Role of Connexins 30, 36, and 43 in Brain Tumors, Neurodegenerative Diseases, and Neuroprotection

 , ,
, ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Connexins: Gene Structure and Transcriptional Regulation via Epigenetic Mechanisms
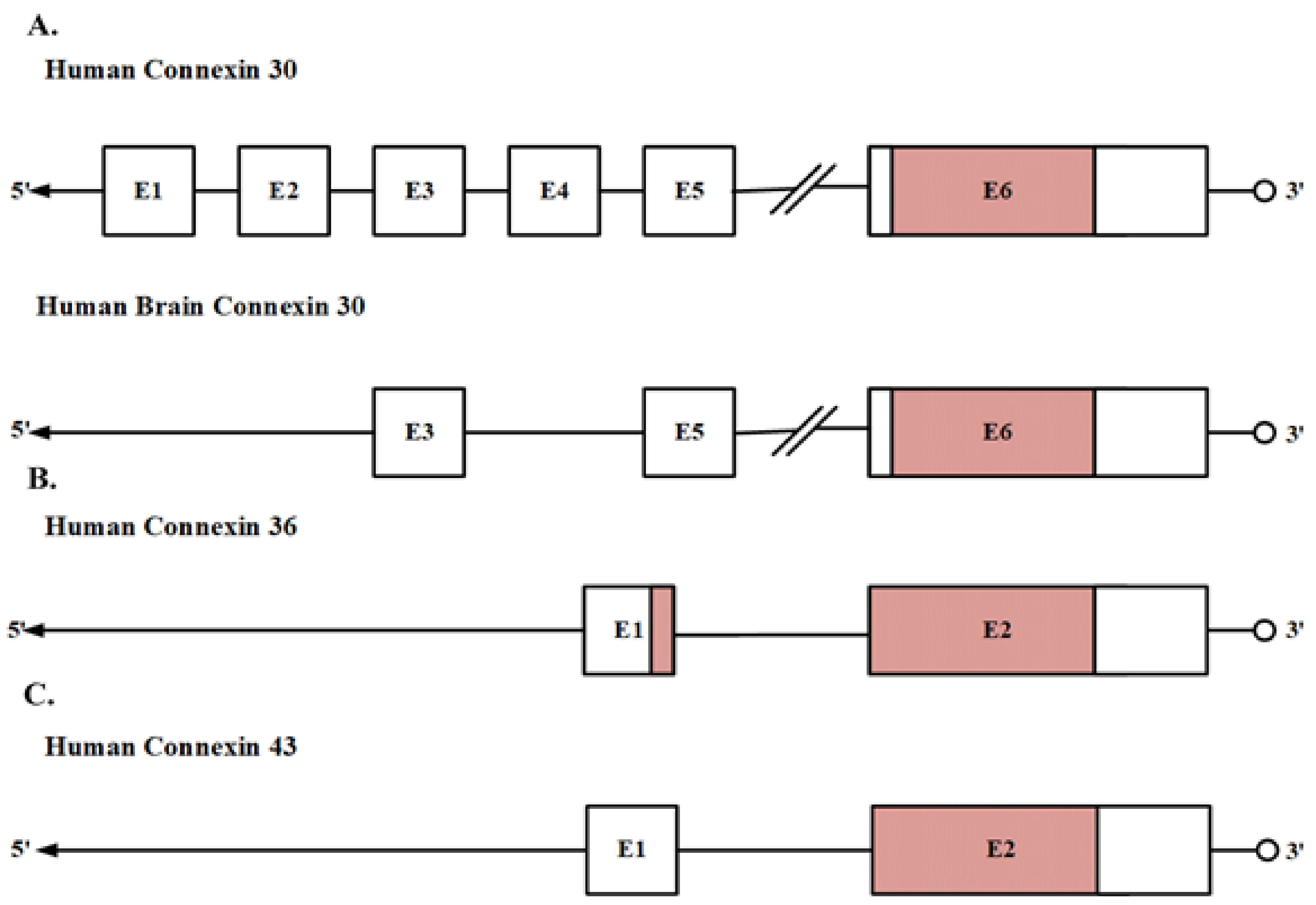
2.1. Gene Structure
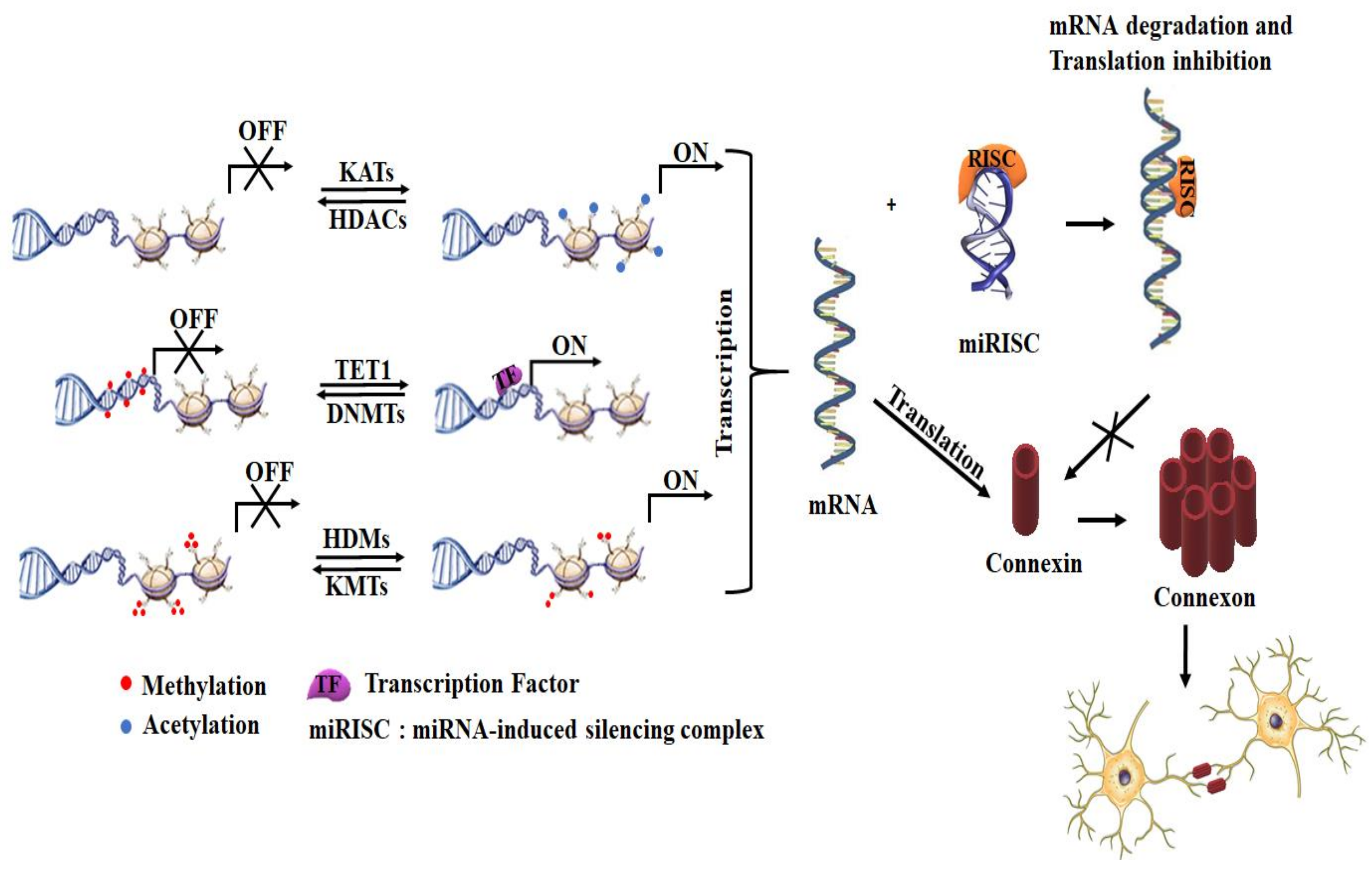
2.2. Transcription Regulation via Epigenetic Mechanisms
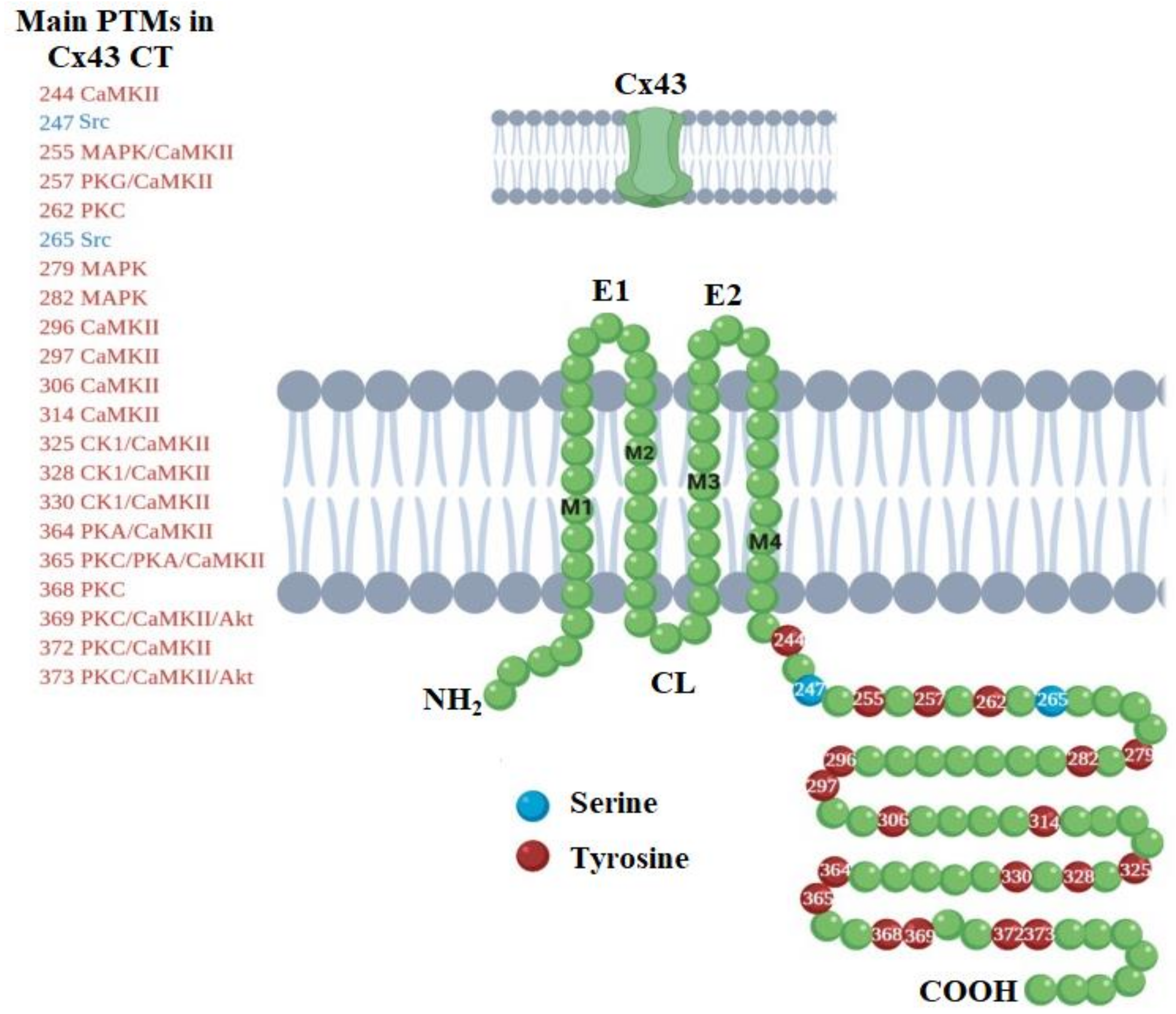
3. Life-Cycle Modulation of Connexins by Post-Translational Modifications
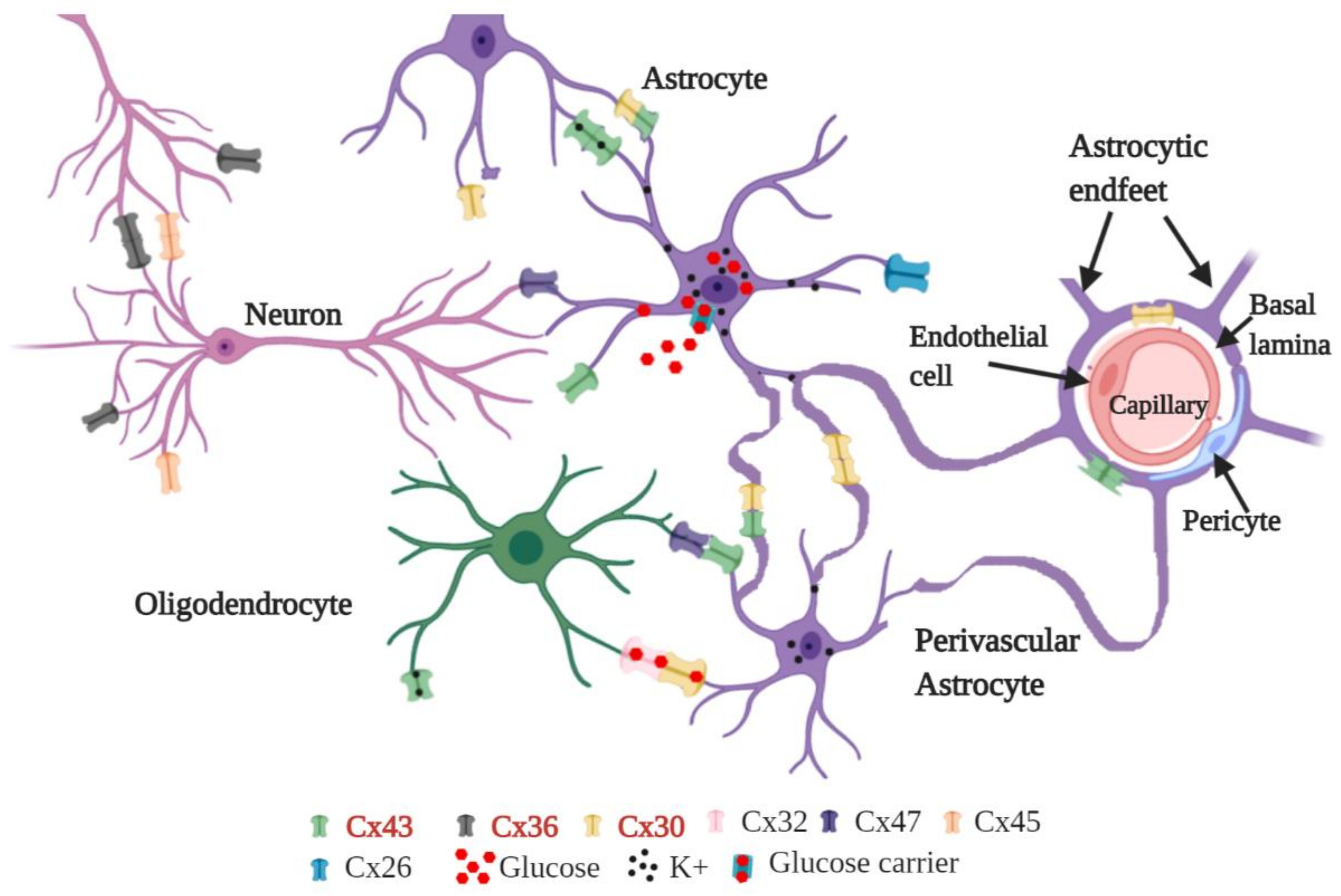
4. Connexins in Diseases of the Nervous System
4.1. Role of Connexins in Glioblastoma
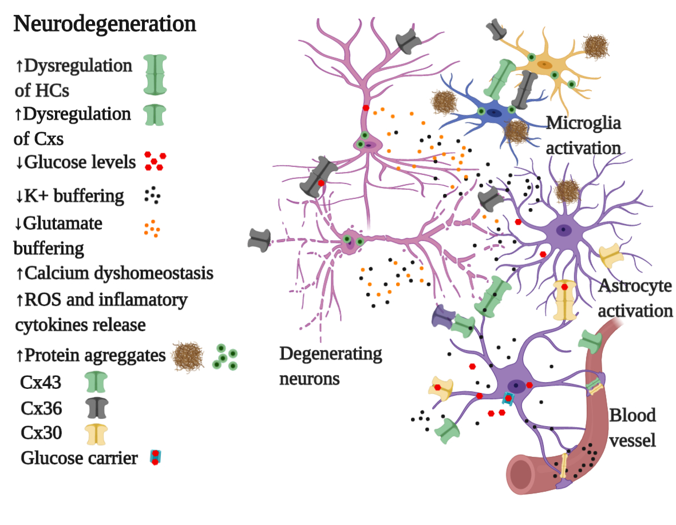
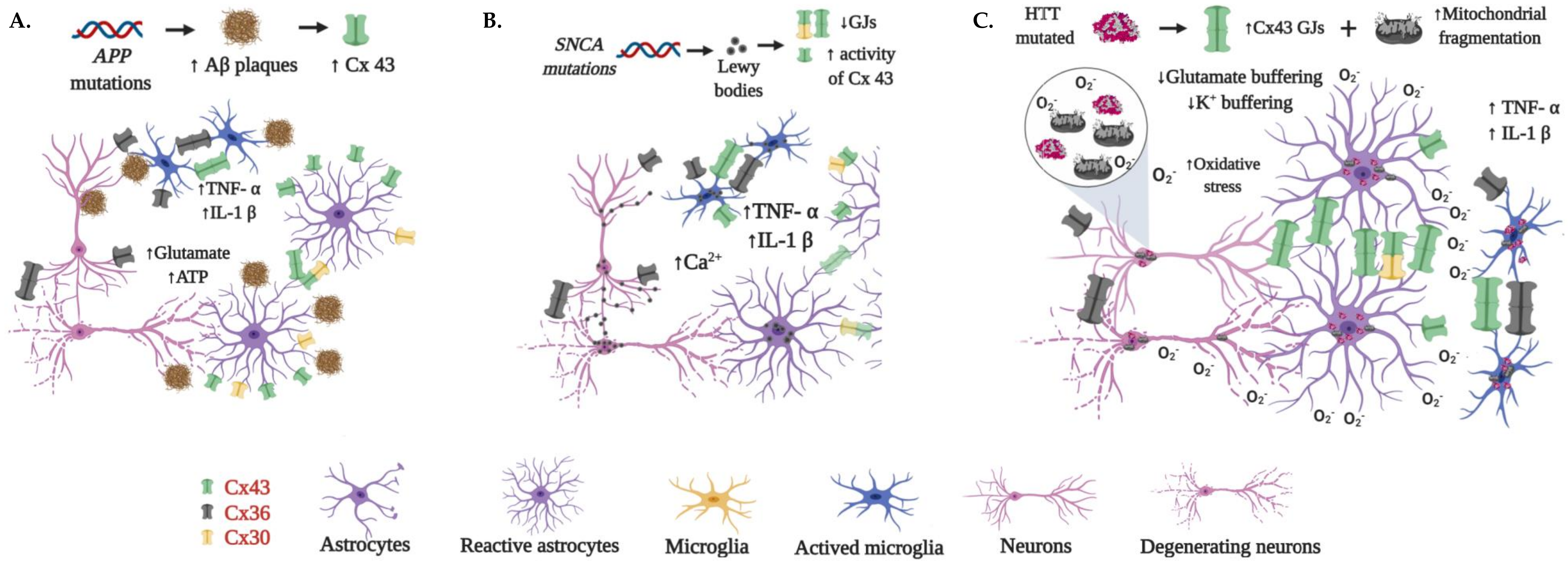
4.2. Connexins in Neurodegeneration
4.2.1. Alzheimer’s Disease
4.2.2. Parkinson’s Disease
4.2.3. Huntington’s Disease
5. Connexins in Neuroprotection
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Murray, L.M.; Krasnodembskaya, A.D. Concise review: intercellular communication via organelle transfer in the biology and therapeutic applications of stem cells. Stem Cells 2019, 37, 14–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sáez, J.C.; Berthoud, V.M.; Branes, M.C.; Martinez, A.D.; Beyer, E.C. Plasma membrane channels formed by connexins: their regulation and functions. Physiol. Rev. 2003, 83, 1359–1400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, B.C.; Purdy, M.D.; Baker, K.A.; Acharya, C.; McIntire, W.E.; Stevens, R.C.; Zhang, Q.; Harris, A.L.; Abagyan, R.; Yeager, M. An electrostatic mechanism for Ca 2+-mediated regulation of gap junction channels. Nat. Commun. 2016, 7, 8770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, D.B.; Goldberg, G.S. Transfer of biologically important molecules between cells through gap junction channels. Curr. Med. Chem. 2003, 10, 2045–2058. [Google Scholar] [CrossRef] [PubMed]
- Kielian, T.; Esen, N. Effects of neuroinflammation on glia–glia gap junctional intercellular communication: a perspective. Neurochem. Int. 2004, 45, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Garrett, F.G.; Durham, P.L. Differential expression of connexins in trigeminal ganglion neurons and satellite glial cells in response to chronic or acute joint inflammation. Neuron Glia Biol 2008, 4, 295–306. [Google Scholar] [CrossRef] [Green Version]
- Swayne, L.A.; Bennett, S.A. Connexins and pannexins in neuronal development and adult neurogenesis. BMC Cell Biol. 2016, 17, 39–49. [Google Scholar] [CrossRef] [Green Version]
- Gajardo-Gómez, R.; Labra, V.C.; Orellana, J.A. Connexins and pannexins: new insights into microglial functions and dysfunctions. Front. Mol. Neurosci. 2016, 9, 86. [Google Scholar] [CrossRef]
- Vicario, N.; Zappalà, A.; Calabrese, G.; Gulino, R.; Parenti, C.; Gulisano, M.; Parenti, R. Connexins in the central nervous system: physiological traits and neuroprotective targets. Front. Physiol. 2017, 8, 1060. [Google Scholar] [CrossRef] [Green Version]
- Nagy, J.I.; Dudek, F.E.; Rash, J.E. Update on connexins and gap junctions in neurons and glia in the mammalian nervous system. Brain Res. Rev. 2004, 47, 191–215. [Google Scholar] [CrossRef]
- Nagy, J.; Patel, D.; Ochalski, P.; Stelmack, G. Connexin30 in rodent, cat and human brain: selective expression in gray matter astrocytes, co-localization with connexin43 at gap junctions and late developmental appearance. Neuroscience 1999, 88, 447–468. [Google Scholar] [CrossRef]
- Richter, N.; Wendt, S.; Georgieva, P.B.; Hambardzumyan, D.; Nolte, C.; Kettenmann, H. Glioma-associated microglia and macrophages/monocytes display distinct electrophysiological properties and do not communicate via gap junctions. Neurosci. Lett. 2014, 583, 130–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaikh, S.B.; Uy, B.; Perera, A.; Nicholson, L.F. AGEs–RAGE mediated up-regulation of connexin43 in activated human microglial CHME-5 cells. Neurochem. Int. 2012, 60, 640–651. [Google Scholar] [CrossRef] [PubMed]
- Maezawa, I.; Jin, L.-W. Rett syndrome microglia damage dendrites and synapses by the elevated release of glutamate. J. Neurosci. 2010, 30, 5346–5356. [Google Scholar] [CrossRef] [Green Version]
- Dobrenis, K.; Chang, H.Y.; Pina-Benabou, M.; Woodroffe, A.; Lee, S.; Rozental, R.; Spray, D.C.; Scemes, E. Human and mouse microglia express connexin36, and functional gap junctions are formed between rodent microglia and neurons. J. Neurosci. Res. 2005, 82, 306–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parenti, R.; Campisi, A.; Vanella, A.; Cicirata, F. Immunocytochemical and RT-PCR analysis of connexin36 in cultures of mammalian glial cells. Arch. Ital. De Biol. 2002, 140, 101–108. [Google Scholar]
- Eugenín, E.A.; Eckardt, D.; Theis, M.; Willecke, K.; Bennett, M.V.; Sáez, J.C. Microglia at brain stab wounds express connexin 43 and in vitro form functional gap junctions after treatment with interferon-γ and tumor necrosis factor-α. Proc. Natl. Acad. Sci. 2001, 98, 4190–4195. [Google Scholar] [CrossRef] [Green Version]
