Targeting GSK3 and Associated Signaling Pathways Involved in Cancer

 , ,
, ,  ,
, 
 , ,
, ,  ,
,  , and
, and
Abstract
1. Introduction
1.1. The GSK-3 Family Consists of GSK-3α and GSK-3β
1.2. Expression of the GSK-3 Isoforms in Different Human Tissues
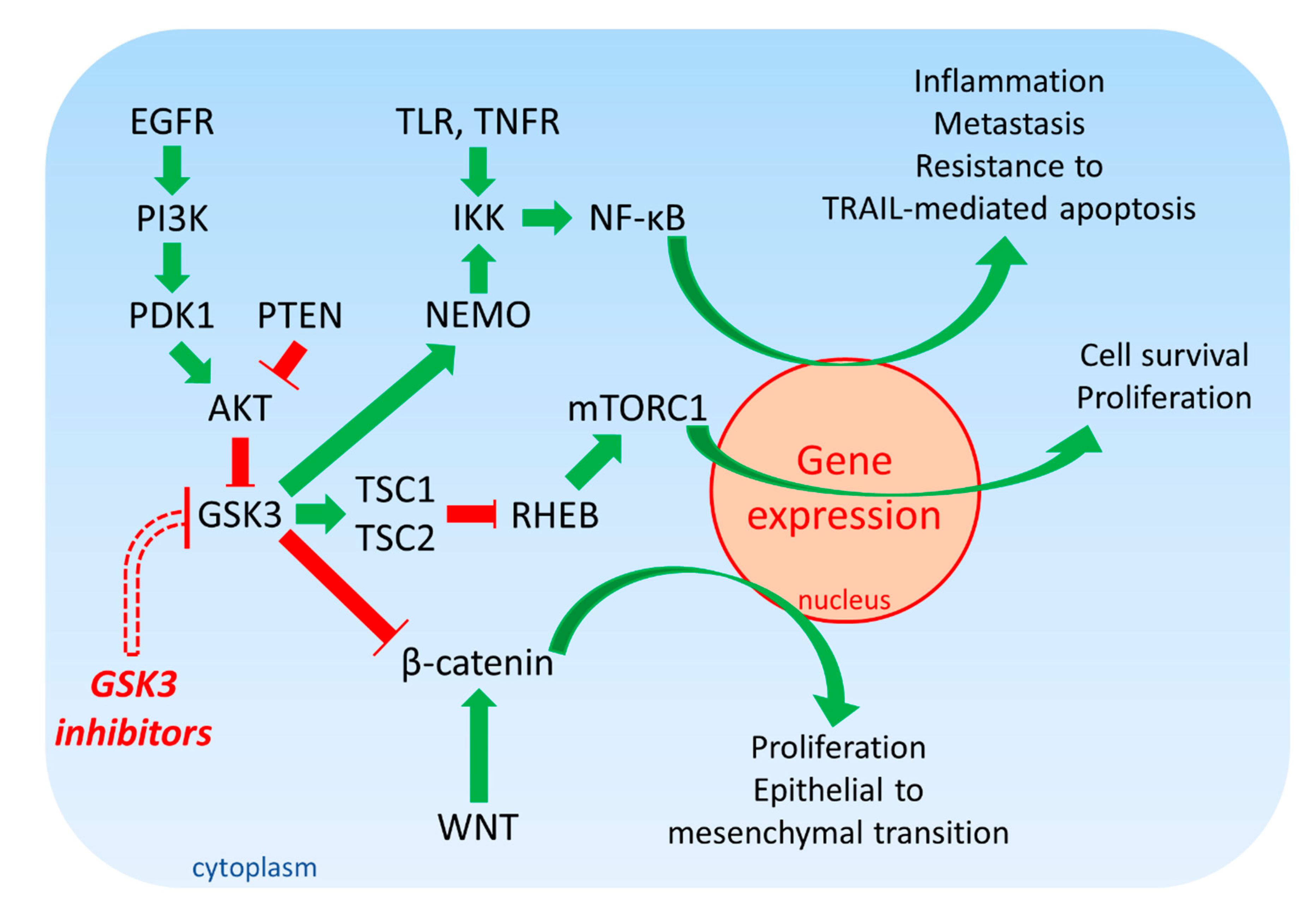
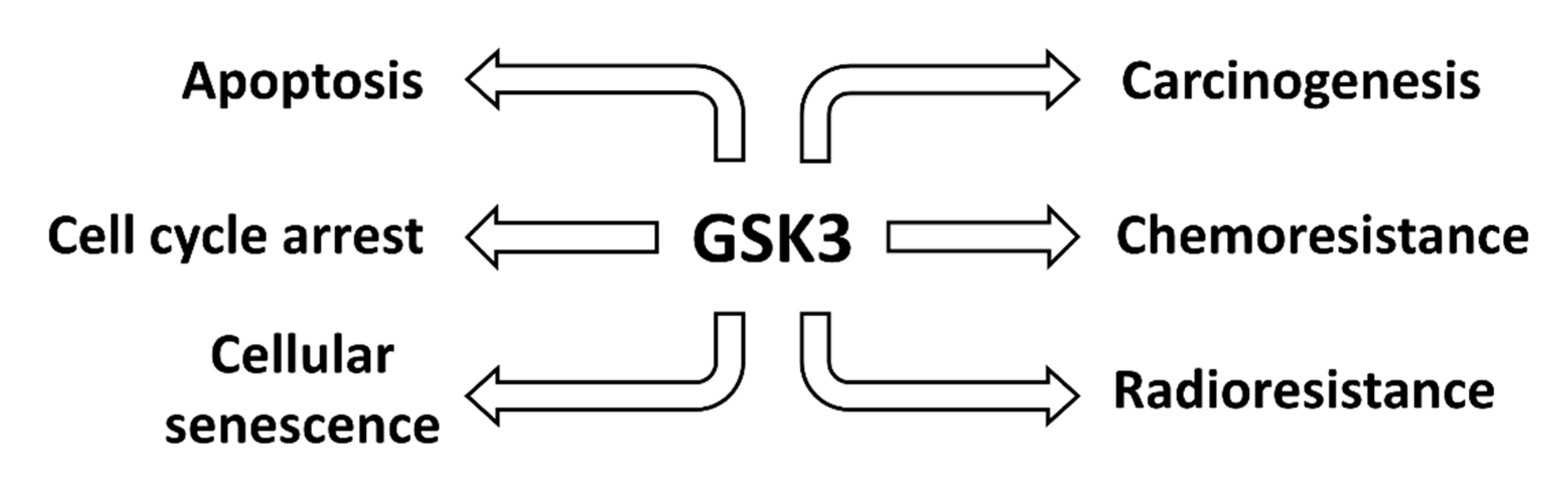
1.3. Biochemical Roles of GSK-3 in Cancer
1.4. Examples of Studies Documenting the Roles GSK-3 Isoforms in Human Cancer
2. GSK-3 Inhibitors
2.1. Combining GSK-3 Inhibitors with Chemotherapeutics, Radiation or Autophagy Inhibitors
2.2. Combining GSK-3 Inhibitors and Immunotherapy
2.3. Combining GSK-3 Inhibitors with Other Inhibitors or Agonists
2.4. Effects of GSK-3 Inhibitors on Drug Resistance in Cancer Cells
2.5. Other Inhibitors Which Also Influence GSK-3 Activity
2.6. Small Molecule Inhibitors and miRrs Which Interact to Regulate GSK-3 Activity in Drug Resistance
3. Natural Products Which Modify GSK-3 Activity
4. GSK-3 Inhibitors in Cancer Clinical Trials
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kockeritz, L.; Doble, B.; Patel, S.; Woodgett, J.R. Glycogen synthase kinase-3-an overview of an over-achieving protein kinase. Curr. Drug Targets 2006, 7, 1377–1388. [Google Scholar] [CrossRef]
- Sutherland, C. What Are the bona fide GSK3 Substrates? Int. J. Alzheimer’s Dis. 2011, 2011, 505607. [Google Scholar]
- McCubrey, J.A.; Rakus, D.; Gizak, A.; Steelman, L.S.; Abrams, S.L.; Lertpiriyapong, K.; Fitzgerald, T.L.; Yang, L.V.; Montalto, G.; Cervello, M.; et al. Effects of mutations in Wnt/β-catenin, hedgehog, Notch and PI3K pathways on GSK-3 activity-Diverse effects on cell growth, metabolism and cancer. Biochim. Biophys. Acta 2016, 1863, 2942–2976. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Fitzgerald, T.L.; Yang, L.V.; Lertpiriyapong, K.; Steelman, L.S.; Abrams, S.L.; Montalto, G.; Cervello, M.; Neri, L.M.; Cocco, L.; et al. Roles of GSK-3 and microRNAs on epithelial mesenchymal transition and cancer stem cells. Oncotarget 2017, 8, 14221–14250. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Franklin, R.A.; Montalto, G.; Cervello, M.; Libra, M.; Candido, S.; Malaponte, G.; et al. Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascade inhibitors: How mutations can result in therapy resistance and how to overcome resistance. Oncotarget 2012, 3, 1068–1111. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Montalto, G.; Cervello, M.; Nicoletti, F.; Fagone, P.; Malaponte, G.; Mazzarino, M.C.; et al. Mutations and deregulation of Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascades which alter therapy response. Oncotarget 2012, 3, 954–987. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Steelman, L.S.; Bertrand, F.E.; Davis, N.M.; Sokolosky, M.; Abrams, S.L.; Montalto, G.; D’Assoro, A.B.; Libra, M.; Nicoletti, F.; et al. GSK-3 as potential target for therapeutic intervention in cancer. Oncotarget 2014, 5, 2881–2911. [Google Scholar] [CrossRef]
- Davis, N.M.; Sokolosky, M.; Stadelman, K.; Abrams, S.L.; Libra, M.; Candido, S.; Nicoletti, F.; Polesel, J.; Maestro, R.; D’Assoro, A.; et al. Deregulation of the EGFR/PI3K/PTEN/Akt/mTORC1 pathway in breast cancer: Possibilities for therapeutic intervention. Oncotarget 2014, 5, 4603–4650. [Google Scholar] [CrossRef]
- Sutherland, C.; Leighton, I.A.; Cohen, P. Inactivation of glycogen synthase kinase-3 beta by phosphorylation: New kinase connections in insulin and growth-factor signalling. Biochem. J. 1993, 296, 15–19. [Google Scholar] [CrossRef]
- Cross, D.A.; Alessi, D.R.; Cohen, P.; Andjelkovich, M.; Hemmings, B.A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995, 378, 785–789. [Google Scholar] [CrossRef]
- Stambolic, V.; Woodgett, J.R. Mitogen inactivation of glycogen synthase kinase-3 beta in intact cells via serine 9 phosphorylation. Biochem. J. 1994, 303, 701–704. [Google Scholar] [CrossRef]
- Medunjanin, S.; Schleithoff, L.; Fiegehenn, C.; Weinert, S.; Zuschratter, W.; Braun-Dullaeus, R.C. GSK-3β controls NF-kappaB activity via IKKγ/NEMO. Sci. Rep. 2016, 6, 38553. [Google Scholar] [CrossRef]
- Martin, M.; Rehani, K.; Jope, R.S.; Michalek, S.M. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat. Immunol. 2005, 6, 777–784. [Google Scholar] [CrossRef]
- Gotschel, F.; Kern, C.; Lang, S.; Sparna, T.; Markmann, C.; Schwager, J.; McNelly, S.; von Weizsäcker, F.; Laufer, S.; Hecht, A.; et al. Inhibition of GSK3 differentially modulates NF-kappaB, CREB, AP-1 and beta-catenin signaling in hepatocytes, but fails to promote TNF-alpha-induced apoptosis. Exp. Cell Res. 2008, 314, 1351–1366. [Google Scholar] [CrossRef]
