.B.; Schmidt-Kittler, O.; Zhu, J.; Cheong, I.; Huang, C.-H.; Kinzler, K.W.; Vogelstein, B.; Amzel, L.M. A frequent kinase domain mutation that changes the interaction between PI3K and the membrane. Proc. Natl. Acad. Sci. USA 2009, 106, 16996–17001. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Sequence |
|---|---|
| WT-PIK3CA-F | 5′-ATGCCTCCACGACCATCATCAGGTGAACTG-3′ |
| WT-PIK3CA-R | 5′-TCAGTTCAATGCATGCTGTTTAATTGTGTGGAAG-3′ |
| E545K-F | 5′CTCTCTCTGAAATCACTAAGCAGGAGAAAGATTTTCTATG-3′ |
| E545K-R | 5′-CATAGAAAATCTTTCTCCTGCTTAGTGATTTCAGAGAGAG-3′ |
| H1047R-R | 5′-GTGACTACTAGTTCAGTTCAATGCATGCTGTTTAATTGTGTGGA AGATCCAATCCATTTTTGTTGTCCAGCCACCATGACGTGCATC-3′ |
| Q661K-F | 5′-GAAAGCATTGACTAATAAAAGGATTGGGCACTTTTTCTTTTG-3′ |
| Q661K-R | 5′-GAAAAAGTGCCCAATCCTTTTATTAGTCAATGCTTTCTTC-3′ |
| C901R-F | 5′-CCTGTTTACACGTTCACGTGCTGGATACTGTGTAGCTACC-3′ |
| C901R-R | 5′-GGTAGCTACACAGTATCCAGCACGTGAACGTGTAAACAGG-3′ |
| Mutation | Polyphen-2 | Mutation Assessor | FATHMM | SIFT |
|---|---|---|---|---|
| Q661K a | Benign | Neutral | Cancer | Tolerated |
| C901R b | Probably damaging c | High | Cancer | Affect protein function |
| Buried area, Å2 | No. of H-Bonds | No. of Salt Bridges | No. of Interfacing Residues in PIK3CA | % | No. of Interfacing Residues in p85 Alpha | % | |
|---|---|---|---|---|---|---|---|
| WT | 3756.4 | 40 | 31 | 106 | 10.70% | 106 | 38.30% |
| E545K | 3673.5 | 42 | 27 | 102 | 9.70% | 105 | 37.90% |
| H1047R | 4107.8 | 48 | 27 | 119 | 11.30% | 115 | 41.50% |
| Q661K | 4097.6 | 46 | 26 | 120 | 11.40% | 116 | 41.90% |
| C901R | 4078.4 | 44 | 28 | 114 | 10.80% | 115 | 41.50% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghodsinia, A.A.; Lego, J.-A.M.T.; Garcia, R.L. Mutation-Associated Phenotypic Heterogeneity in Novel and Canonical PIK3CA Helical and Kinase Domain Mutants. Cells 2020, 9, 1116. https://doi.org/10.3390/cells9051116
Ghodsinia AA, Lego J-AMT, Garcia RL. Mutation-Associated Phenotypic Heterogeneity in Novel and Canonical PIK3CA Helical and Kinase Domain Mutants. Cells. 2020; 9(5):1116. https://doi.org/10.3390/cells9051116
Chicago/Turabian StyleGhodsinia, Arman Ali, J-Ann Marie T. Lego, and Reynaldo L. Garcia. 2020. "Mutation-Associated Phenotypic Heterogeneity in Novel and Canonical PIK3CA Helical and Kinase Domain Mutants" Cells 9, no. 5: 1116. https://doi.org/10.3390/cells9051116
APA StyleGhodsinia, A. A., Lego, J.-A. M. T., & Garcia, R. L. (2020). Mutation-Associated Phenotypic Heterogeneity in Novel and Canonical PIK3CA Helical and Kinase Domain Mutants. Cells, 9(5), 1116. https://doi.org/10.3390/cells9051116