Activation of the PI3K-AKT Pathway by Old World Alphaviruses


Abstract
1. Introduction
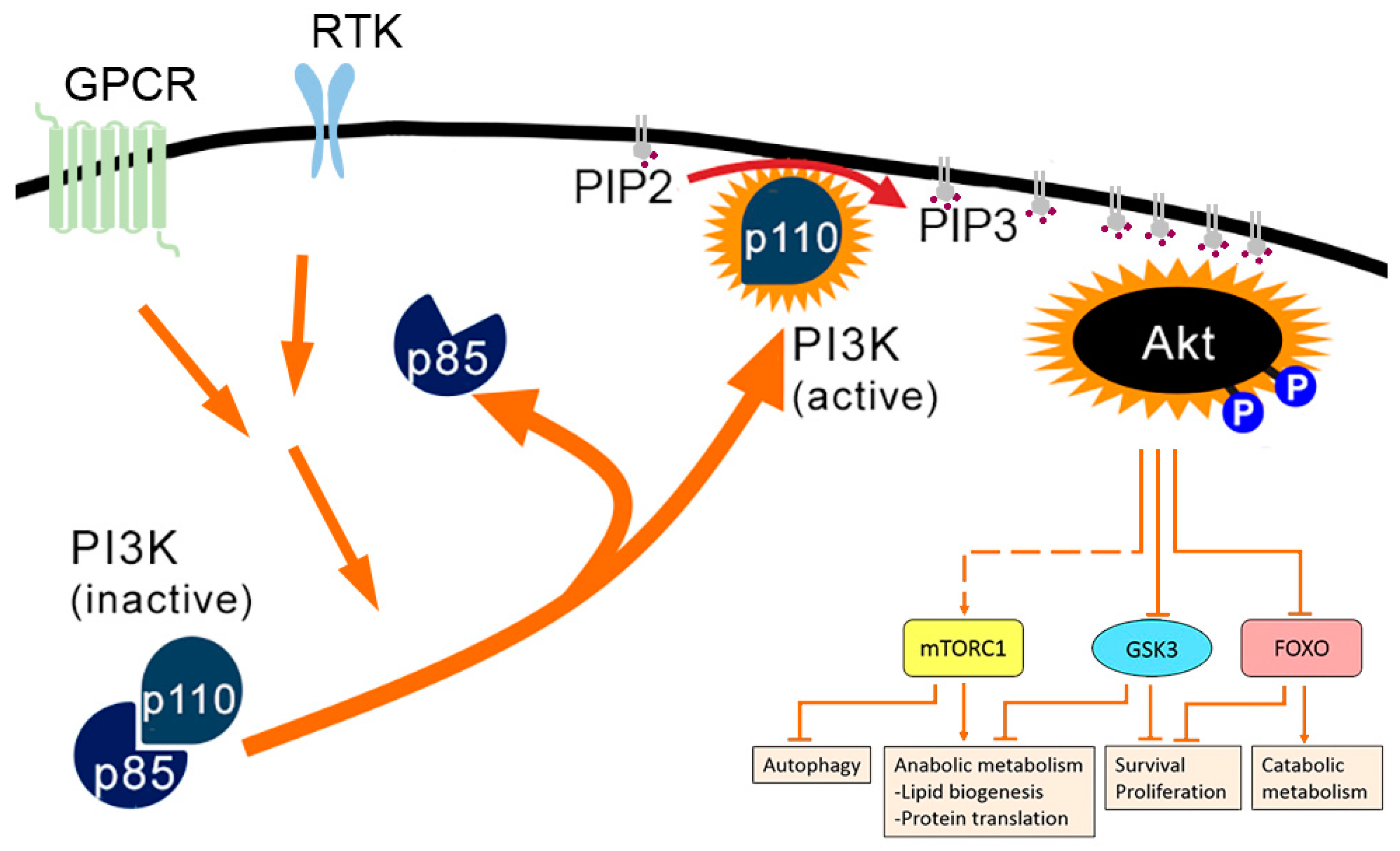
1.1. PI3K-AKT Pathway
1.2. Alphaviruses
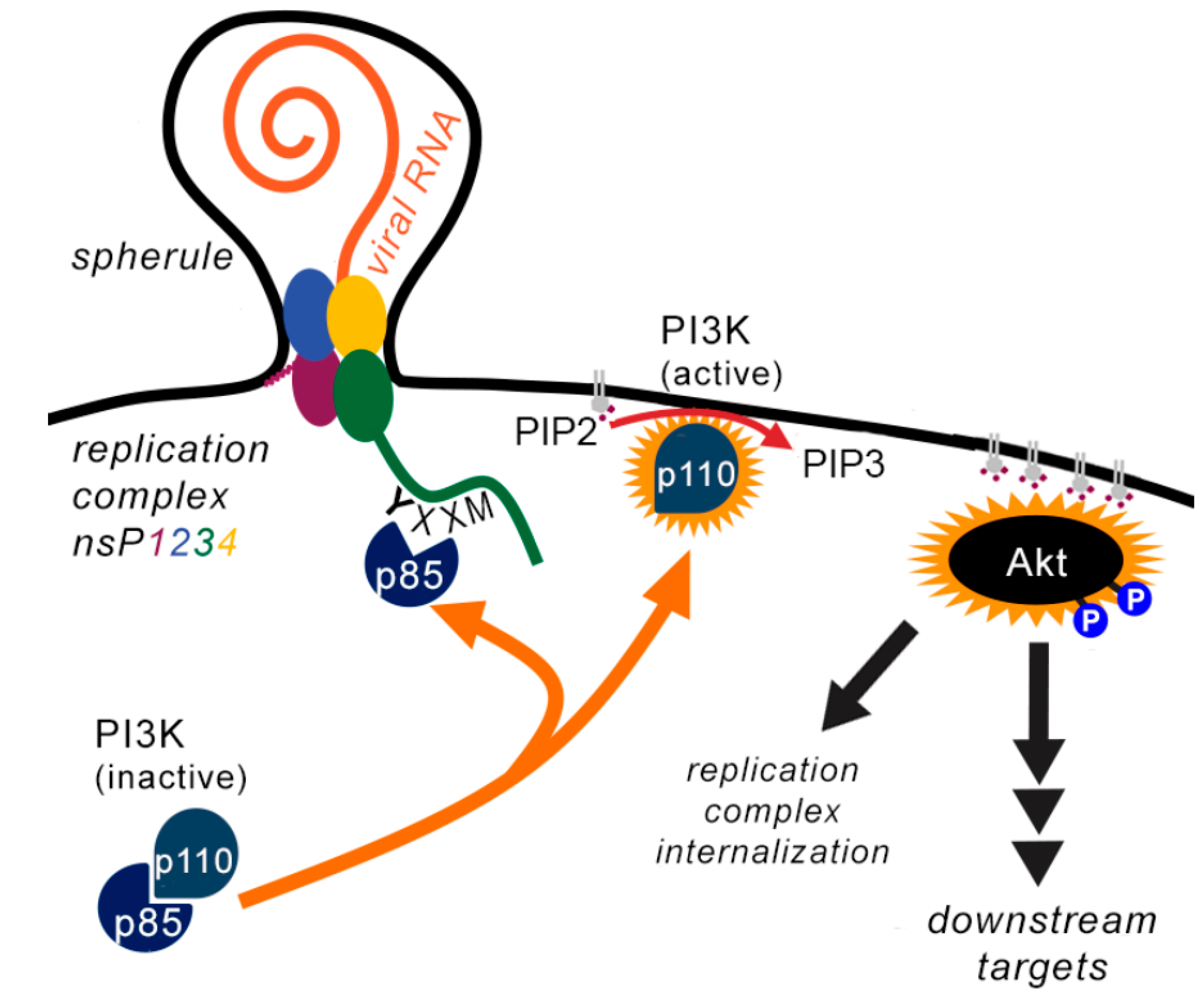
1.3. Effect of Alphaviruses on PI3K-AKT Pathway During Infection
1.4. SFV and RRV Hyperactivate the PI3K-AKT-mTOR Pathway and Downstream Effectors
1.5. CHIKV Activates PI3K-AKT Moderately
1.6. SINV Differentially Activates PI3K-AKT in Different Species
1.7. Benefits of Activation of PI3K Pathway for Alphaviruses
2. Metabolic Change
Alphaviruses Influence Cellular Metabolism
3. Interaction with Autophagy
3.1. Various Viruses Influence Autophagy
3.2. CHIKV Induce Autophagy, Whereas SFV Blocks It
4. Promotion of Cell Survival
Unknown Whether Alphaviruses Activate PI3K-AKT to Promote Cell Survival
5. The Strange Case of the Trafficking of Replication Complexes
Some Alphaviruses Stimulate Internalisation of Replication Complexes
6. Remarks in Conclusion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Diehl, N.; Schaal, H. Make yourself at home: Viral hijacking of the PI3K/Akt signaling pathway. Viruses 2013, 5, 3192–3212. [Google Scholar] [CrossRef] [PubMed]
- Dunn, E.F.; Connor, J.H. HijAkt: The PI3K/Akt pathway in virus replication and pathogenesis. Prog. Mol. Biol. Transl. Sci. 2012, 106, 223–250. [Google Scholar] [PubMed]
- Buchkovich, N.J.; Yu, Y.; Zampieri, C.A.; Alwine, J.C. The TORrid affairs of viruses: Effects of mammalian DNA viruses on the PI3K-Akt-mTOR signalling pathway. Nat. Rev. Microbiol. 2008, 6, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Cooray, S. The pivotal role of phosphatidylinositol 3-kinase-Akt signal transduction in virus survival. J. Gen. Virol. 2004, 85, 1065–1076. [Google Scholar] [CrossRef]
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341. [Google Scholar] [CrossRef]
- Ersahin, T.; Tuncbag, N.; Cetin-Atalay, R. The PI3K/AKT/mTOR interactive pathway. Mol. Biosyst. 2015, 11, 1946–1954. [Google Scholar] [CrossRef]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef]
- Rupp, J.C.; Sokoloski, K.J.; Gebhart, N.N.; Hardy, R.W. Alphavirus RNA synthesis and non-structural protein functions. J. Gen. Virol. 2015, 96, 2483–2500. [Google Scholar] [CrossRef]
