Lack of Overt Retinal Degeneration in a K42E Dhdds Knock-In Mouse Model of RP59

 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. PCR Genotyping and DNA Analysis
2.3. Spectral Domain Optical Coherence Tomography (SD-OCT)
2.4. Immunohistochemistry (IHC)
2.5. Lectin Cytochemistry
3. Results
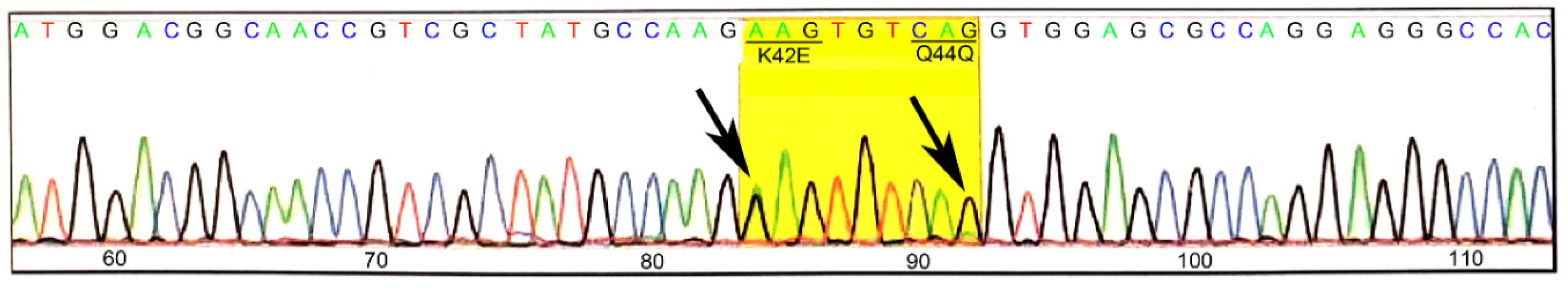
3.1. Generation and Validation of K42E DHDDS Knock-In Mutation
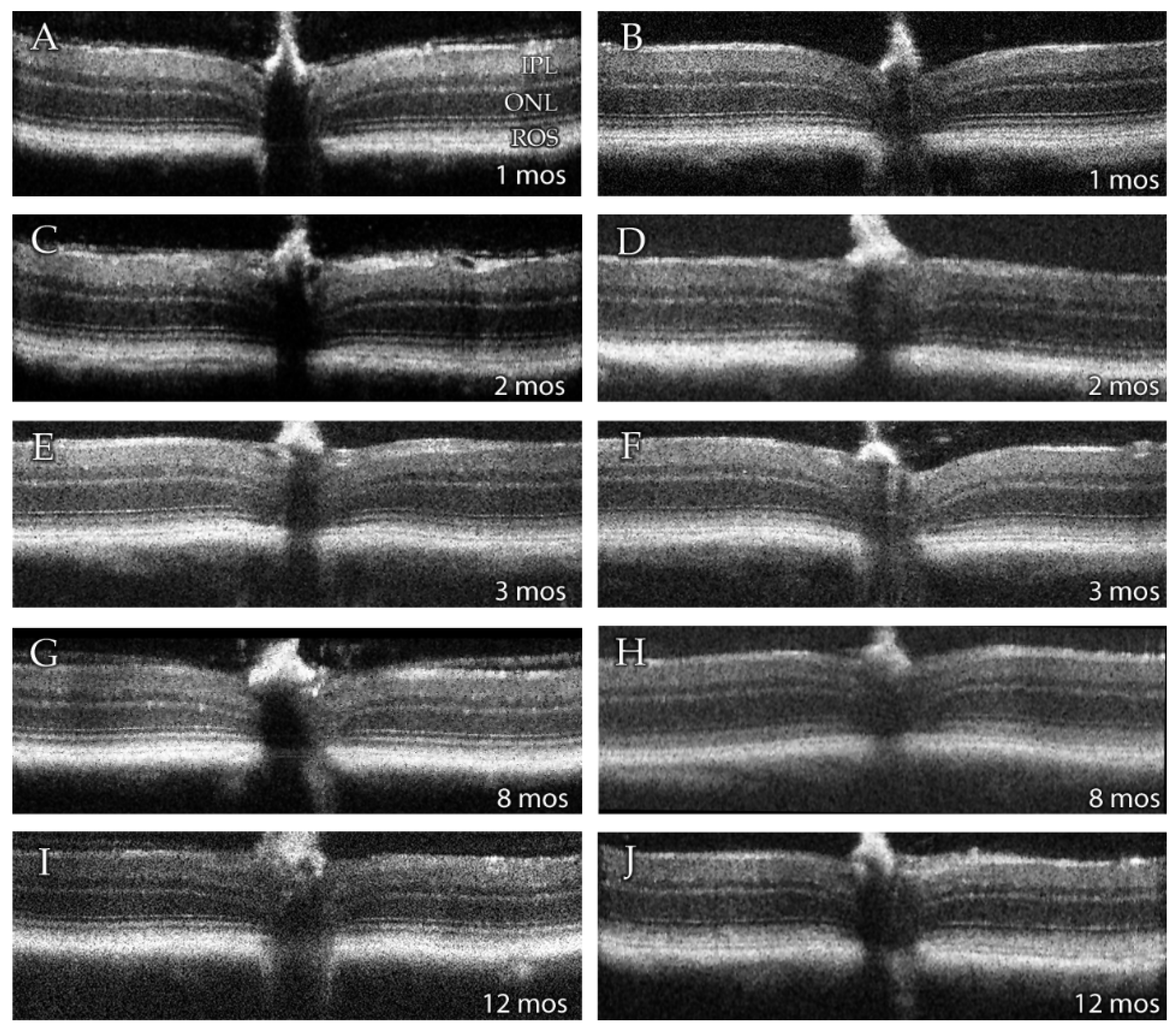
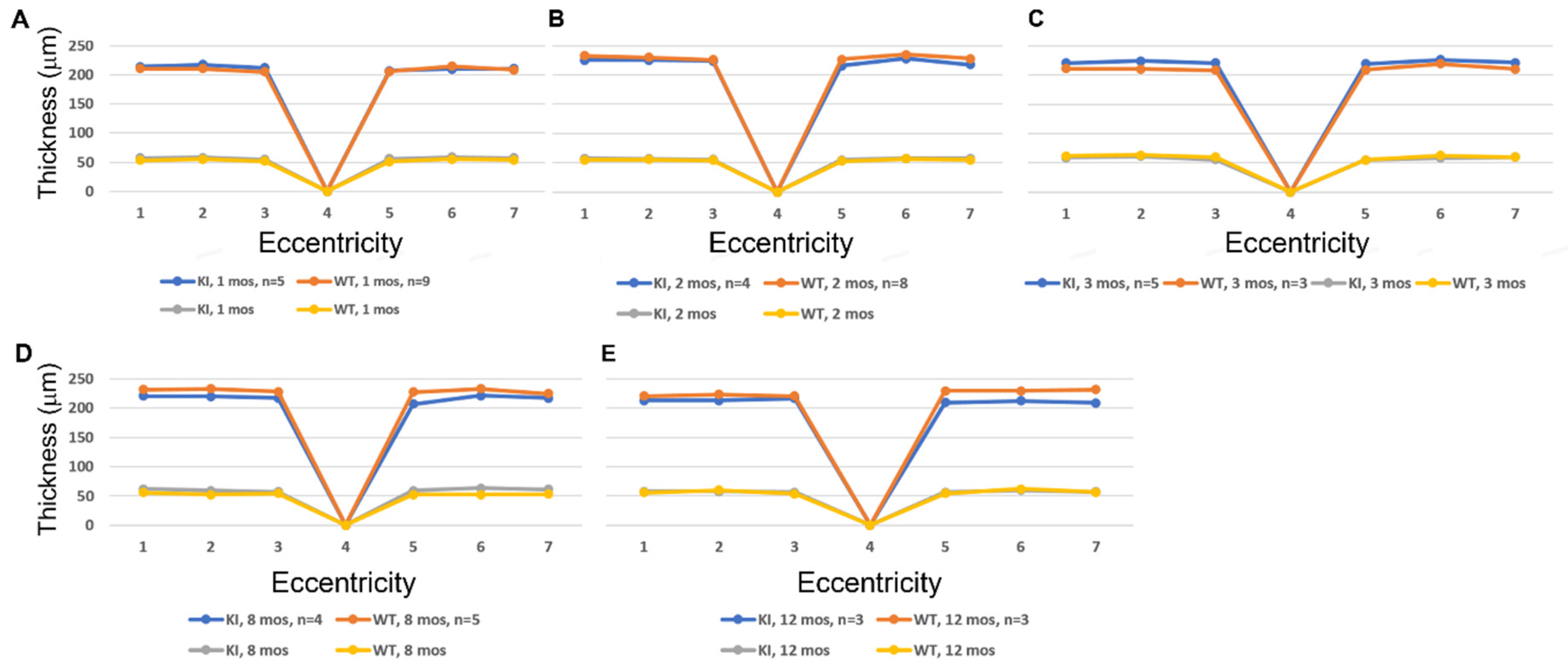
3.2. SD-OCT Analysis Reveals No Evidence for Retinal Degeneration in DhddsK42E/K42E Mice
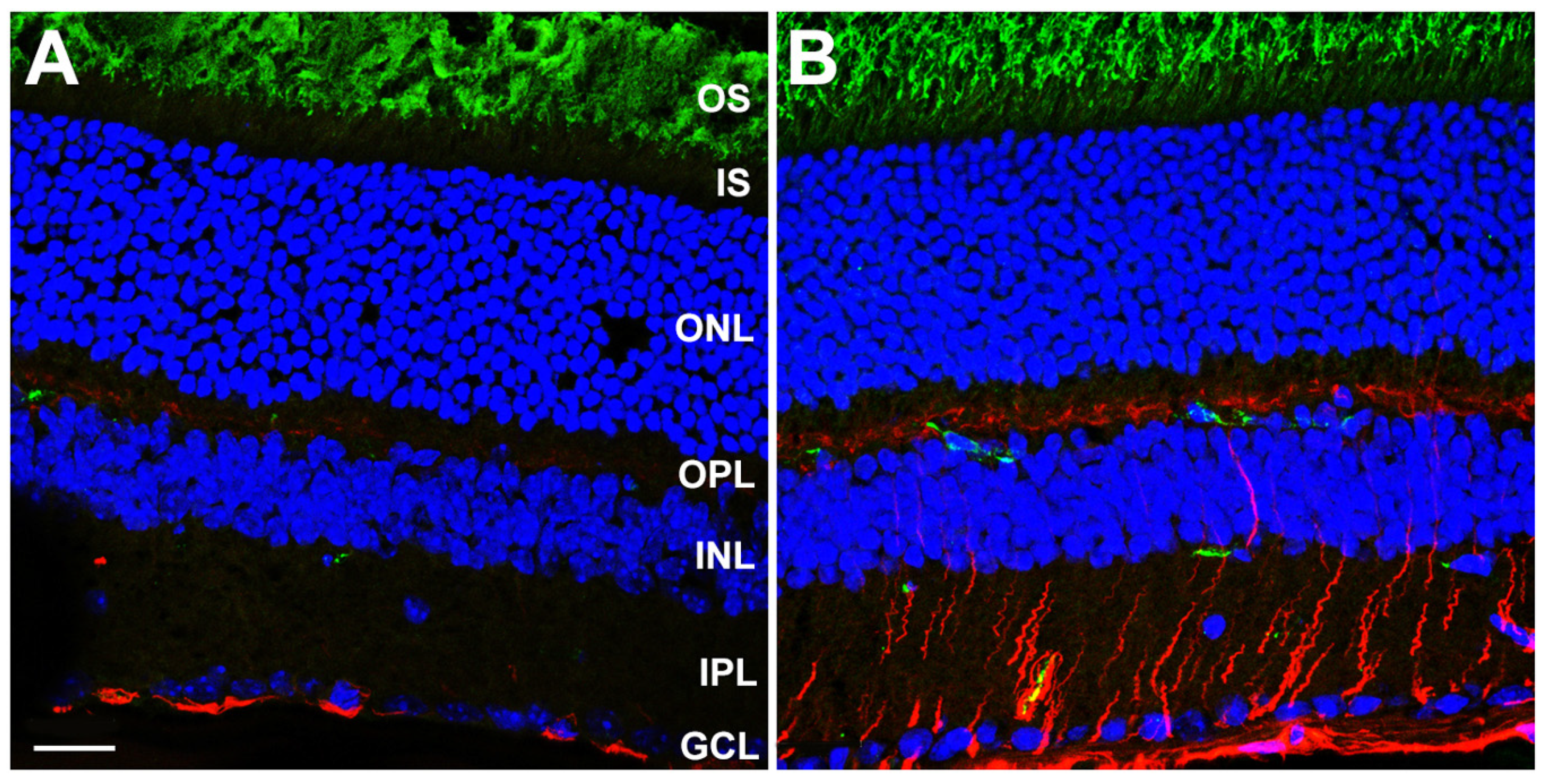
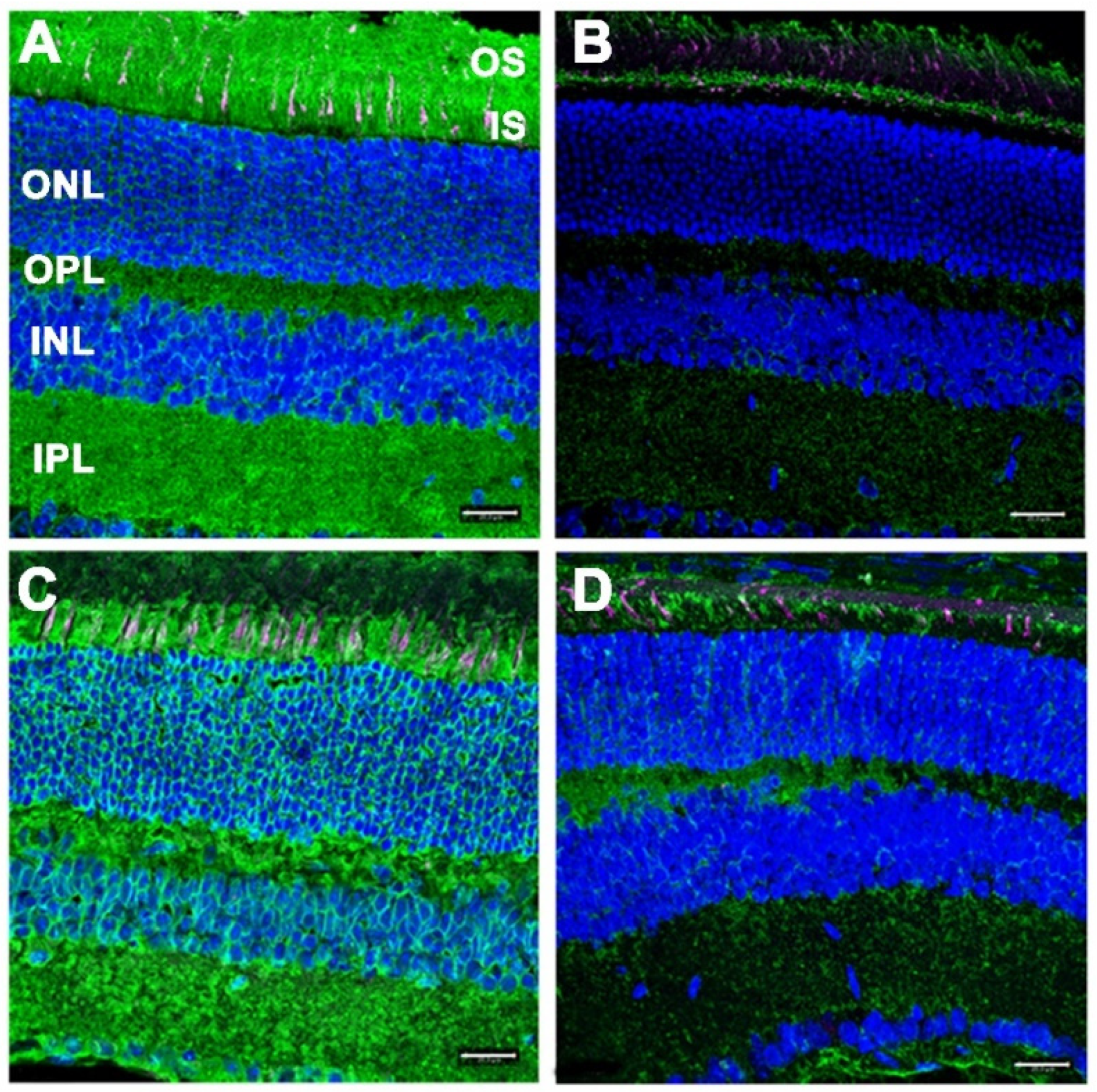
3.3. Gliotic Reactivity, Despite Lack of Overt Neural Retina Degeneration, in DhddsK42E/K42E Mice
3.4. Lack of Defective Protein Glycosylation in DhddsK42E/K42E Mouse Retinas
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hartong, D.T.; Berson, E.L.; Dryja, T.P. Retinitis pigmentosa. Lancet 2006, 368, 1795–1809. [Google Scholar] [CrossRef]
- Zhang, Q. Retinitis Pigmentosa: Progress and Perspective. Asia Pac. J. Ophthalmol. (Phila) 2016, 5, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Zuchner, S.; Dallman, J.; Wen, R.; Beecham, G.; Naj, A.; Farooq, A.; Kohli, M.A.; Whitehead, P.L.; Hulme, W.; Konidari, I.; et al. Whole-exome sequencing links a variant in DHDDS to retinitis pigmentosa. Am. J. Hum. Genet. 2011, 88, 201–206. [Google Scholar] [CrossRef]