- Zhang, J.S.; Herreros-Villanueva, M.; Koenig, A.; Deng, Z.; de Narvajas, A.A.; Gomez, T.S.; Meng, X.; Bujanda, L.; Ellenrieder, V.; Li, X.K.; et al. Differential activity of GSK-3 isoforms regulates NF-κB and TRAIL- or TNFα induced apoptosis in pancreatic cancer cells. Cell Death Dis. 2014, 5, e1142. [Google Scholar] [CrossRef]
- Liu, C.; Li, Y.; Semenov, M.; Han, C.; Baeg, G.H.; Tan, Y.; Zhang, Z.; Lin, X.; He, X. Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell 2002, 108, 837–847. [Google Scholar] [CrossRef]
- Webster, M.T.; Rozycka, M.; Sara, E.; Davis, E.; Smalley, M.; Young, N.; Dale, T.C.; Wooster, R. Sequence variants of the axin gene in breast, colon, and other cancers: An analysis of mutations that interfere with GSK3 binding. Genes Chromosom. Cancer 2000, 28, 443–453. [Google Scholar] [CrossRef]
- Silva-Garcia, O.; Rico-Mata, R.; Maldonado-Pichardo, M.C.; Bravo-Patino, A.; Valdez-Alarcon, J.J.; Aguirre-Gonzalez, J.; Baizabal-Aguirre, V.M. Glycogen synthase kinase 3α is the main isoform that regulates the transcription factors nuclear factor-Kappa B and cAMP response element binding in bovine endothelial cells infected with Staphylococcus aureus. Front. Immunol. 2018, 9, 92. [Google Scholar] [CrossRef]
- Cortes-Vieyra, R.; Silva-Garcia, O.; Oviedo-Boyso, J.; Huante-Mendoza, A.; Bravo-Patino, A.; Valdez-Alarcon, J.J.; Finlay, B.B.; Baizabal-Aguirre, V.M. The glycogen synthase kinase 3α and β isoforms differentially regulates interleukin- 12p40 expression in endothelial cells stimulated with peptidoglycan from Staphylococcus aureus. PLoS ONE 2015, 10, e0132867. [Google Scholar] [CrossRef]
- Bhattacharjee, R.; Goswami, S.; Dudiki, T.; Popkie, A.P.; Phiel, C.J.; Kline, D.; Vijayaraghavan, S. Targeted Disruption of Glycogen Synthase Kinase 3a (Gsk3a) in Mice Affects Sperm Motility Resulting in Male Infertility1. Biol. Reprod. 2015, 92, 65. [Google Scholar] [CrossRef]
- Guezguez, B.; Almakadi, M.; Benoit, Y.D.; Shapovalova, Z.; Rahmig, S.; Fiebig-Comyn, A.; Casado, F.L.; Tanasijevic, B.; Bresolin, S.; Masetti, R.; et al. GSK3 Deficiencies in Hematopoietic Stem Cells Initiate Pre-neoplastic State that Is Predictive of Clinical Outcomes of Human Acute Leukemia. Cancer Cell 2016, 29, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Ma, T. GSK3 in Alzheimer’s disease: Mind the isoforms. J. Alzheimer’s Dis. 2014, 39, 707–710. [Google Scholar] [CrossRef] [PubMed]
- Duda, P.; Wiśniewski, J.; Wójtowicz, T.; Wójcicka, O.; Jaśkiewicz, M.; Drulis-Fajdasz, D.; Rakus, D.; McCubrey, J.A.; Gizak, A. Targeting GSK3 signaling as a potential therapy of neurodegenerative diseases and aging. Expert Opin. Ther. Targets 2018, 22, 833–848. [Google Scholar] [CrossRef] [PubMed]
- Fagerberg, L.; Hallstrom, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell Proteom. 2014, 13, 397–406. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Davis, N.M.; Abrams, S.L.; Montalto, G.; Cervello, M.; Basecke, J.; Libra, M.; Nicoletti, F.; Cocco, L.; Martelli, A.M.; et al. Diverse roles of GSK-3: Tumor promoter-tumor suppressor, target in cancer therapy. Adv. Biol. Regul. 2014, 54, 176–196. [Google Scholar] [CrossRef]
- Sokolosky, M.; Chappell, W.H.; Stadelman, K.; Abrams, S.L.; Davis, N.M.; Steelman, L.S.; McCubrey, J.A. Inhibition of GSK-3β activity can result in drug and hormonal resistance and alter sensitivity to targeted therapy in MCF-7 breast cancer cells. Cell Cycle 2014, 13, 820–833. [Google Scholar] [CrossRef]
- Nagini, S.; Sophia, J.; Mishra, R. Glycogen synthase kinases: Moonlighting proteins with theranostic potential in cancer. Semin. Cancer Biol. 2019, 56, 25–36. [Google Scholar] [CrossRef]
- Walz, A.; Ugolkov, A.; Chandra, S.; Kozikowski, A.; Carneiro, B.A.; O’Halloran, T.V.; Giles, F.J.; Billadeau, D.D.; Mazar, A.P. Molecular pathways: Revisiting glycogen synthase kinase-3beta as a target for the treatment of cancer. Clin. Cancer Res. 2017, 23, 1891–1897. [Google Scholar] [CrossRef]
- Wang, J.S.; Wang, C.L.; Wen, J.F.; Wang, Y.J.; Hu, Y.B.; Ren, H.Z. Lithium inhibits proliferation of human esophageal cancer cell line Eca-109 by inducing a G2/M cell cycle arrest. World J. Gastroenterol. 2008, 14, 3982–3989. [Google Scholar] [CrossRef]
- Kazi, A.; Xiang, S.; Yang, H.; Delitto, D.; Trevino, J.; Jiang, R.H.Y.; Ayaz, M.; Lawrence, H.R.; Kennedy, P.; Sebti, S.M. GSK3 suppression upregulates beta-catenin and c-Myc to abrogate KRas-dependent tumors. Nat. Commun. 2018, 9, 5154. [Google Scholar] [CrossRef]
- Acikgoz, E.; Güler, G.; Camlar, M.; Oktem, G.; Aktug, H. Glycogen synthase kinase-3 inhibition in glioblastoma multiforme cells induces apoptosis, cell cycle arrest and changing biomolecular structure. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2019, 209, 150–164. [Google Scholar] [CrossRef] [PubMed]