- Talaverón, R.; Matarredona, E.R.; de la Cruz, R.R.; Macías, D.; Gálvez, V.; Pastor, A.M. Implanted neural progenitor cells regulate glial reaction to brain injury and establish gap junctions with host glial cells. Glia 2014, 62, 623–638. [Google Scholar] [CrossRef]
- Cepeda, C.; Chang, J.W.; Owens, G.C.; Huynh, M.N.; Chen, J.Y.; Tran, C.; Vinters, H.V.; Levine, M.S.; Mathern, G.W. In Rasmussen encephalitis, hemichannels associated with microglial activation are linked to cortical pyramidal neuron coupling: a possible mechanism for cellular hyperexcitability. Cns Neurosci. Ther. 2015, 21, 152–163. [Google Scholar] [CrossRef]
- Moon, Y.; Choi, S.Y.; Kim, K.; Kim, H.; Sun, W. Expression of connexin29 and 32 in the penumbra region after traumatic brain injury of mice. Neuroreport 2010, 21, 1135–1139. [Google Scholar] [CrossRef]
- Pannasch, U.; Rouach, N. Emerging role for astroglial networks in information processing: from synapse to behavior. Trends Neurosci. 2013, 36, 405–417. [Google Scholar] [CrossRef] [PubMed]
- Rampon, C.; Jiang, C.H.; Dong, H.; Tang, Y.-P.; Lockhart, D.J.; Schultz, P.G.; Tsien, J.Z.; Hu, Y. Effects of environmental enrichment on gene expression in the brain. Proc. Natl. Acad. Sci. 2000, 97, 12880–12884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dere, E.; De Souza-Silva, M.; Frisch, C.; Teubner, B.; Söhl, G.; Willecke, K.; Huston, J. Connexin30-deficient mice show increased emotionality and decreased rearing activity in the open-field along with neurochemical changes. Eur. J. Neurosci. 2003, 18, 629–638. [Google Scholar] [CrossRef] [PubMed]
- Pannasch, U.; Freche, D.; Dallérac, G.; Ghézali, G.; Escartin, C.; Ezan, P.; Cohen-Salmon, M.; Benchenane, K.; Abudara, V.; Dufour, A. Connexin 30 sets synaptic strength by controlling astroglial synapse invasion. Nat. Neurosci. 2014, 17, 549. [Google Scholar] [CrossRef]
- Belluardo, N.; Mudò, G.; Trovato-Salinaro, A.; Le Gurun, S.; Charollais, A.; Serre-Beinier, V.; Amato, G.; Haefliger, J.-A.; Meda, P.; Condorelli, D.F. Expression of connexin36 in the adult and developing rat brain. Brain Res. 2000, 865, 121–138. [Google Scholar] [CrossRef]
- De Zeeuw, C.I.; Chorev, E.; Devor, A.; Manor, Y.; Van Der Giessen, R.S.; De Jeu, M.T.; Hoogenraad, C.C.; Bijman, J.; Ruigrok, T.J.; French, P. Deformation of network connectivity in the inferior olive of connexin 36-deficient mice is compensated by morphological and electrophysiological changes at the single neuron level. J. Neurosci. 2003, 23, 4700–4711. [Google Scholar] [CrossRef] [Green Version]
- Deans, M.R.; Gibson, J.R.; Sellitto, C.; Connors, B.W.; Paul, D.L. Synchronous activity of inhibitory networks in neocortex requires electrical synapses containing connexin36. Neuron 2001, 31, 477–485. [Google Scholar] [CrossRef] [Green Version]
- Hormuzdi, S.G.; Pais, I.; LeBeau, F.E.; Towers, S.K.; Rozov, A.; Buhl, E.H.; Whittington, M.A.; Monyer, H. Impaired electrical signaling disrupts gamma frequency oscillations in connexin 36-deficient mice. Neuron 2001, 31, 487–495. [Google Scholar] [CrossRef] [Green Version]
- Frisch, C.; Souza-Silva, M.A.D.; Söhl, G.; Güldenagel, M.; Willecke, K.; Huston, J.P.; Dere, E. Stimulus complexity dependent memory impairment and changes in motor performance after deletion of the neuronal gap junction protein connexin36 in mice. Behav. Brain Res. 2005, 157, 177–185. [Google Scholar] [CrossRef]
- Maier, N.; Güldenagel, M.; Söhl, G.; Siegmund, H.; Willecke, K.; Draguhn, A. Reduction of high-frequency network oscillations (ripples) and pathological network discharges in hippocampal slices from connexin 36-deficient mice. J. Physiol. 2002, 541, 521–528. [Google Scholar] [CrossRef]
- Long, M.A.; Deans, M.R.; Paul, D.L.; Connors, B.W. Rhythmicity without synchrony in the electrically uncoupled inferior olive. J. Neurosci. 2002, 22, 10898–10905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marshall, S.P.; Van Der Giessen, R.S.; De Zeeuw, C.I.; Lang, E.J. Altered olivocerebellar activity patterns in the connexin36 knockout mouse. Cerebellum 2007, 6, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Placantonakis, D.G.; Bukovsky, A.A.; Aicher, S.A.; Kiem, H.-P.; Welsh, J.P. Continuous electrical oscillations emerge from a coupled network: a study of the inferior olive using lentiviral knockdown of connexin36. J. Neurosci. 2006, 26, 5008–5016. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.M.; Roach, J.P.; Hu, A.; Stamatovic, S.M.; Zochowski, M.R.; Keep, R.F.; Andjelkovic, A.V. Connexin 43 gap junctions contribute to brain endothelial barrier hyperpermeability in familial cerebral cavernous malformations type III by modulating tight junction structure. Faseb J. 2018, 32, 2615–2629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouach, N.; Avignone, E.; Meme, W.; Koulakoff, A.; Venance, L.; Blomstrand, F.; Giaume, C. Gap junctions and connexin expression in the normal and pathological central nervous system. Biol. Cell 2002, 94, 457–475. [Google Scholar] [CrossRef]