- Zelinger, L.; Banin, E.; Obolensky, A.; Mizrahi-Meissonnier, L.; Beryozkin, A.; Bandah-Rozenfeld, D.; Frenkel, S.; Ben-Yosef, T.; Merin, S.; Schwartz, S.B.; et al. A missense mutation in DHDDS, encoding dehydrodolichyl diphosphate synthase, is associated with autosomal-recessive retinitis pigmentosa in Ashkenazi Jews. Am. J. Hum. Genet. 2011, 88, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Lam, B.L.; Zuchner, S.L.; Dallman, J.; Wen, R.; Alfonso, E.C.; Vance, J.M.; Pericak-Vance, M.A. Mutation K42E in dehydrodolichol diphosphate synthase (DHDDS) causes recessive retinitis pigmentosa. Adv. Exp Med. Biol. 2014, 801, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Parodi, A.J.; Leloir, L.F. The role of lipid intermediates in the glycosylation of proteins in the eucaryotic cell. Biochim. Biophys. Acta 1979, 559, 1–37. [Google Scholar] [CrossRef]
- Hemming, F.W. Control and manipulation of the phosphodolichol pathway of protein N-glycosylation. Biosci. Rep. 1982, 2, 203–221. [Google Scholar] [CrossRef]
- Giladi, M.; Edri, I.; Goldenberg, M.; Newman, H.; Strulovich, R.; Khananshvili, D.; Haitin, Y.; Loewenstein, A. Purification and characterization of human dehydrodolychil diphosphate synthase (DHDDS) overexpressed in E. coli. Protein Expr. Purif. 2017, 132, 138–142. [Google Scholar] [CrossRef]
- Lisnyansky Bar-El, M.; Lee, S.Y.; Ki, A.Y.; Kapelushnik, N.; Loewenstein, A.; Chung, K.Y.; Schneidman-Duhovny, D.; Giladi, M.; Newman, H.; Haitin, Y. Structural Characterization of Full-Length Human Dehydrodolichyl Diphosphate Synthase Using an Integrative Computational and Experimental Approach. Biomolecules 2019, 9. [Google Scholar] [CrossRef]
- Behrens, N.H.; Leloir, L.F. Dolichol monophosphate glucose: An intermediate in glucose transfer in liver. Proc. Natl. Acad. Sci. USA 1970, 66, 153–159. [Google Scholar] [CrossRef]
- Tam, B.M.; Moritz, O.L. The role of rhodopsin glycosylation in protein folding, trafficking, and light-sensitive retinal degeneration. J. Neurosci. 2009, 29, 15145–15154. [Google Scholar] [CrossRef] [PubMed]
- Murray, A.R.; Vuong, L.; Brobst, D.; Fliesler, S.J.; Peachey, N.S.; Gorbatyuk, M.S.; Naash, M.I.; Al-Ubaidi, M.R. Glycosylation of rhodopsin is necessary for its stability and incorporation into photoreceptor outer segment discs. Hum. Mol. Genet. 2015, 24, 2709–2723. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fliesler, S.J.; Rayborn, M.E.; Hollyfield, J.G. Membrane morphogenesis in retinal rod outer segments: Inhibition by tunicamycin. J. Cell Biol. 1985, 100, 574–587. [Google Scholar] [CrossRef] [PubMed]