- Pal, K.; Cao, Y.; Gaisina, I.N.; Bhattacharya, S.; Dutta, S.K.; Wang, E.; Gunosewoyo, H.; Kozikowski, A.P.; Billadeau, D.D.; Mukhopadhyay, D. Inhibition of GSK-3 induces differentiation and impaired glucose metabolism in renal cancer. Mol. Cancer Ther. 2014, 13, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhang, L.; Fu, X.; Jiang, Z.; Tong, R.; Shi, J.; Li, J.; Zhong, L. Design, synthesis and in vitro tumor cytotoxicity evaluation of 3,5-diamino-N-substituted benzamide derivatives as novel GSK-3beta small molecule inhibitors. Chem. Biodivers. 2019, 16, e1900304. [Google Scholar] [CrossRef]
- Schrecengost, R.S.; Green, C.L.; Zhuang, Y.; Keller, S.N.; Smith, R.A.; Maines, L.W.; Smith, C.D. In vitro and in vivo antitumor and anti-inflammatory capabilities of the novel GSK3 and CDK9 inhibitor ABC1183. J. Pharmacol. Exp. Ther. 2009, 365, 107–116. [Google Scholar] [CrossRef]
- Shao, J.; Teng, Y.; Padia, R.; Hong, S.; Noh, H.; Xie, X.; Mumm, J.S.; Dong, Z.; Ding, H.F.; Cowell, J.; et al. COP1 and GSK3beta cooperate to promote c-Jun degradation and inhibit breast cancer cell tumorigenesis. Neoplasia 2013, 15, 1075–1085. [Google Scholar] [CrossRef]
- Rinnab, L.; Schutz, S.V.; Diesch, J.; Schmid, E.; Kufer, R.; Hautmann, R.E.; Spindler, K.D.; Cronauer, M.V. Inhibition of glycogen synthase kinase-3 in androgen-responsive prostate cancer cell lines: Are GSK inhibitors therapeutically useful? Neoplasia 2008, 10, 624–634. [Google Scholar] [CrossRef]
- Kobayashi, T.; Hino, S.; Oue, N.; Asahara, T.; Zollo, M.; Yasui, W.; Kikuchi, A. Glycogen synthase kinase 3 and h-prune regulate cell migration by modulating focal adhesions. Mol. Cell. Biol. 2006, 26, 898–911. [Google Scholar] [CrossRef]
- Wang, F.; Lin, J.; Hou, W.; Huang, M.Y.; Sun, P.H.; Chen, W.M. 5-Benzylidene-3,4-dihalo-furan-2-one derivatives inhibit human leukemia cancer cells through suppression of NF-kappaB and GSK-3beta. Anti Cancer Agents Med. Chem. 2015, 15, 744–754. [Google Scholar] [CrossRef]
- Zhang, H.; Hou, W.; Wang, H.L.; Liu, H.J.; Jia, X.Y.; Zheng, X.Z.; Zou, Y.X.; Li, X.; Hou, L.; McNutt, M.A.; et al. GSK-3β-regulated N-acetyltransferase 10 is involved in colorectal cancer invasion. Clin. Cancer Res. 2014, 20, 4717–4729. [Google Scholar] [CrossRef]
- Guo, R.; Abdelmohsen, K.; Morin, P.J.; Gorospe, M. Novel MicroRNA reporter uncovers repression of Let-7 by GSK-3beta. PLoS ONE 2013, 8, e66330. [Google Scholar]
- Romagnani, P.; Lasagni, L.; Mazzinghi, B.; Lazzeri, E.; Romagnani, S. Pharmacological modulation of stem cell function. Curr. Med. Chem. 2007, 14, 1129–1139. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Smith, K.S.; Murphy, M.; Piloto, O.; Somervaille, T.C.; Cleary, M.L. Glycogen synthase kinase 3 in MLL leukaemia maintenance and targeted therapy. Nature 2008, 455, 1205–1209. [Google Scholar] [CrossRef] [PubMed]
- Yeung, J.; Esposito, M.T.; Gandillet, A.; Zeisig, B.B.; Griessinger, E.; Bonnet, D.; So, C.W. beta-Catenin mediates the establishment and drug resistance of MLL leukemic stem cells. Cancer Cell. 2010, 18, 606–618. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Ueda, M.; Stetson, L.; Ignatz-Hoover, J.; Moreton, S.; Chakrabarti, A.; Xia, Z.; Karan, G.; De Lima, M.; Agrawal, M.K.; et al. A novel glycogen synthase kinase-3 inhibitor optimized for acute myeloid leukemia differentiation activity. Mol. Cancer Ther. 2016, 15, 1485–1494. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Uddin, S.; Zimmerman, T.; Kang, J.A.; Ulaszek, J.; Wickrema, A. Growth control of multiple myeloma cells through inhibition of glycogen synthase kinase-3. Leuk. Lymphoma 2008, 49, 1945–1953. [Google Scholar] [CrossRef]
- Lin, J.; Song, T.; Li, C.; Mao, W. GSK-3β in DNA repair, apoptosis, and resistance of chemotherapy, radiotherapy of cancer. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118659. [Google Scholar] [CrossRef]
- O’Flaherty, L.; Shnyder, S.D.; Cooper, P.A.; Cross, S.J.; Wakefield, J.G.; Pardo, O.E.; Seckl, M.J.; Tavare, J.M. Tumor growth suppression using a combination of taxol-based therapy and GSK3 inhibition in non-small cell lung cancer. PLoS ONE 2019, 14, e0214610. [Google Scholar] [CrossRef]
- Rashid, M.S.; Mazur, T.; Ji, W.; Liu, S.T.; Taylor, W.R. Analysis of the role of GSK3 in the mitotic checkpoint. Sci. Rep. 2018, 8, 14259. [Google Scholar] [CrossRef]
- Santoro, R.; Zanotto, M.; Simionato, F.; Zecchetto, C.; Merz, V.; Cavallini, C.; Piro, G.; Sabbadini, F.; Boschi, F.; Scarpa, A.; et al. Modulating TAK1 Expression Inhibits YAP and TAZ Oncogenic Functions in Pancreatic Cancer. Mol. Cancer Ther. 2020, 19, 247–257. [Google Scholar] [CrossRef]
- Ugolkov, A.V.; Bondarenko, G.I.; Dubrovskyi, O.; Berbegall, A.P.; Navarro, S.; Noguera, R.; O’Halloran, T.V.; Hendrix, M.J.; Giles, F.J.; Mazar, A.P. 9-ING-41, a small-molecule glycogen synthase kinase-3 inhibitor, is active in neuroblastoma. Anticancer Drugs 2018, 29, 717–724. [Google Scholar] [CrossRef]