- Gotte, B.; Panas, M.D.; Hellstrom, K.; Liu, L.; Samreen, B.; Larsson, O.; Ahola, T.; McInerney, G.M. Separate domains of G3BP promote efficient clustering of alphavirus replication complexes and recruitment of the translation initiation machinery. PLoS Pathog. 2019, 15, e1007842. [Google Scholar] [CrossRef] [PubMed]
- Lark, T.; Keck, F.; Narayanan, A. Interactions of Alphavirus nsP3 Protein with Host Proteins. Front. Microbiol. 2017, 8, 2652. [Google Scholar] [CrossRef] [PubMed]
- Gotte, B.; Liu, L.; McInerney, G.M. The Enigmatic Alphavirus Non-Structural Protein 3 (nsP3) Revealing Its Secrets at Last. Viruses 2018, 10, 105. [Google Scholar] [CrossRef] [PubMed]
- Mazzon, M.; Castro, C.; Thaa, B.; Liu, L.; Mutso, M.; Liu, X.; Mahalingam, S.; Griffin, J.L.; Marsh, M.; McInerney, G.M. Alphavirus-induced hyperactivation of PI3K/AKT directs pro-viral metabolic changes. PLoS Pathog. 2018, 14, e1006835. [Google Scholar] [CrossRef]
- Wu, H.; Windmiller, D.A.; Wang, L.; Backer, J.M. YXXM motifs in the PDGF-beta receptor serve dual roles as phosphoinositide 3-kinase binding motifs and tyrosine-based endocytic sorting signals. J. Biol. Chem. 2003, 278, 40425–40428. [Google Scholar] [CrossRef] [PubMed]
- Songyang, Z.; Shoelson, S.E.; Chaudhuri, M.; Gish, G.; Pawson, T.; Haser, W.G.; King, F.; Roberts, T.; Ratnofsky, S.; Lechleider, R.J.; et al. SH2 domains recognize specific phosphopeptide sequences. Cell 1993, 72, 767–778. [Google Scholar] [PubMed]
- Thaa, B.; Biasiotto, R.; Eng, K.; Neuvonen, M.; Gotte, B.; Rheinemann, L.; Mutso, M.; Utt, A.; Varghese, F.; Balistreri, G.; et al. Differential Phosphatidylinositol-3-Kinase-Akt-mTOR Activation by Semliki Forest and Chikungunya Viruses Is Dependent on nsP3 and Connected to Replication Complex Internalization. J. Virol. 2015, 89, 11420–11437. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.K.; Liu, Q.; Tikoo, S.K.; Babiuk, L.A.; Zhou, Y. Influenza A virus NS1 protein activates the phosphatidylinositol 3-kinase (PI3K)/Akt pathway by direct interaction with the p85 subunit of PI3K. J. Gen. Virol. 2007, 88, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Strunk, U.; Saffran, H.A.; Wu, F.W.; Smiley, J.R. Role of herpes simplex virus VP11/12 tyrosine-based motifs in binding and activation of the Src family kinase Lck and recruitment of p85, Grb2, Shc. J. Virol. 2013, 87, 11276–11286. [Google Scholar] [CrossRef]