- De Bock, M.; Leybaert, L.; Giaume, C. Connexin channels at the glio-vascular interface: gatekeepers of the brain. Neurochem. Res. 2017, 42, 2519–2536. [Google Scholar] [CrossRef]
- Charron, G.; Doudnikoff, E.; Canron, M.-H.; Li, Q.; Véga, C.; Marais, S.; Baufreton, J.; Vital, A.; Oliet, S.H.; Bezard, E. Astrocytosis in parkinsonism: considering tripartite striatal synapses in physiopathology? Front. Aging Neurosci. 2014, 6, 258. [Google Scholar] [CrossRef] [Green Version]
- Fujita, A.; Yamaguchi, H.; Yamasaki, R.; Cui, Y.; Matsuoka, Y.; Yamada, K.-i.; Kira, J.-i. Connexin 30 deficiency attenuates A2 astrocyte responses and induces severe neurodegeneration in a 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine hydrochloride Parkinson’s disease animal model. J. Neuroinflammation 2018, 15, 227. [Google Scholar] [CrossRef] [Green Version]
- Belousov, A.B.; Fontes, J.D. Neuronal gap junctions: making and breaking connections during development and injury. Trends Neurosci. 2013, 36, 227–236. [Google Scholar] [CrossRef] [Green Version]
- Orellana, J.A.; Shoji, K.F.; Abudara, V.; Ezan, P.; Amigou, E.; Sáez, P.J.; Jiang, J.X.; Naus, C.C.; Sáez, J.C.; Giaume, C. Amyloid β-induced death in neurons involves glial and neuronal hemichannels. J. Neurosci. 2011, 31, 4962–4977. [Google Scholar] [CrossRef]
- Aasen, T.; Johnstone, S.; Vidal-Brime, L.; Lynn, K.S.; Koval, M. Connexins: Synthesis, post-translational modifications, and trafficking in health and disease. Int. J. Mol. Sci. 2018, 19, 1296. [Google Scholar] [CrossRef] [Green Version]
- Oyamada, M.; Takebe, K.; Oyamada, Y. Regulation of connexin expression by transcription factors and epigenetic mechanisms. Biochim. Et Biophys. Acta (Bba) Biomembr. 2013, 1828, 118–133. [Google Scholar] [CrossRef] [Green Version]
- Berson, A.; Nativio, R.; Berger, S.L.; Bonini, N.M. Epigenetic regulation in neurodegenerative diseases. Trends Neurosci. 2018, 41, 587–598. [Google Scholar] [CrossRef]
- Carlberg, C.; Molnár, F. Cancer Epigenetics. In Human Epigenetics: How Science Works; Springer: New York, NY, USA, 2019; pp. 89–99. [Google Scholar]
- Oyamada, M.; Oyamada, Y.; Takamatsu, T. Regulation of connexin expression. Biochim. Et Biophys. Acta (Bba) Biomembr. 2005, 1719, 6–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Essenfelder, G.M.; Larderet, G.; Waksman, G.; Lamartine, J. Gene structure and promoter analysis of the human GJB6 gene encoding connexin30. Gene 2005, 350, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Belluardo, N.; Trovato-Salinaro, A.; Mudo, G.; Hurd, Y.; Condorelli, D. Structure, chromosomal localization, and brain expression of human Cx36 gene. J. Neurosci. Res. 1999, 57, 740–752. [Google Scholar] [CrossRef]
- Paznekas, W.A.; Boyadjiev, S.A.; Shapiro, R.E.; Daniels, O.; Wollnik, B.; Keegan, C.E.; Innis, J.W.; Dinulos, M.B.; Christian, C.; Hannibal, M.C. Connexin 43 (GJA1) mutations cause the pleiotropic phenotype of oculodentodigital dysplasia. Am. J. Hum. Genet. 2003, 72, 408–418. [Google Scholar] [CrossRef] [Green Version]
- Račkauskas, M.; Neverauskas, V.; Skeberdis, V.A. Diversity and properties of connexin gap junction channels. Medicina 2010, 46, 1. [Google Scholar] [CrossRef] [Green Version]
- Li, E. Chromatin modification and epigenetic reprogramming in mammalian development. Nat. Rev. Genet. 2002, 3, 662. [Google Scholar] [CrossRef]
- Bhutani, N.; Burns, D.M.; Blau, H.M. DNA Demethylation Dynamics. Cell 2011, 146, 866–872. [Google Scholar] [CrossRef] [Green Version]
- Karmodiya, K.; Krebs, A.R.; Oulad-Abdelghani, M.; Kimura, H.; Tora, L. H3K9 and H3K14 acetylation co-occur at many gene regulatory elements, while H3K14ac marks a subset of inactive inducible promoters in mouse embryonic stem cells. Bmc Genom. 2012, 13, 424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381. [Google Scholar] [CrossRef] [PubMed]
- Hon, G.C.; Hawkins, R.D.; Ren, B. Predictive chromatin signatures in the mammalian genome. Hum. Mol. Genet. 2009, 18, R195–R201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janssen, A.; Colmenares, S.U.; Karpen, G.H. Heterochromatin: Guardian of the Genome. Annu. Rev. Cell Dev. Biol. 2018, 34, 265–288. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.K.; Workman, J.L. Histone acetyltransferase complexes: one size doesn’t fit all. Nat. Rev. Mol. Cell Biol. 2007, 8, 284. [Google Scholar] [CrossRef]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [Green Version]
- Heintzman, N.D.; Stuart, R.K.; Hon, G.; Fu, Y.; Ching, C.W.; Hawkins, R.D.; Barrera, L.O.; Van Calcar, S.; Qu, C.; Ching, K.A. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 2007, 39, 311. [Google Scholar] [CrossRef] [PubMed]
- Youn, H.-D. Methylation and demethylation of DNA and histones in chromatin: the most complicated epigenetic marker. Exp. Mol. Med. 2017, 49, e321. [Google Scholar] [CrossRef] [Green Version]