- Fliesler, S.J.; Rapp, L.M.; Hollyfield, J.G. Photoreceptor-specific degeneration caused by tunicamycin. Nature 1984, 311, 575–577. [Google Scholar] [CrossRef]
- DeRamus, M.L.; Stacks, D.A.; Zhang, Y.; Huisingh, C.E.; McGwin, G.; Pittler, S.J. GARP2 accelerates retinal degeneration in rod cGMP-gated cation channel beta-subunit knockout mice. Sci. Rep. 2017, 7, 42545. [Google Scholar] [CrossRef]
- Ramachandra Rao, S.; Pfeffer, B.A.; Mas Gomez, N.; Skelton, L.A.; Keiko, U.; Sparrow, J.R.; Rowsam, A.M.; Mitchell, C.H.; Fliesler, S.J. Compromised phagosome maturation underlies RPE pathology in cell culture and whole animal models of Smith-Lemli-Opitz Syndrome. Autophagy 2018, 1–22. [Google Scholar] [CrossRef]
- Goldstein, I.J.; Hayes, C.E. The lectins: Carbohydrate-binding proteins of plants and animals. Adv. Carbohydr. Chem. Biochem. 1978, 35, 127–340. [Google Scholar] [CrossRef]
- Wang, T.; Voglmeir, J. PNGases as valuable tools in glycoprotein analysis. Protein Pept. Lett. 2014, 21, 976–985. [Google Scholar] [CrossRef]
- Blanks, J.C.; Johnson, L.V. Specific binding of peanut lectin to a class of retinal photoreceptor cells. A species comparison. Invest. Ophthalmol. Vis. Sci. 1984, 25, 546–557. [Google Scholar]