- Anraku, T.; Kuroki, H.; Kazama, A.; Bilim, V.; Tasaki, M.; Schmitt, D.; Mazar, A.; Giles, F.J.; Ugolkov, A.; Tomita, Y. Clinically relevant GSK-3beta inhibitor 9-ING-41 is active as a single agent and in combination with other antitumor therapies in human renal cancer. Int. J. Mol. Med. 2020, 45, 315–323. [Google Scholar] [PubMed]
- Taylor, A.; Rudd, C.E. Small Molecule Inhibition of Glycogen Synthase Kinase-3 in Cancer Immunotherapy. Adv. Exp. Med. Biol. 2019, 1164, 225–233. [Google Scholar]
- Taylor, A.; Rothstein, D.; Rudd, C.E. Small-molecule inhibition of PD-1 transcription is an effective alternative to antibody blockade in cancer therapy. Cancer Res. 2018, 78, 706–717. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.; Harker, J.A.; Chanthong, K.; Stevenson, P.G.; Zuniga, E.I.; Rudd, C.E. Glycogen Synthase Kinase 3 Inactivation Drives T-bet-Mediated Downregulation of Co-receptor PD-to Enhance CD8+ Cytolytic T Cell Responses. Immunity 2016, 44, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Katz, S.C.; Sengupta, S.; Sampath, P. Glycogen synthase kinase 3 inhibition lowers PD-1 expression, promotes long-term survival and memory generation in antigen-specific CAR-T cells. Cancer Lett. 2018, 433, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Parameswaran, R.; Ramakrishnan, P.; Moreton, S.A.; Xia, Z.; Hou, Y.; Lee, D.A.; Gupta, K.; Delima, M.; Beck, R.C.; Wald, D.N. Repression of GSK3 restores NK cell cytotoxicity in AML patients. Nat. Commun. 2016, 7, 11154. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.Y.; Hsieh, C.T.; Chiu, Y.M.; Chou, S.C.; Kao, J.T.; Shieh, D.C.; Lee, Y.J. GSK-3 inhibitors enhance TRAIL-mediated apoptosis in human gastric adenocarcinoma cells. PLoS ONE 2018, 13, e0208094. [Google Scholar] [CrossRef]
- Nwankwo, N.; Zhang, Z.; Wang, T.; Collins, C.; Resta, L.; Ermisch, S.; Day, J.; Decker, R.; Kornberg, L.; Nicol, S.; et al. Phase I study of enzastaurin and bevacizumab in patients with advanced cancer: Safety, efficacy and pharmacokinetics. Invest. N. Drugs 2013, 31, 653–660. [Google Scholar] [CrossRef]
- Odia, Y.; Iwamoto, F.M.; Moustakas, A.; Fraum, T.J.; Salgado, C.A.; Li, A.; Kreisl, T.N.; Sul, J.; Butman, J.A.; Fine, H.A. A phase II trial of enzastaurin (LY317615) in combination with bevacizumab in adults with recurrent malignant gliomas. J. Neurooncol. 2016, 127, 127–135. [Google Scholar] [CrossRef]
- Tang, Q.L.; Xie, X.B.; Wang, J.; Chen, Q.; Han, A.J.; Zou, C.Y.; Yin, J.Q.; Liu, D.W.; Liang, Y.; Zhao, Z.Q.; et al. Glycogen synthase kinase-3beta, NF-kappaB signaling, and tumorigenesis of human osteosarcoma. J. Nat. Cancer Inst. 2012, 104, 749–763. [Google Scholar] [CrossRef]
- Ban, J.O.; Oh, J.H.; Son, S.M.; Won, D.; Song, H.S.; Han, S.B.; Moon, D.C.; Kang, K.W.; Song, M.J.; Hong, J.T. Troglitazone, a PPAR agonist, inhibits human prostate cancer cell growth through inactivation of NFkappaB via suppression of GSK-3beta expression. Cancer Biol. Ther. 2011, 12, 288–296. [Google Scholar] [PubMed]
- Salamone, S.; Colin, C.; Grillier-Vuissoz, I.; Kuntz, S.; Mazerbourg, S.; Flament, S.; Martin, H.; Richert, L.; Chapleur, Y.; Boisbrun, M. Synthesis of new troglitazone derivatives: Anti-proliferative activity in breast cancer cell lines and preliminary toxicological study. Eur. J. Med. Chem. 2012, 51, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Hu, D.; Qiu, J.; Xie, X.; Ye, F.; Lu, W.G. Overexpression of glycogen synthase kinase-3 in ovarian carcinoma cells with acquired paclitaxel resistance. Int. J. Gynecol. Cancer 2011, 21, 439–444. [Google Scholar] [CrossRef]
- Zhang, X.; Castanotto, D.; Nam, S.; Horne, D.; Stein, C. 6BIO enhances oligonucleotide activity in cells: A potential combinatorial anti-androgen receptor therapy in prostate cancer cells. Mol. Ther. 2017, 25, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Shigeishi, H.; Biddle, A.; Gammon, L.; Emich, H.; Rodini, C.O.; Gemenetzidis, E.; Fazil, B.; Sugiyama, M.; Kamata, N.; Mackenzie, I.C. Maintenance of stem cell self-renewal in head and neck cancers requires actions of GSK3β influenced by CD44 and RHAMM. Stem Cells 2013, 31, 2073–2083. [Google Scholar] [CrossRef] [PubMed]
- Shigeishi, H.; Biddle, A.; Gammon, L.; Rodini, C.O.; Yamasaki, M.; Seino, S.; Sugiyama, M.; Takechi, M.; Mackenzie, I.C. Elevation in 5-FU-induced apoptosis in head and neck cancer stem cells by a combination of CDHP and GSK3beta inhibitors. J. Oral Pathol. Med. 2015, 44, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Ugolkov, A.; Gaisina, I.; Zhang, J.S.; Billadeau, D.D.; White, K.; Kozikowski, A.; Jain, S.; Cristofanilli, M.; Giles, F.; O’Halloran, T.; et al. GSK-3 inhibition overcomes chemoresistance in human breast cancer. Cancer Lett. 2016, 380, 384–392. [Google Scholar] [CrossRef]