- Hata, A.; Lieberman, J. Dysregulation of microRNA biogenesis and gene silencing in cancer. Sci. Signal. 2015, 8, re3. [Google Scholar] [CrossRef] [Green Version]
- Quévillon Huberdeau, M.; Simard, M.J. A guide to microRNA-mediated gene silencing. Febs J. 2019, 286, 642–652. [Google Scholar] [CrossRef]
- Vinken, M. Regulation of connexin signaling by the epigenetic machinery. Biochim. Et Biophys. Acta (Bba) Gene Regul. Mech. 2016, 1859, 262–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hohl, M.; Thiel, G. Cell type-specific regulation of RE-1 silencing transcription factor (REST) target genes. Eur. J. Neurosci. 2005, 22, 2216–2230. [Google Scholar] [CrossRef] [PubMed]
- Kaut, O.; Sharma, A.; Schmitt, I.; Hurlemann, R.; Wüllner, U. DNA methylation of DLG4 and GJA-1 of human hippocampus and prefrontal cortex in major depression is unchanged in comparison to healthy individuals. J. Clin. Neurosci. 2017, 43, 261–263. [Google Scholar] [CrossRef] [PubMed]
- Ernst, C.; Nagy, C.; Kim, S.; Yang, J.P.; Deng, X.; Hellstrom, I.C.; Choi, K.H.; Gershenfeld, H.; Meaney, M.J.; Turecki, G. Dysfunction of Astrocyte Connexins 30 and 43 in Dorsal Lateral Prefrontal Cortex of Suicide Completers. Biol. Psychiatry 2011, 70, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Jayalakshmi, J.; Vanisree, A.J.; Ravisankar, S.; K, R. Site specific hypermethylation of CpGs in Connexin genes 30, 26 and 43 in different grades of glioma and attenuated levels of their mRNAs. Int. J. Neurosci. 2019, 129, 273–282. [Google Scholar]
- Nagy, C.; Torres-Platas, S.G.; Mechawar, N.; Turecki, G. Repression of astrocytic connexins in cortical and subcortical brain regions and prefrontal enrichment of H3K9me3 in depression and suicide. Int. J. Neuropsychopharmacol. 2016, 20, 50–57. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Krichevsky, A.; Grad, Y.; Hayes, G.D.; Kosik, K.S.; Church, G.M.; Ruvkun, G. Identification of many microRNAs that copurify with polyribosomes in mammalian neurons. Proc. Natl. Acad. Sci. 2004, 101, 360–365. [Google Scholar] [CrossRef] [Green Version]
- Smirnova, L.; Gräfe, A.; Seiler, A.; Schumacher, S.; Nitsch, R.; Wulczyn, F.G. Regulation of miRNA expression during neural cell specification. Eur. J. Neurosci. 2005, 21, 1469–1477. [Google Scholar] [CrossRef]
- Neumann, E.; Hermanns, H.; Barthel, F.; Werdehausen, R.; Brandenburger, T. Expression changes of microRNA-1 and its targets Connexin 43 and brain-derived neurotrophic factor in the peripheral nervous system of chronic neuropathic rats. Mol. Pain 2015, 11, 39. [Google Scholar] [CrossRef] [Green Version]
- Hao, J.; Zhang, C.; Zhang, A.; Wang, K.; Jia, Z.; Wang, G.; Han, L.; Kang, C.; Pu, P. miR-221/222 is the regulator of Cx43 expression in human glioblastoma cells. Oncol. Rep. 2012, 27, 1504–1510. [Google Scholar]
- Zhang, C.; Liu, C.-F.; Chen, A.-B.; Yao, Z.; Li, W.-G.; Xu, S.-J.; Ma, X. Prognostic and clinic pathological value of Cx43 expression in glioma: a meta-analysis. Front. Oncol. 2019, 9, 1209. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Du, J.; Jiang, H. Post-Translational Modifications to Regulate Protein Function. Wiley Encycl. Chem. Biol. 2007, 1–31. [Google Scholar]
- Jürgen Dohmen, R. SUMO protein modification. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2004, 1695, 113–131. [Google Scholar]
- Kresge, N.; Simoni, R.D.; Hill, R.L. The Discovery of Ubiquitin-mediated Proteolysis by Aaron Ciechanover, Avram Hershko, and Irwin Rose. J. Biol. Chem. 2006, 281, e32. [Google Scholar]
- Axelsen, L.N.; Calloe, K.; Holstein-Rathlou, N.-H.; Nielsen, M.S. Managing the complexity of communication: regulation of gap junctions by post-translational modification. Front. Pharmacol. 2013, 4, 130. [Google Scholar] [CrossRef] [Green Version]
- Leithe, E. Regulation of connexins by the ubiquitin system: Implications for intercellular communication and cancer. Biochim. Et Biophys. Acta (Bba) Rev. Cancer 2016, 1865, 133–146. [Google Scholar] [CrossRef]
- Pogoda, K.; Kameritsch, P.; Retamal, M.A.; Vega, J.L. Regulation of gap junction channels and hemichannels by phosphorylation and redox changes: a revision. Bmc Cell Biol. 2016, 17, 11. [Google Scholar] [CrossRef] [Green Version]
- Solan, J.L.; Lampe, P.D. Specific Cx43 phosphorylation events regulate gap junction turnover in vivo. Febs Lett. 2014, 588, 1423–1429. [Google Scholar] [CrossRef] [Green Version]
- Totland, M.Z.; Rasmussen, N.L.; Knudsen, L.M.; Leithe, E. Regulation of gap junction intercellular communication by connexin ubiquitination: physiological and pathophysiological implications. Cell. Mol. Life Sci. 2019, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Leithe, E.; Mesnil, M.; Aasen, T. The connexin 43 C-terminus: A tail of many tales. Biochim. Et Biophys. Acta (Bba) Biomembr. 2018, 1860, 48–64. [Google Scholar] [CrossRef]