- Sharma, A.; Bhomia, M.; Yeh, T.J.; Singh, J.; Maheshwari, R.K. Miltefosine inhibits Chikungunya virus replication in human primary dermal fibroblasts. F1000Res 2018, 7, 9. [Google Scholar] [CrossRef]
- Broeckel, R.; Sarkar, S.; May, N.A.; Totonchy, J.; Kreklywich, C.N.; Smith, P.; Graves, L.; DeFilippis, V.R.; Heise, M.T.; Morrison, T.E.; et al. Src Family Kinase Inhibitors Block Translation of Alphavirus Subgenomic mRNAs. Antimicrob. Agents Chemother. 2019, 63, e02325-18. [Google Scholar] [CrossRef]
- Das, I.; Basantray, I.; Mamidi, P.; Nayak, T.K.; Pratheek, B.M.; Chattopadhyay, S. Heat shock protein 90 positively regulates Chikungunya virus replication by stabilizing viral non-structural protein nsP2 during infection. PLoS ONE 2014, 9, e100531. [Google Scholar] [CrossRef]
- Sharma, A.; Balakathiresan, N.S.; Maheshwari, R.K. Chikungunya Virus Infection Alters Expression of MicroRNAs Involved in Cellular Proliferation, Immune Response and Apoptosis. Intervirology 2015, 58, 332–341. [Google Scholar] [CrossRef] [PubMed]
- Wikan, N.; Khongwichit, S.; Phuklia, W.; Ubol, S.; Thonsakulprasert, T.; Thannagith, M.; Tanramluk, D.; Paemanee, A.; Kittisenachai, S.; Roytrakul, S.; et al. Comprehensive proteomic analysis of white blood cells from chikungunya fever patients of different severities. J. Transl. Med. 2014, 12, 96. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Qiu, Y. Interaction between Src and a C-terminal proline-rich motif of Akt is required for Akt activation. J. Biol. Chem. 2003, 278, 15789–15793. [Google Scholar] [CrossRef] [PubMed]
- Yori, J.L.; Lozada, K.L.; Seachrist, D.D.; Mosley, J.D.; Abdul-Karim, F.W.; Booth, C.N.; Flask, C.A.; Keri, R.A. Combined SFK/mTOR inhibition prevents rapamycin-induced feedback activation of AKT and elicits efficient tumor regression. Cancer Res. 2014, 74, 4762–4771. [Google Scholar] [CrossRef] [PubMed]
- Joubert, P.E.; Stapleford, K.; Guivel-Benhassine, F.; Vignuzzi, M.; Schwartz, O.; Albert, M.L. Inhibition of mTORC1 Enhances the Translation of Chikungunya Proteins via the Activation of the MnK/eIF4E Pathway. PLoS Pathog. 2015, 11, e1005091. [Google Scholar] [CrossRef]