- Shimasaki, T.; Ishigaki, Y.; Nakamura, Y.; Takata, T.; Nakaya, N.; Nakajima, H.; Sato, I.; Zhao, X.; Kitano, A.; Kawakami, K.; et al. Glycogen synthase kinase 3beta inhibition sensitizes pancreatic cancer cells to gemcitabine. J. Gastroenterol. 2012, 47, 321–333. [Google Scholar] [CrossRef]
- Remsing Rix, L.L.; Kuenzi, B.M.; Luo, Y.; Remily-Wood, E.; Kinose, F.; Wright, G.; Li, J.; Koomen, J.M.; Haura, E.B.; Lawrence, H.R.; et al. GSK3 alpha and beta are new functionally relevant targets of tivantinib in lung cancer cells. ACS Chem. Biol. 2014, 9, 353–358. [Google Scholar] [CrossRef]
- Shi, F.; Guo, H.; Zhang, R.; Liu, H.; Wu, L.; Wu, Q.; Liu, J.; Liu, T.; Zhang, Q. The PI3K inhibitor GDC-0941 enhances radiosensitization and reduces chemoresistance to temozolomide in GBM cell lines. Neuroscience 2017, 346, 298–308. [Google Scholar] [CrossRef]
- Atkins, R.J.; Dimou, J.; Paradiso, L.; Morokoff, A.P.; Kaye, A.H.; Drummond, K.J.; Hovens, C.M. Regulation of glycogen synthase kinase-3 beta (GSK-3β) by the Akt pathway in gliomas. J. Clin. Neurosci. 2012, 19, 1558–1563. [Google Scholar] [CrossRef]
- Namba, T.; Kodama, R.; Moritomo, S.; Hoshino, T.; Mizushima, T. Zidovudine, an anti-viral drug, resensitizes gemcitabine-resistant pancreatic cancer cells to gemcitabine by inhibition of the Akt-GSK3beta-Snail pathway. Cell Death Dis. 2015, 6, e1795. [Google Scholar] [CrossRef]
- Gaelzer, M.M.; Coelho, B.P.; De Quadros, A.H.; Hoppe, J.B.; Terra, S.R.; Guerra, M.C.B.; Usach, V.; Guma, F.C.R.; Gonçalves, C.A.S.; Setton-Avruj, P.; et al. Phosphatidylinositol 3-kinase/AKT pathway inhibition by doxazosin promotes glioblastoma cells death, upregulation of p53 and triggers low neurotoxicity. PLoS ONE 2016, 11, e0154612. [Google Scholar] [CrossRef]
- Botting, G.M.; Rastogi, I.; Chhabra, G.; Nlend, M.; Puri, N. Mechanism of resistance and novel targets mediating resistance to EGFR and c-Met tyrosine kinase inhibitors in non-small cell lung cancer. PLoS ONE 2015, 10, e0136155. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Mingyi, M.; Qiu, X.; Qiu, Y. MicroRNA-101 reverses temozolomide resistance by inhibition of GSK3β in glioblastoma. Oncotarget 2016, 7, 79584–79595. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Lertpiriyapong, K.; Steelman, L.S.; Abrams, S.L.; Cocco, L.; Ratti, S.; Martelli, A.M.; Candido, S.; Libra, M.; Montalto, G.; et al. Regulation of GSK-3 activity by curcumin, berberine and resveratrol: Potential effects on multiple diseases. Adv. Biol. Regul. 2017, 65, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Gururajan, M.; Dasu, T.; Shahidain, S.; Jennings, C.D.; Robertson, D.A.; Rangnekar, V.M.; Bondada, S. Spleen tyrosine kinase (Syk), a novel target of curcumin, is required for B lymphoma growth. J. Immunol. 2007, 178, 111–121. [Google Scholar] [CrossRef]
- Lai, C.S.; Wu, J.C.; Yu, S.F.; Badmaev, V.; Nagabhushanam, K.; Ho, C.T.; Pan, M.H. Tetrahydrocurcumin is more effective than curcumin in preventing azoxymethane-induced colon carcinogenesis. Mol. Nutr. Food Res. 2011, 55, 1819–1828. [Google Scholar] [CrossRef]
- Song, Y.C.; Lee, Y.; Kim, H.M.; Hyun, M.Y.; Lim, Y.Y.; Song, K.Y.; Kim, B.J. Berberine regulates melanin synthesis by activating PI3K/AKT, ERK and GSK3β in B16F10 melanocytes. Int. J. Mol. Med. 2015, 35, 1011–1016. [Google Scholar] [CrossRef]
- Zhang, R.; Qiao, H.; Chen, S.; Chen, X.; Dou, K.; Wei, L.; Zhang, J. Berberine reverses lapatinib resistance of HER2-positive breast cancer cells by increasing the level of ROS. Cancer Biol. Ther. 2016, 17, 925–934. [Google Scholar] [CrossRef]
- Tsai, J.H.; Hsu, L.S.; Lin, C.L.; Hong, H.M.; Pan, M.H.; Way, T.D.; Chen, W.J. 3,5,4′-Trimethoxystilbene, a natural methoxylated analog of resveratrol, inhibits breast cancer cell invasiveness by downregulation of PI3K/Akt and Wnt/β-catenin signaling cascades and reversal of epithelial-mesenchymal transition. Toxicol. Appl. Pharmacol. 2013, 272, 746–756. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Zhou, M.; Huang, D.; Wasan, H.S.; Zhang, K.; Sun, L.; Huang, H.; Ma, S.; Shen, M.; Ruan, S. Resveratrol inhibits the invasion and metastasis of colon cancer through reversal of epithelial-mesenchymal transition via the AKT/GSK-3β/Snail signaling pathway. Mol. Med. Rep. 2019, 20, 2783–2795. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Zhang, Y.; Wang, J.; Wei, L.; Li, H.; Hanley, G.; Zhao, M.; Li, Y.; Yin, D. Beta-arrestin 2 modulates resveratrol-induced apoptosis and regulation of Akt/GSK3ß pathways. Biochim. Biophys. Acta 2010, 1800, 912–918. [Google Scholar] [CrossRef] [PubMed]