- Johnson, L.V.; Hageman, G.S.; Blanks, J.C. Interphotoreceptor matrix domains ensheath vertebrate cone photoreceptor cells. Invest. Ophthalmol. Vis. Sci. 1986, 27, 129–135. [Google Scholar]
- Fariss, R.N.; Anderson, D.H.; Fisher, S.K. Comparison of photoreceptor-specific matrix domains in the cat and monkey retinas. Exp. Eye Res. 1990, 51, 473–485. [Google Scholar] [CrossRef]
- Gemmill, T.R.; Trimble, R.B. Overview of N- and O-linked oligosaccharide structures found in various yeast species. Biochim. Biophys. Acta 1999, 1426, 227–237. [Google Scholar] [CrossRef]
- Li, Y.; Lam, B.L.; Guan, Z.; Wang, Z.; Wang, N.; Chen, Y.; Wen, R. Photoreceptor degeneration in the DHDDSK42E/K42E mouse. Invest. Ophthalmol. Vis. Sci. 2014, 55, 4371. [Google Scholar]
- Sabry, S.; Vuillaumier-Barrot, S.; Mintet, E.; Fasseu, M.; Valayannopoulos, V.; Heron, D.; Dorison, N.; Mignot, C.; Seta, N.; Chantret, I.; et al. A case of fatal Type I congenital disorders of glycosylation (CDG I) associated with low dehydrodolichol diphosphate synthase (DHDDS) activity. Orphanet. J. Rare Dis. 2016, 11, 84. [Google Scholar] [CrossRef]