- Joubert, P.E.; Werneke, S.W.; De la Calle, C.; Guivel-Benhassine, F.; Giodini, A.; Peduto, L.; Levine, B.; Schwartz, O.; Lenschow, D.J.; Albert, M.L. Chikungunya virus-induced autophagy delays caspase-dependent cell death. J. Exp. Med. 2012, 209, 1029–1047. [Google Scholar] [CrossRef]
- Varghese, F.S.; Thaa, B.; Amrun, S.N.; Simarmata, D.; Rausalu, K.; Nyman, T.A.; Merits, A.; McInerney, G.M.; Ng, L.F.; Ahola, T. The antiviral alkaloid berberine reduces chikungunya virus-induced mitogen-activated protein kinase (MAPK) signaling. J. Virol. 2015, 90, 9743–9757. [Google Scholar] [CrossRef]
- Oh, W.J.; Jacinto, E. mTOR complex 2 signaling and functions. Cell Cycl. 2011, 10, 2305–2316. [Google Scholar] [CrossRef]
- Scherbik, S.V.; Brinton, M.A. Virus-induced Ca2+ influx extends survival of west nile virus-infected cells. J. Virol. 2010, 84, 8721–8731. [Google Scholar] [CrossRef]
- Mohankumar, V.; Dhanushkodi, N.R.; Raju, R. Sindbis virus replication, is insensitive to rapamycin and torin1, suppresses Akt/mTOR pathway late during infection in HEK cells. Biochem. Biophys. Res. Commun. 2011, 406, 262–267. [Google Scholar] [CrossRef]
- Gharbi, S.I.; Zvelebil, M.J.; Shuttleworth, S.J.; Hancox, T.; Saghir, N.; Timms, J.F.; Waterfield, M.D. Exploring the specificity of the PI3K family inhibitor LY294002. Biochem. J. 2007, 404, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Findlay, J.S.; Ulaeto, D. Semliki Forest virus and Sindbis virus, but not vaccinia virus, require glycolysis for optimal replication. J. Gen. Virol. 2015, 96, 2693–2696. [Google Scholar] [CrossRef] [PubMed]
- Silva da Costa, L.; Da Silva, A.P.P.; Da Poian, A.T.; El-Bacha, T. Mitochondrial bioenergetic alterations in mouse neuroblastoma cells infected with Sindbis virus: Implications to viral replication and neuronal death. PLoS ONE 2012, 7, e33871. [Google Scholar] [CrossRef] [PubMed]
- Dhanwani, R.; Khan, M.; Lomash, V.; Rao, P.V.; Ly, H.; Parida, M. Characterization of chikungunya virus induced host response in a mouse model of viral myositis. PLoS ONE 2014, 9, e92813. [Google Scholar] [CrossRef] [PubMed]
- Thio, C.L.; Yusof, R.; Abdul-Rahman, P.S.; Karsani, S.A. Differential proteome analysis of chikungunya virus infection on host cells. PLoS ONE 2013, 8, e61444. [Google Scholar] [CrossRef] [PubMed]