- Cecchinato, V.; Chiaramonte, R.; Nizzardo, M.; Cristofaro, B.; Basile, A.; Sherbet, G.V.; Comi, P. Resveratrol-induced apoptosis in human T-cell acute lymphoblastic leukaemia MOLT-4 cells. Biochem. Pharmacol. 2007, 74, 1568–1574. [Google Scholar] [CrossRef]
- Kato, A.; Naiki-Ito, A.; Nakazawa, T.; Hayashi, K.; Naitoh, I.; Miyabe, K.; Shimizu, S.; Kondo, H.; Nishi, Y.; Yoshida, M.; et al. Chemopreventive effect of resveratrol and apocynin on pancreatic carcinogenesis via modulation of nuclear phosphorylated GSK3β and ERK1/2. Oncotarget 2015, 6, 42963–42975. [Google Scholar] [CrossRef]
- Benitez, D.A.; Pozo-Guisado, E.; Clementi, M.; Castellón, E.; Fernandez-Salguero, P.M. Non-genomic action of resveratrol on androgen and oestrogen receptors in prostate cancer: Modulation of the phosphoinositide 3-kinase pathway. Br. J. Cancer. 2007, 96, 1595–1604. [Google Scholar] [CrossRef]
- Benitez, D.A.; Hermoso, M.A.; Pozo-Guisado, E.; Fernández-Salguero, P.M.; Castellón, E.A. Regulation of cell survival by resveratrol involves inhibition of NF kappa B-regulated gene expression in prostate cancer cells. Prostate 2009, 69, 1045–1054. [Google Scholar] [CrossRef]
- Hsieh, T.C.; Yang, C.J.; Lin, C.Y.; Lee, Y.S.; Wu, J.M. Control of stability of cyclin D1 by quinone reductase 2 in CWR22Rv1 prostate cancer cells. Carcinogenesis 2012, 33, 670–677. [Google Scholar] [CrossRef]
- Vergara, D.; Simeone, P.; Toraldo, D.; Del Boccio, P.; Vergaro, V.; Leporatti, S.; Pieragostino, D.; Tinelli, A.; De Domenico, S.; Alberti, S.; et al. Resveratrol downregulates Akt/GSK and ERK signalling pathways in OVCAR-3 ovarian cancer cells. Mol. Biosyst. 2012, 8, 1078–1087. [Google Scholar] [CrossRef]
- Vergara, D.; De Domenico, S.; Tinelli, A.; Stanca, E.; Del Mercato, L.L.; Giudetti, A.M.; Simeone, P.; Guazzelli, N.; Lessi, M.; Manzini, C.; et al. Anticancer effects of novel resveratrol analogues on human ovarian cancer cells. Mol. Biosyst. 2017, 13, 1131–1141. [Google Scholar] [CrossRef]
- Guzman, E.A.; Maers, K.; Roberts, J.; Kemami-Wangun, H.V.; Harmody, D.; Wright, A.E. The marine natural product microsclerodermin A is a novel inhibitor of the nuclear factor kappa B and induces apoptosis in pancreatic cancer cells. Investig. New Drugs 2015, 33, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; He, Z.; Ma, W.Y.; Schmid, P.C.; Bode, A.M.; Yang, C.S.; Dong, Z. Caffeine inhibits cell proliferation by G0/G1 phase arrest in JB6 cells. Cancer Res. 2004, 64, 3344–3349. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hoi, P.M.; Chan, J.Y.; Lee, S.M. New perspective on the dual functions of indirubins in cancer therapy and neuroprotection. Anticancer Agents Med. Chem. 2014, 14, 1213–1219. [Google Scholar] [CrossRef]
- Chen, X.L.; Ren, K.H.; He, H.W.; Shao, R.G. Involvement of PI3K/AKT/GSK3beta pathway in tetrandrine-induced G1 arrest and apoptosis. Cancer Biol. Ther. 2008, 7, 1073–1078. [Google Scholar] [CrossRef]
- Takahashi-Yanaga, F.; Sasaguri, T. Drug development targeting the glycogen synthase kinase-3beta (GSK-3beta)-mediated signal transduction pathway: Inhibitors of the Wnt/beta-catenin signaling pathway as novel anticancer drugs. J. Pharm. Sci. 2009, 109, 179–183. [Google Scholar] [CrossRef]
- Jingushi, K.; Nakamura, T.; Takahashi-Yanaga, F.; Matsuzaki, E.; Watanabe, Y.; Yoshihara, T.; Morimoto, S.; Sasaguri, T. Differentiation-inducing factor-1 suppresses the expression of c-Myc in the human cancer cell lines. J. Pharm. Sci. 2013, 121, 103–109. [Google Scholar] [CrossRef]
- Liu, W.; Zhao, Z.; Wang, Y.; Li, W.; Su, Q.; Jia, Q.; Zhang, J.; Zhang, X.; Shen, J.; Yin, J. Dioscin inhibits stem-cell-like properties and tumor growth of osteosarcoma through Akt/GSK3/beta-catenin signaling pathway. Cell Death Dis. 2018, 9, 343. [Google Scholar] [CrossRef]
- Sophia, J.; Kiran Kishore, T.K.; Kowshik, J.; Mishra, R.; Nagini, S. Nimbolide, a neem limonoid inhibits Phosphatidyl Inositol-3 Kinase to activate Glycogen Synthase Kinase-3β in a hamster model of oral oncogenesis. Sci. Rep. 2016, 6, 22192. [Google Scholar] [CrossRef]
- Sophia, J.; Kowshik, J.; Dwivedi, A.; Bhutia, S.K.; Manavathi, B.; Mishra, R.; Nagini, S. Nimbolide, a neem limonoid inhibits cytoprotective autophagy to activate apoptosis via modulation of the PI3K/Akt/GSK-3β signalling pathway in oral cancer. Cell Death Dis. 2018, 9, 1087. [Google Scholar] [CrossRef]
- Huang, H.L.; Weng, H.Y.; Wang, L.Q.; Yu, C.H.; Huang, Q.J.; Zhao, P.P.; Wen, J.Z.; Zhou, H.; Qu, L.H. Triggering Fbw7-mediated proteasomal degradation of c-Myc by oridonin induces cell growth inhibition and apoptosis. Mol. Cancer Ther. 2012, 11, 1155–1165. [Google Scholar] [CrossRef]
- Tu, C.C.; Cheng, L.H.; Hsu, H.H.; Chen, L.M.; Lin, Y.M.; Chen, M.C.; Lee, N.H.; Tsai, F.J.; Huang, C.Y.; Wu, W.J. Activation of snail and EMT-like signaling via the IKKalphabeta/NF-kappaB pathway in Apicidin-resistant HA22T hepatocellular carcinoma cells. Chin. J. Physiol. 2013, 56, 326–333. [Google Scholar] [CrossRef]