- Wen, R.; Lam, B.L.; Guan, Z. Aberrant dolichol chain lengths as biomarkers for retinitis pigmentosa caused by impaired dolichol biosynthesis. J. Lipid Res. 2013, 54, 3516–3522. [Google Scholar] [CrossRef]
- Wen, R.; Dallman, J.E.; Li, Y.; Zuchner, S.L.; Vance, J.M.; Pericak-Vance, M.A.; Lam, B.L. Knock-down DHDDS expression induces photoreceptor degeneration in zebrafish. Adv. Exp. Med. Biol. 2014, 801, 543–550. [Google Scholar] [CrossRef]
- DeRamus, M.L.; Davis, S.J.; Rao, S.R.; Nyankerh, C.; Stacks, D.; Kraft, T.W.; Fliesler, S.J.; Pittler, S.J. Selective Ablation of Dehydrodolichyl Diphosphate Synthase in Murine Retinal Pigment Epithelium (RPE) Causes RPE Atrophy and Retinal Degeneration. Cells 2020, 9, 771. [Google Scholar] [CrossRef]
- Harrison, K.D.; Park, E.J.; Gao, N.; Kuo, A.; Rush, J.S.; Waechter, C.J.; Lehrman, M.A.; Sessa, W.C. Nogo-B receptor is necessary for cellular dolichol biosynthesis and protein N-glycosylation. EMBO J. 2011, 30, 2490–2500. [Google Scholar] [CrossRef]
- Park, E.J.; Grabinska, K.A.; Guan, Z.; Stranecky, V.; Hartmannova, H.; Hodanova, K.; Baresova, V.; Sovova, J.; Jozsef, L.; Ondruskova, N.; et al. Mutation of Nogo-B receptor, a subunit of cis-prenyltransferase, causes a congenital disorder of glycosylation. Cell Metab. 2014, 20, 448–457. [Google Scholar] [CrossRef]