- El-Bacha, T.; Menezes, M.M.; Silva, M.C.A.e.; Sola-Penna, M.; Da Poian, A.T. Mayaro virus infection alters glucose metabolism in cultured cells through activation of the enzyme 6-phosphofructo 1-kinase. Mol. Cell Biochem. 2004, 266, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, E.L.; Lagunoff, M. Viral activation of cellular metabolism. Virology 2015, 479–480, 609–618. [Google Scholar]
- Burke, J.D.; Platanias, L.C.; Fish, E.N. Beta interferon regulation of glucose metabolism is PI3K/Akt dependent and important for antiviral activity against coxsackievirus B3. J. Virol. 2014, 88, 3485–3495. [Google Scholar] [CrossRef]
- Chun, Y.; Kim, J. Autophagy: An Essential Degradation Program for Cellular Homeostasis and Life. Cells 2018, 7, E278. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Echavarria-Consuegra, L.; Smit, J.M.; Reggiori, F. Role of autophagy during the replication and pathogenesis of common mosquito-borne flavi- and alphaviruses. Open Biol. 2019, 9, 190009. [Google Scholar] [CrossRef]
- Choi, Y.; Bowman, J.W.; Jung, J.U. Autophagy during viral infection—A double-edged sword. Nat. Rev. Microbiol. 2018, 16, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Luo, Z.; Zeng, J.; Chen, W.; Foo, S.S.; Lee, S.A.; Ge, J.; Wang, S.; Goldman, S.A.; Zlokovic, B.V.; et al. Zika Virus NS4A and NS4B Proteins Deregulate Akt-mTOR Signaling in Human Fetal Neural Stem Cells to Inhibit Neurogenesis and Induce Autophagy. Cell Stem Cell 2016, 19, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Parnell, L.A.; Diamond, M.S.; Mysorekar, I.U. Inhibition of autophagy limits vertical transmission of Zika virus in pregnant mice. J. Exp. Med. 2017, 214, 2303–2313. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Geng, P.; Liu, Y.; Wu, J.; Qiao, H.; Xie, Y.; Yin, N.; Chen, L.; Lin, X.; Liu, Y.; et al. Rotavirus-encoded virus-like small RNA triggers autophagy by targeting IGF1R via the PI3K/Akt/mTOR pathway. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 60–68. [Google Scholar] [CrossRef]
- Yin, Y.; Dang, W.; Zhou, X.; Xu, L.; Wang, W.; Cao, W.; Chen, S.; Su, J.; Cai, X.; Xiao, S.; et al. PI3K-Akt-mTOR axis sustains rotavirus infection via the 4E-BP1 mediated autophagy pathway and represents an antiviral target. Virulence 2018, 9, 83–98. [Google Scholar] [CrossRef]