- Chen, X.M.; Bai, Y.; Zhong, Y.J.; Xie, X.L.; Long, H.W.; Yang, Y.Y.; Wu, S.G.; Jia, Q.; Wang, X.H. Wogonin has multiple anti-cancer effects by regulating c-Myc/SKP2/Fbw7alpha and HDAC1/HDAC2 pathways and inducing apoptosis in human lung adenocarcinoma cell line A549. PLoS ONE 2013, 8, e79201. [Google Scholar]
- Zhu, J.; Wang, S.; Chen, Y.; Li, X.; Jiang, Y.; Yang, X.; Li, Y.; Wang, X.; Meng, Y.; Zhu, M.; et al. miR-19 targeting of GSK3β mediates sulforaphane suppression of lung cancer stem cells. J. Nutr. Biochem. 2017, 44, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Pant, K.; Yadav, A.K.; Gupta, P.; Islam, R.; Saraya, A.; Venugopal, S.K. Butyrate induces ROS-mediated apoptosis by modulating miR-22/SIRT-1 pathway in hepatic cancer cells. Redox Biol. 2017, 12, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Kwon, H.Y.; Sohn, E.J.; Kim, K.A.; Kim, B.; Jeong, S.J.; Song, J.H.; Koo, J.S.; Kim, S.H. Inhibition of Wnt/beta-catenin signaling mediates ursolic acid-induced apoptosis in PC-3 prostate cancer cells. Pharmacol. Rep. 2013, 65, 1366–1374. [Google Scholar] [CrossRef]
- Song, Y.H.; Jeong, S.J.; Kwon, H.Y.; Kim, B.; Kim, S.H.; Yoo, D.Y. Ursolic acid from Oldenlandia diffusa induces apoptosis via activation of caspases and phosphorylation of glycogen synthase kinase 3 beta in SK-OV-3 ovarian cancer cells. Biol. Pharm. Bull. 2012, 35, 1022–1028. [Google Scholar] [CrossRef]
- Chen, H.B.; Zhou, L.Z.; Mei, L.; Shi, X.J.; Wang, X.S.; Li, Q.L.; Huang, L. Gambogenic acid-induced time- and dose-dependent growth inhibition and apoptosis involving Akt pathway inactivation in U251 glioblastoma cells. J. Nat. Med. 2012, 66, 62–69. [Google Scholar] [CrossRef]
- Lynce, F.; Wang, H.; Petricoin, E.F.; Pohlmann, P.R.; Smaglo, B.; Hwang, J.; He, A.R.; Subramaniam, D.S.; Deeken, J.; Marshall, J.; et al. A phase I study of HER1, HER2 dual kinase inhibitor lapatinib plus the proteasome inhibitor bortezomib in patients with advanced malignancies. Cancer Chemother. Pharmacol. 2019, 84, 1145–1151. [Google Scholar] [CrossRef]
- Algazi, A.P.; Esteve-Puig, R.; Nosrati, A.; Hinds, B.; Hobbs-Muthukumar, A.; Nandoskar, P.; Ortiz-Urda, S.; Chapman, P.B.; Daud, A. Dual MEK/AKT inhibition with trametinib and GSK2141795 does not yield clinical benefit in metastatic NRAS-mutant and wild-type melanoma. Pigment Cell Melanoma Res. 2018, 31, 110–114. [Google Scholar] [CrossRef]
- Furuta, T.; Sabit, H.; Dong, Y.; Miyashita, K.; Kinoshita, M.; Uchiyama, N.; Hayashi, Y.; Hayashi, Y.; Minamoto, T.; Nakada, M. Biological basis and clinical study of glycogen synthase kinase- 3β-targeted therapy by drug repositioning for glioblastoma. Oncotarget 2017, 8, 22811–22824. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tissue Type | GSK-3α | GSK-3β | Ratio GSK-3α/ GSK-3β |
|---|---|---|---|
| Adrenal | 12.5 ± 0.7 | 7.3 ± 0.1 | 1.7 |
| Appendix | 11 ± 1.2 | 6.2 ± 0.8 | 1.8 |
| Bone Marrow | 10.4 ± 1.1 | 3.7 ± 0.5 | 2.8 |
| Brain | 26.3 ± 6.8 | 13.9 ± 1.2 | 1.9 |
| Colon | 10.8 ± 1.3 | 6.4 ± 0.5 | 1.7 |
| Duodenum | 16.2 ± 0.07 | 5 ± 0.3 | 3.2 |
| Endometrium | 11.7 ± 0.6 | 6.5 ± 1.5 | 1.8 |
| Esophagus | 11.9 ± 0.5 | 5.9 ± 0.6 | 2 |
| Fat | 10 ± 2 | 5.7 ± 0.6 | 1.8 |
| Gall bladder | 10.8 ± 0.2 | 6.6 ± 1.3 | 1.6 |
| Heart | 9.6 ± 0.8 | 6.7 ± 0.7 | 1.4 |
| Kidney | 12.8 ± 1.2 | 5.9 ± 0.5 | 2.2 |
| Liver | 4.6 ± 0.6 | 2.8 ± 0.1 | 1.6 |
| Lung | 11.6 ± 1.5 | 7.9 ± 1.1 | 1.5 |
| Lymph node | 10.5 ± 1.6 | 5.5 ± 1.4 | 1.9 |
| Ovary | 12.1 ± 0.3 | 4.6 ± 0.07 | 2.6 |
| Pancreas | 3.1 ± 0.04 | 1.4 ± 0.05 | 2.2 |
| Placenta | 12.4 ± 1.3 | 7.7 ± 1.7 | 1.6 |
| Prostate | 12.4 ± 0.6 | 5.5 ± 0.5 | 2.3 |
| Salivary gland | 5.1 ± 1.1 | 2.9 ± 0.5 | 1.8 |
| Skin | 11.8 ± 1 | 7.4 ± 2.3 | 1.6 |
| Small intestine | 14.4 ± 0.7 | 5.7 ± 0.5 | 2.5 |
| Spleen | 11.6 ± 0.6 | 7.1 ± 0.9 | 1.6 |
| Stomach | 8.7 ± 2.1 | 4.9 ± 1.2 | 1.8 |
| Thyroid | 13.3 ± 1.2 | 9.9 ± 1.7 | 1.3 |
| Urinary bladder | 10.9 ± 0.7 | 6.5 ± 1.2 | 1.7 |
| Molecule | Result | Clinical Trial | Number/Reference |
|---|---|---|---|
| 9-ING-41, GSK-3β inhibitor | Ongoing, recruiting | 9-ING-41 in Patients With Advanced Cancers, 29 advanced cancer types also including chemotherapeutic drugs. | NCT03678883 |
| 9-ING-41, GSK-3β inhibitor | Not yet recruiting | 9-ING-41 in Pediatric Patients With Refractory Malignancies, 10 different types of pediatric cancers also including chemotherapeutic drugs | NCT04239092 |
| LY2090314, GSK-3β inhibitor | Terminated, due to slow recruitment | A Study of LY2090314 and Chemotherapy in Participants With Metastatic Pancreatic Cancer | NCT01632306, results are presented on ClinicalTrials.gov |