- Haeuptle, M.A.; Hennet, T. Congenital disorders of glycosylation: An update on defects affecting the biosynthesis of dolichol-linked oligosaccharides. Hum. Mutat. 2009, 30, 1628–1641. [Google Scholar] [CrossRef]
- Ng, B.G.; Freeze, H.H. Perspectives on Glycosylation and Its Congenital Disorders. Trends Genet. 2018, 34, 466–476. [Google Scholar] [CrossRef] [PubMed]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramachandra Rao, S.; Fliesler, S.J.; Kotla, P.; Nguyen, M.N.; Pittler, S.J. Lack of Overt Retinal Degeneration in a K42E Dhdds Knock-In Mouse Model of RP59. Cells 2020, 9, 896. https://doi.org/10.3390/cells9040896
Ramachandra Rao S, Fliesler SJ, Kotla P, Nguyen MN, Pittler SJ. Lack of Overt Retinal Degeneration in a K42E Dhdds Knock-In Mouse Model of RP59. Cells. 2020; 9(4):896. https://doi.org/10.3390/cells9040896
Chicago/Turabian StyleRamachandra Rao, Sriganesh, Steven J. Fliesler, Pravallika Kotla, Mai N. Nguyen, and Steven J. Pittler. 2020. "Lack of Overt Retinal Degeneration in a K42E Dhdds Knock-In Mouse Model of RP59" Cells 9, no. 4: 896. https://doi.org/10.3390/cells9040896
APA StyleRamachandra Rao, S., Fliesler, S. J., Kotla, P., Nguyen, M. N., & Pittler, S. J. (2020). Lack of Overt Retinal Degeneration in a K42E Dhdds Knock-In Mouse Model of RP59. Cells, 9(4), 896. https://doi.org/10.3390/cells9040896