- Johnson, K.E.; Mitra, S.; Katoch, P.; Kelsey, L.S.; Johnson, K.R.; Mehta, P.P. Phosphorylation on Ser-279 and Ser-282 of connexin43 regulates endocytosis and gap junction assembly in pancreatic cancer cells. Mol. Biol. Cell 2013, 24, 715–733. [Google Scholar] [CrossRef] [PubMed]
- Smyth, J.W.; Zhang, S.S.; Sanchez, J.M.; Lamouille, S.; Vogan, J.M.; Hesketh, G.G.; Hong, T.; Tomaselli, G.F.; Shaw, R.M. A 14-3-3 mode-1 binding motif initiates gap junction internalization during acute cardiac ischemia. Traffic 2014, 15, 684–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, C.D.; Lampe, P.D. Casein kinase 1 regulates connexin-43 gap junction assembly. J. Biol. Chem. 2002, 277, 44962–44968. [Google Scholar] [CrossRef] [Green Version]
- Kanemitsu, M.; Jiang, W.; Eckhart, W. Cdc2-mediated phosphorylation of the gap junction protein, connexin43, during mitosis. Cell Growth Differ. Mol. Biol. J. Am. Assoc. Cancer Res. 1998, 9, 13–21. [Google Scholar]
- Lampe, P.D.; Kurata, W.E.; Warn-Cramer, B.J.; Lau, A.F. Formation of a distinct connexin43 phosphoisoform in mitotic cells is dependent upon p34cdc2 kinase. J. Cell Sci. 1998, 111, 833–841. [Google Scholar]
- Li, H.; Spagnol, G.; Zheng, L.; Stauch, K.L.; Sorgen, P.L. Regulation of connexin43 function and expression by tyrosine kinase 2. J. Biol. Chem. 2016, 291, 15867–15880. [Google Scholar] [CrossRef] [Green Version]
- Alev, C.; Urschel, S.; Sonntag, S.; Zoidl, G.; Fort, A.G.; Höher, T.; Matsubara, M.; Willecke, K.; Spray, D.C.; Dermietzel, R. The neuronal connexin36 interacts with and is phosphorylated by CaMKII in a way similar to CaMKII interaction with glutamate receptors. Proc. Natl. Acad. Sci. 2008, 105, 20964–20969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- del Corsso, C.; Iglesias, R.; Zoidl, G.; Dermietzel, R.; Spray, D.C. Calmodulin dependent protein kinase increases conductance at gap junctions formed by the neuronal gap junction protein connexin36. Brain Res. 2012, 1487, 69–77. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Chuang, A.Z.; O’Brien, J. Photoreceptor coupling is controlled by connexin 35 phosphorylation in zebrafish retina. J. Neurosci. 2009, 29, 15178–15186. [Google Scholar] [CrossRef] [Green Version]
- Ribelayga, C.; Cao, Y.; Mangel, S.C. The circadian clock in the retina controls rod-cone coupling. Neuron 2008, 59, 790–801. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, X.; Winbow, V.M.; Patel, L.S.; Burr, G.S.; Mitchell, C.K.; O’Brien, J. Protein kinase A mediates regulation of gap junctions containing connexin35 through a complex pathway. Mol. Brain Res. 2005, 135, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, L.S.; Mitchell, C.K.; Dubinsky, W.P.; O’Brien, J. Regulation of gap junction coupling through the neuronal connexin Cx35 by nitric oxide and cGMP. Cell Commun. Adhes. 2006, 13, 41–54. [Google Scholar] [CrossRef] [Green Version]
- Stamler, J.S.; Simon, D.I.; Osborne, J.A.; Mullins, M.E.; Jaraki, O.; Michel, T.; Singel, D.J.; Loscalzo, J. S-nitrosylation of proteins with nitric oxide: synthesis and characterization of biologically active compounds. Proc. Natl. Acad. Sci. 1992, 89, 444–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Straub, A.C.; Billaud, M.; Johnstone, S.R.; Best, A.K.; Yemen, S.; Dwyer, S.T.; Looft-Wilson, R.; Lysiak, J.J.; Gaston, B.; Palmer, L. Compartmentalized connexin 43 s-nitrosylation/denitrosylation regulates heterocellular communication in the vessel wall. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 399–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, X.; Reuss, L.; Altenberg, G.A. Regulation of purified and reconstituted connexin 43 hemichannels by protein kinase C-mediated phosphorylation of Serine 368. J. Biol. Chem. 2004, 279, 20058–20066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Retamal, M.A.; Cortés, C.J.; Reuss, L.; Bennett, M.V.L.; Sáez, J.C. S-nitrosylation and permeation through connexin 43 hemichannels in astrocytes: Induction by oxidant stress and reversal by reducing agents. Proc. Natl. Acad. Sci. USA 2006, 103, 4475–4480. [Google Scholar] [CrossRef] [Green Version]
- Illi, B.; Colussi, C.; Grasselli, A.; Farsetti, A.; Capogrossi, M.C.; Gaetano, C. NO sparks off chromatin: tales of a multifaceted epigenetic regulator. Pharmacol. Ther. 2009, 123, 344–352. [Google Scholar] [CrossRef]
- Rahman, I.; Marwick, J.; Kirkham, P. Redox modulation of chromatin remodeling: impact on histone acetylation and deacetylation, NF-κB and pro-inflammatory gene expression. Biochem. Pharmacol. 2004, 68, 1255–1267. [Google Scholar] [CrossRef]
- Colussi, C.; Rosati, J.; Straino, S.; Spallotta, F.; Berni, R.; Stilli, D.; Rossi, S.; Musso, E.; Macchi, E.; Mai, A. Nε-lysine acetylation determines dissociation from GAP junctions and lateralization of connexin 43 in normal and dystrophic heart. Proc. Natl. Acad. Sci. 2011, 108, 2795–2800. [Google Scholar] [CrossRef] [Green Version]
- Laguesse, S.; Close, P.; Van Hees, L.; Chariot, A.; Malgrange, B.; Nguyen, L. Loss of Elp3 impairs the acetylation and distribution of connexin-43 in the developing cerebral cortex. Front. Cell. Neurosci. 2017, 11, 122. [Google Scholar] [CrossRef]