| AKT inhibitor AZD5363, which will result in GSK-3 activation, and combination of the PARP inhibitor olaparib | Phase I trial of effects of combining Olaparib and AZD5362. Completed | Trial of Olaparib in Combination With AZD5363 (ComPAKT) (ComPAKT) in advanced cancer patients. | NCT02338622, no results posted. |
| Combining the EGFR/HER2 inhibitor with the proteasomal inhibitor bortezomib. Lapatinib should inhibit AKT activity which will lead to GSK-3 activity. | Phase I study was terminated due to withdrawal of sponsor support. | A Phase I Study of the HER1, HER2 Dual Kinase Inhibitor, Lapatinib Plus the Proteasomal Inhibitor Bortezomib in Patients With Advanced Malignancies | NCT01497626, [108]. |
| EGFR inhibitor panitumumab in combination with lycopene which is a potent antioxidant. EGFR inhibitor should suppress downstream AKT which lead to GSK-3 activity. | Recruiting | Panitumumab Skin Toxicity Prevention Trial (PaSTo). Colorectal cancer patients. | NCT03167268, no results posted |
| The protein kinase C beta inhibitor Enzastaurin result in inhibition of AKT which leads to activation of GSK-3. The effects of Enzataurin and the vascular endothelial growth factor A (VEGFa) inhibitor Bevacizumab were examined in advanced or metastatic cancer patients. | Finished Phase I study. 67 patients were evaluable for safety and efficacy. Good results with patients with ovarian cancers. 50.4 % of ovarian cancer patients remained without disease progression after 6 months. | Enzastaurin and Bevacizumab in Treating Patients With Locally Advanced or Metastatic Cancer | NCT00550927, [58]. |
| To determine effects of combination of enzataurin and bevacizumab in adults with glioma. | Finished Phase II study with 81 patients with glioblastomas (GBM, n = 40) and anaplastic gliomas (AG, n = 41). Early response was associated with longer progression free survival for glioblastomas. Combined treatment was well tolerated, and survival time was similar to that observed in patients treated with bevacizumab. | Phase II study with enzastaurin (LY317615) in combination with bevacizumab in adults with recurrent malignant gliomas. | NCT00586508, [59]. |
| Effects of combining Enzastaurin (LY317615) With Carboplatin on recurrent glioma patients | Completed | A Phase I Trial of Enzastaurin (LY317615) in Combination With Carboplatin in Adults With Recurrent Gliomas | NCT01445119, No results with combining Enzastaurin (LY317615) with Carboplatin. |
| Effects of Trametinib MEK inhibitor and pan AKT inhibitor (GSK2141795) treatment in melanoma. Suppression of AKT should result in increased levels of active GSK-3 | Completed, Phase II clinical study did not reveal any clinical benefit of trametinib and GSK2141795 treatment in melanoma patients with NRAS mutations or wild-type melanoma. GSK 2141795 inhibited phosphorylation of GSK-3β. | Trametinib With GSK2141795 in BRAF Wild-type Melanoma | NCT01941927, [109]. |
| Treatment of humans and mouse model of recurrent GBM with temozolomide (TMZ) and other drugs which suppress GSK-3β (cimetidine, lithium, olanzapine, and valproate, (CLOVA) cocktail. The safety and efficacy of the CLOVA cocktail) in combination with TMZ were performed to human and murine studies. | Inhibition of active GSK-3β in the tumor resulted in increased patient survival. The combination of TMZ and the CLOVA cocktail significantly inhibited cell invasion and TMZ increased survival compared to patients treated with TMZ alone. Active GSK-3β was associated with a poor prognosis | Clinical study in Japan completed with 7 GBM patients. | [110]. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duda, P.; Akula, S.M.; Abrams, S.L.; Steelman, L.S.; Martelli, A.M.; Cocco, L.; Ratti, S.; Candido, S.; Libra, M.; Montalto, G.; et al. Targeting GSK3 and Associated Signaling Pathways Involved in Cancer. Cells 2020, 9, 1110. https://doi.org/10.3390/cells9051110
Duda P, Akula SM, Abrams SL, Steelman LS, Martelli AM, Cocco L, Ratti S, Candido S, Libra M, Montalto G, et al. Targeting GSK3 and Associated Signaling Pathways Involved in Cancer. Cells. 2020; 9(5):1110. https://doi.org/10.3390/cells9051110
Chicago/Turabian StyleDuda, Przemysław, Shaw M. Akula, Stephen L. Abrams, Linda S. Steelman, Alberto M. Martelli, Lucio Cocco, Stefano Ratti, Saverio Candido, Massimo Libra, Giuseppe Montalto, and et al. 2020. "Targeting GSK3 and Associated Signaling Pathways Involved in Cancer" Cells 9, no. 5: 1110. https://doi.org/10.3390/cells9051110
APA StyleDuda, P., Akula, S. M., Abrams, S. L., Steelman, L. S., Martelli, A. M., Cocco, L., Ratti, S., Candido, S., Libra, M., Montalto, G., Cervello, M., Gizak, A., Rakus, D., & McCubrey, J. A. (2020). Targeting GSK3 and Associated Signaling Pathways Involved in Cancer. Cells, 9(5), 1110. https://doi.org/10.3390/cells9051110