- Wang, R.; Zhu, Y.; Zhao, J.; Ren, C.; Li, P.; Chen, H.; Jin, M.; Zhou, H. Autophagy Promotes Replication of Influenza A Virus In Vitro. J. Virol. 2016, 2019, 93. [Google Scholar] [CrossRef]
- Krejbich-Trotot, P.; Gay, B.; Li-Pat-Yuen, G.; Hoarau, J.J.; Jaffar-Bandjee, M.C.; Briant, L.; Gasque, P.; Denizot, M. Chikungunya triggers an autophagic process which promotes viral replication. Virol. J. 2011, 8, 432. [Google Scholar] [CrossRef]
- Khongwichit, S.; Wikan, N.; Abere, B.; Thepparit, C.; Kuadkitkan, A.; Ubol, S.; Smith, D.R. Cell-type specific variation in the induction of ER stress and downstream events in chikungunya virus infection. Microb. Pathog. 2016, 101, 104–118. [Google Scholar] [CrossRef]
- Judith, D.; Mostowy, S.; Bourai, M.; Gangneux, N.; Lelek, M.; Lucas-Hourani, M.; Cayet, N.; Jacob, Y.; Prevost, M.C.; Pierre, P.; et al. Species-specific impact of the autophagy machinery on Chikungunya virus infection. EMBO Rep. 2013, 14, 534–544. [Google Scholar] [CrossRef]
- Abraham, R.; Mudaliar, P.; Padmanabhan, A.; Sreekumar, E. Induction of cytopathogenicity in human glioblastoma cells by chikungunya virus. PLoS ONE 2013, 8, e75854. [Google Scholar] [CrossRef]
- Shoji-Kawata, S.; Sumpter, R.; Leveno, M.; Campbell, G.R.; Zou, Z.; Kinch, L.; Wilkins, A.D.; Sun, Q.; Pallauf, K.; MacDuff, D.; et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature 2013, 494, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Eng, K.E.; Panas, M.D.; Murphy, D.; Hedestam, G.B.K.; McInerney, G.M. Accumulation of autophagosomes in Semliki Forest virus infected cells is dependent on the expression of the viral glycoproteins. J. Virol. 2012, 86, 5674–5685. [Google Scholar] [CrossRef] [PubMed]
- Dahal, B.; Lin, S.C.; Carey, B.D.; Jacobs, J.L.; Dinman, J.D.; Van Hoek, M.L.; Adams, A.A.; Kehn-Hall, K. EGR1 upregulation following Venezuelan equine encephalitis viaarus infection is regulated by ERK and PERK pathways contributing to cell death. Virology 2020, 539, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Cooray, S.; Jin, L.; Best, J.M. The involvement of survival signaling pathways in rubella-virus induced apoptosis. Virol. J. 2005, 2, 1. [Google Scholar] [CrossRef]