- Tielens, S.; Huysseune, S.; Godin, J.D.; Chariot, A.; Malgrange, B.; Nguyen, L. Elongator controls cortical interneuron migration by regulating actomyosin dynamics. Cell Res. 2016, 26, 1131. [Google Scholar] [CrossRef] [PubMed]
- Winkler, G.S.; Kristjuhan, A.; Erdjument-Bromage, H.; Tempst, P.; Svejstrup, J.Q. Elongator is a histone H3 and H4 acetyltransferase important for normal histone acetylation levels in vivo. Proc. Natl. Acad. Sci. 2002, 99, 3517–3522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willis, M.S.; Townley-Tilson, W.D.; Kang, E.Y.; Homeister, J.W.; Patterson, C. Sent to destroy: the ubiquitin proteasome system regulates cell signaling and protein quality control in cardiovascular development and disease. Circ. Res. 2010, 106, 463–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geiss-Friedlander, R.; Melchior, F. Concepts in sumoylation: a decade on. Nat. Rev. Mol. Cell Biol. 2007, 8, 947. [Google Scholar] [CrossRef] [PubMed]
- Lynn, B.D.; Li, X.; Hormuzdi, S.G.; Griffiths, E.K.; McGlade, C.J.; Nagy, J.I. E3 ubiquitin ligases LNX 1 and LNX 2 localize at neuronal gap junctions formed by connexin36 in rodent brain and molecularly interact with connexin36. Eur. J. Neurosci. 2018, 48, 3062–3081. [Google Scholar] [CrossRef] [Green Version]
- Chen, V.C.; Kristensen, A.R.; Foster, L.J.; Naus, C.C. Association of Connexin43 with E3 Ubiquitin Ligase TRIM21 Reveals a Mechanism for Gap Junction Phosphodegron Control. J. Proteome Res. 2012, 11, 6134–6146. [Google Scholar] [CrossRef]
- Zhang, F.F.; Morioka, N.; Kitamura, T.; Hisaoka-Nakashima, K.; Nakata, Y. Proinflammatory cytokines downregulate connexin 43-gap junctions via the ubiquitin-proteasome system in rat spinal astrocytes. Biochem. Biophys. Res. Commun. 2015, 464, 1202–1208. [Google Scholar] [CrossRef]
- Kjenseth, A.; Fykerud, T.A.; Sirnes, S.; Bruun, J.; Yohannes, Z.; Kolberg, M.; Omori, Y.; Rivedal, E.; Leithe, E. The gap junction channel protein connexin 43 is covalently modified and regulated by SUMOylation. J. Biol. Chem. 2012, 287, 15851–15861. [Google Scholar] [CrossRef] [Green Version]
- Mayorquin, L.C.; Rodriguez, A.V.; Sutachan, J.J.; Albarracín, S.L. Connexin-Mediated Functional and Metabolic Coupling Between Astrocytes and Neurons. Front. Mol. Neurosci 2018, 11, 118. [Google Scholar] [CrossRef]
- Princen, F.; Robe, P.; Gros, D.; Jarry-Guichard, T.r.s.; Gielen, J.; Merville, M.-P.; Bours, V. Rat gap junction connexin-30 inhibits proliferation of glioma cell lines. Carcinogenesis 2001, 22, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Orellana, J.A.; Froger, N.; Ezan, P.; Jiang, J.X.; Bennett, M.V.; Naus, C.C.; Giaume, C.; Sáez, J.C. ATP and glutamate released via astroglial connexin 43 hemichannels mediate neuronal death through activation of pannexin 1 hemichannels. J. Neurochem. 2011, 118, 826–840. [Google Scholar] [CrossRef] [Green Version]
- Yamasaki, H. Gap junctional intercellular communication and carcinogenesis. In Parallels in Cell to Cell Junctions in Plants and Animals; Springer: Berlin/Heidelberg, Germany, 1990; pp. 115–127. [Google Scholar]
- Brenner, D.R.; Scherer, D.; Muir, K.; Schildkraut, J.; Boffetta, P.; Spitz, M.R.; Le Marchand, L.; Chan, A.T.; Goode, E.L.; Ulrich, C.M. A review of the application of inflammatory biomarkers in epidemiologic cancer research. Cancer Epidemiol. Prev. Biomark. 2014, 23, 1729–1751. [Google Scholar] [CrossRef] [Green Version]
- Zitvogel, L.; Apetoh, L.; Ghiringhelli, F.; Kroemer, G. Immunological aspects of cancer chemotherapy. Nat. Rev. Immunol. 2008, 8, 59. [Google Scholar] [CrossRef]
- Graham, S.V.; Jiang, J.X.; Mesnil, M. Connexins and pannexins: Important players in tumorigenesis, metastasis and potential therapeutics. Int. J. Mol. Sci. 2018, 19, 1645. [Google Scholar] [CrossRef] [Green Version]
- Louis, D.N.; Perry, A.; Reifenberger, G.; Von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [Green Version]
- Galli, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; De Vitis, S.; Fiocco, R.; Foroni, C.; Dimeco, F.; Vescovi, A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004, 64, 7011–7021. [Google Scholar] [CrossRef] [Green Version]
- Ignatova, T.N.; Kukekov, V.G.; Laywell, E.D.; Suslov, O.N.; Vrionis, F.D.; Steindler, D.A. Human cortical glial tumors contain neural stem-like cells expressing astroglial and neuronal markers in vitro. Glia 2002, 39, 193–206. [Google Scholar] [CrossRef]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. nature 2004, 432, 396. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756. [Google Scholar] [CrossRef]
- Liu, G.; Yuan, X.; Zeng, Z.; Tunici, P.; Ng, H.; Abdulkadir, I.R.; Lu, L.; Irvin, D.; Black, K.L.; John, S.Y. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol. Cancer 2006, 5, 67. [Google Scholar] [CrossRef] [Green Version]
- Huang, R.; Lin, Y.; Wang, C.C.; Gano, J.; Lin, B.; Shi, Q.; Boynton, A.; Burke, J.; Huang, R.-P. Connexin 43 suppresses human glioblastoma cell growth by down-regulation of monocyte chemotactic protein 1, as discovered using protein array technology. Cancer Res. 2002, 62, 2806–2812. [Google Scholar]
- Soroceanu, L.; Manning Jr, T.J.; Sontheimer, H. Reduced expression of connexin-43 and functional gap junction coupling in human gliomas. Glia 2001, 33, 107–117. [Google Scholar] [CrossRef]
- Yao, J.; Morioka, T.; Oite, T. PDGF regulates gap junction communication and connexin43 phosphorylation by PI 3-kinase in mesangial cells. Kidney Int. 2000, 57, 1915–1926. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.C.; Xiao, H.L.; Jiang, X.F.; Wang, Q.L.; Li, Y.; Yang, X.J.; Ping, Y.F.; Duan, J.J.; Jiang, J.Y.; Ye, X.Z. Connexin 43 reverses malignant phenotypes of glioma stem cells by modulating E-cadherin. Stem Cells 2012, 30, 108–120. [Google Scholar] [CrossRef]
- Hitomi, M.; Deleyrolle, L.P.; Mulkearns-Hubert, E.E.; Jarrar, A.; Li, M.; Sinyuk, M.; Otvos, B.; Brunet, S.; Flavahan, W.A.; Hubert, C.G. Differential connexin function enhances self-renewal in glioblastoma. Cell Rep. 2015, 11, 1031–1042. [Google Scholar] [CrossRef] [Green Version]
- Grek, C.L.; Sheng, Z.; Naus, C.C.; Sin, W.C.; Gourdie, R.G.; Ghatnekar, G.G. Novel approach to temozolomide resistance in malignant glioma: connexin43-directed therapeutics. Curr. Opin. Pharmacol. 2018, 41, 79–88. [Google Scholar] [CrossRef]
- Osswald, M.; Jung, E.; Sahm, F.; Solecki, G.; Venkataramani, V.; Blaes, J.; Weil, S.; Horstmann, H.; Wiestler, B.; Syed, M. Brain tumour cells interconnect to a functional and resistant network. Nature 2015, 528, 93. [Google Scholar] [CrossRef]
- De Meulenaere, V.; Bonte, E.; Verhoeven, J.; Okito, J.-P.K.; Pieters, L.; Vral, A.; De Wever, O.; Leybaert, L.; Goethals, I.; Vanhove, C. Adjuvant therapeutic potential of tonabersat in the standard treatment of glioblastoma: A preclinical F98 glioblastoma rat model study. Plos One 2019, 14. [Google Scholar] [CrossRef] [Green Version]
- Ismail, F.S.; Moinfar, Z.; Prochnow, N.; Dambach, H.; Hinkerohe, D.; Haase, C.G.; Förster, E.; Faustmann, P.M. Dexamethasone and levetiracetam reduce hetero-cellular gap-junctional coupling between F98 glioma cells and glial cells in vitro. J. Neuro-Oncol. 2017, 131, 469–476. [Google Scholar] [CrossRef] [Green Version]
- Artesi, M.; Kroonen, J.; Bredel, M.; Nguyen-Khac, M.; Deprez, M.; Schoysman, L.; Poulet, C.; Chakravarti, A.; Kim, H.; Scholtens, D. Connexin 30 expression inhibits growth of human malignant gliomas but protects them against radiation therapy. Neuro-Oncol. 2014, 17, 392–406. [Google Scholar] [CrossRef]
- Giaume, C.; Theis, M. Pharmacological and genetic approaches to study connexin-mediated channels in glial cells of the central nervous system. Brain Res. Rev. 2010, 63, 160–176. [Google Scholar] [CrossRef]
- Zhukova, G.V.; Kirichenko, E.; Sukhov, A.; Atmachidi, D.P.; Grankina, A.; Shikhliarova, A.I.; Sakun, P.G.; Shirnina, E.A.; Bragina, M.I.; Barteneva, T.A. Immunohistochemical characteristics of neuron-glial relationships in human astrocytic brain tumors of different grades of malignancy. J. Clin. Oncol. 2015, 33, e22096. [Google Scholar] [CrossRef]
- Kirichenko, E.; Zhukova, G.; Grigorov, S.; Grankina, A.; Atmachidi, D. The expression of connexin 36 and some neuroglial antigens in human brain astrocytic tumors of different grades. Arkhiv Patol. 2015, 77, 23–29. [Google Scholar] [CrossRef]
- Dong, H.; Zhou, X.W.; Wang, X.; Yang, Y.; Luo, J.W.; Liu, Y.H.; Mao, Q. Complex role of connexin 43 in astrocytic tumors and possible promotion of glioma-associated epileptic discharge. Mol. Med. Rep. 2017, 16, 7890–7900. [Google Scholar] [CrossRef] [Green Version]
- Dermietzel, R.; Gao, Y.; Scemes, E.; Vieira, D.; Urban, M.; Kremer, M.; Bennett, M.V.; Spray, D.C. Connexin43 null mice reveal that astrocytes express multiple connexins. Brain Res. Rev. 2000, 32, 45–56. [Google Scholar] [CrossRef]
- Sin, W.-C.; Crespin, S.; Mesnil, M. Opposing roles of connexin43 in glioma progression. Biochim. Et Biophys. Acta (Bba) Biomembr. 2012, 1818, 2058–2067. [Google Scholar] [CrossRef]
- Crespin, S.; Fromont, G.; Wager, M.; Levillain, P.; Cronier, L.; Monvoisin, A.; Defamie, N.; Mesnil, M. Expression of a gap junction protein, connexin43, in a large panel of human gliomas: new insights. Cancer Med. 2016, 5, 1742–1752. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; de Carvalho, A.C.; Lyu, S.; Li, P.; Li, Y. Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell 2017, 32, 42–56.e46. [Google Scholar] [CrossRef] [Green Version]
- Buniello, A.; Montanaro, D.; Volinia, S.; Gasparini, P.; Marigo, V. An expression atlas of connexin genes in the mouse. Genomics 2004, 83, 812–820. [Google Scholar] [CrossRef]