- Spuul, P.; Balistreri, G.; Kaariainen, L.; Ahola, T. Phosphatidylinositol 3-kinase-, actin-, microtubule-dependent transport of Semliki Forest Virus replication complexes from the plasma membrane to modified lysosomes. J. Virol. 2010, 84, 7543–7557. [Google Scholar] [CrossRef]
- Frolova, E.I.; Gorchakov, R.; Pereboeva, L.; Atasheva, S.; Frolov, I. Functional Sindbis virus replicative complexes are formed at the plasma membrane. J. Virol. 2010, 84, 11679–11695. [Google Scholar] [CrossRef]
- Kaur, S.; Sassano, A.; Joseph, A.M.; Majchrzak-Kita, B.; Eklund, E.A.; Verma, A.; Brachmann, S.M.; Fish, E.N.; Platanias, L.C. Dual regulatory roles of phosphatidylinositol 3-kinase in IFN signaling. J. Immunol. 2008, 181, 7316–7323. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Mechanism of PI3K-AKT Activation | Effects of PI3K-AKT Activation on | ||||
|---|---|---|---|---|---|
| Metabolism | Autophagy | Apoptosis | Trafficking RC | ||
| SFV | Strong activation via YXXM motif in nsP3 | Increases glycolysis and fatty acid synthesis | Blocks degradation of autophagosomes | Small, not significant delay | RCs traffic from PM to CPV-I |
| RRV | Strong activation via YXXM motif in nsP3 | Increases fatty acid synthesis | Unknown | Small, not significant delay | RCs traffic from PM to CPV-I |
| CHIKV | Moderate activation by unknown mechanism | Unknown | Increases production of autophagosomes | Unknown | RC mostly remain at PM |
| SINV | Weak or transient activation by unknown mechanism | Unknown | Unknown | Unknown | RC mostly remain at PM |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van Huizen, E.; McInerney, G.M. Activation of the PI3K-AKT Pathway by Old World Alphaviruses. Cells 2020, 9, 970. https://doi.org/10.3390/cells9040970
Van Huizen E, McInerney GM. Activation of the PI3K-AKT Pathway by Old World Alphaviruses. Cells. 2020; 9(4):970. https://doi.org/10.3390/cells9040970
Chicago/Turabian StyleVan Huizen, Eline, and Gerald M. McInerney. 2020. "Activation of the PI3K-AKT Pathway by Old World Alphaviruses" Cells 9, no. 4: 970. https://doi.org/10.3390/cells9040970
APA StyleVan Huizen, E., & McInerney, G. M. (2020). Activation of the PI3K-AKT Pathway by Old World Alphaviruses. Cells, 9(4), 970. https://doi.org/10.3390/cells9040970