- Phi, L.T.H.; Sari, I.N.; Yang, Y.-G.; Lee, S.-H.; Jun, N.; Kim, K.S.; Lee, Y.K.; Kwon, H.Y. Cancer stem cells (CSCs) in drug resistance and their therapeutic implications in cancer treatment. Stem Cells Int. 2018, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Rich, J.N.; Bao, S. Chemotherapy and cancer stem cells. Cell Stem Cell 2007, 1, 353–355. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, D.; Das, S.; Molina, S.A.; Madgwick, D.; Katz, M.R.; Jena, S.; Bossmann, L.K.; Pal, D.; Takemoto, D.J. Investigation of the reciprocal relationship between the expression of two gap junction connexin proteins, connexin46 and connexin43. J. Biol. Chem. 2011, 286, 24519–24533. [Google Scholar] [CrossRef] [Green Version]
- Gangoso, E.; Thirant, C.; Chneiweiss, H.; Medina, J.; Tabernero, A. A cell-penetrating peptide based on the interaction between c-Src and connexin43 reverses glioma stem cell phenotype. Cell Death Dis. 2014, 5, e1023. [Google Scholar] [CrossRef]
- Aasen, T.; Leithe, E.; Graham, S.V.; Kameritsch, P.; Mayán, M.D.; Mesnil, M.; Pogoda, K.; Tabernero, A. Connexins in cancer: bridging the gap to the clinic. Oncogene 2019, 38, 4429–4451. [Google Scholar] [CrossRef] [Green Version]
- Lampe, P.D.; Lau, A.F. The effects of connexin phosphorylation on gap junctional communication. Int. J. Biochem. Cell Biol. 2004, 36, 1171–1186. [Google Scholar] [CrossRef] [Green Version]
- Mennecier, G.; Derangeon, M.; Coronas, V.; Hervé, J.C.; Mesnil, M. Aberrant expression and localization of connexin43 and connexin30 in a rat glioma cell line. Mol. Carcinog. Publ. Coop. Univ. Tex. Md Anderson Cancer Cent. 2008, 47, 391–401. [Google Scholar] [CrossRef]
- Dere, E. Gap Junctions in the Brain: Physiological and Pathological Roles; Academic Press: San Diego, CA, USA, 2012. [Google Scholar]
- Nakase, T.; Naus, C.C. Gap junctions and neurological disorders of the central nervous system. Biochim. Et Biophys. Acta (Bba) Biomembr. 2004, 1662, 149–158. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Li, J.; Zheng, J.; Qin, S. Reactive Astrocytes in Neurodegenerative Diseases. Aging Dis. 2019, 10, 664. [Google Scholar] [CrossRef] [Green Version]
- Lagos-Cabré, R.; Burgos, F.; Avalos, A.M.; Leyton, L. Connexins in Astrocyte Migration. Front. Pharmacol. 2019, 10, 1546. [Google Scholar] [CrossRef] [Green Version]
- Chun, H.; Lee, J. Reactive astrocytes in Alzheimer’s disease: A double-edged sword. Neurosci. Res. 126 2018, 44–52. [Google Scholar] [CrossRef]
- Dorszewska, J.; Prendecki, M.; Oczkowska, A.; Dezor, M.; Kozubski, W. Molecular basis of familial and sporadic Alzheimer’s disease. Curr. Alzheimer Res. 2016, 13, 952–963. [Google Scholar] [CrossRef]
- Tzioras, M.; Davies, C.; Newman, A.; Jackson, R.; Spires-Jones, T. Invited Review: APOE at the interface of inflammation, neurodegeneration and pathological protein spread in Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2019, 45, 327–346. [Google Scholar] [CrossRef]
- Zhang, Y.-w.; Thompson, R.; Zhang, H.; Xu, H. APP processing in Alzheimer’s disease. Mol. Brain 2011, 4, 3. [Google Scholar] [CrossRef] [Green Version]
- Roher, A.E.; Kokjohn, T.A.; Clarke, S.G.; Sierks, M.R.; Maarouf, C.L.; Serrano, G.E.; Sabbagh, M.S.; Beach, T.G. APP/Aβ structural diversity and Alzheimer’s disease pathogenesis. Neurochem. Int. 2017, 110, 1–13. [Google Scholar] [CrossRef]
- Frozza, R.L.; Horn, A.P.; Hoppe, J.B.; Simão, F.; Gerhardt, D.; Comiran, R.A.; Salbego, C.G. A Comparative Study of β-Amyloid Peptides Aβ1-42 and Aβ25-35 Toxicity in Organotypic Hippocampal Slice Cultures. Neurochem. Res. 2009, 34, 295–303. [Google Scholar] [CrossRef]
- Liu, P.; Reed, M.N.; Kotilinek, L.A.; Grant, M.K.; Forster, C.L.; Qiang, W.; Shapiro, S.L.; Reichl, J.H.; Chiang, A.C.; Jankowsky, J.L. Quaternary structure defines a large class of amyloid-β oligomers neutralized by sequestration. Cell Rep. 2015, 11, 1760–1771. [Google Scholar] [CrossRef] [Green Version]
- Salazar, S.V.; Strittmatter, S.M. Cellular prion protein as a receptor for amyloid-β oligomers in Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2017, 483, 1143–1147. [Google Scholar] [CrossRef] [Green Version]
- Figueiredo, C.P.; Clarke, J.R.; Ledo, J.H.; Ribeiro, F.C.; Costa, C.V.; Melo, H.M.; Mota-Sales, A.P.; Saraiva, L.M.; Klein, W.L.; Sebollela, A. Memantine rescues transient cognitive impairment caused by high-molecular-weight aβ oligomers but not the persistent impairment induced by low-molecular-weight oligomers. J. Neurosci. 2013, 33, 9626–9634. [Google Scholar] [CrossRef] [Green Version]
- Irie, K. New diagnostic method for Alzheimer’s disease based on the toxic conformation theory of amyloid β. Biosci. Biotechnol. Biochem. 2020, 84, 1–16. [Google Scholar] [CrossRef]
- Koulakoff, A. Glial connexin expression and function in the context of Alzheimer’s disease. Biochim. Et Biophys. Acta 2012, 1818, 2048–2057. [Google Scholar] [CrossRef] [Green Version]
- Yi, C.; Mei, X.; Ezan, P. Astroglial connexin43 contributes to neuronal suffering in a mouse model of Alzheimer’s disease. Cell Death Differ. 2016, 1691–1701. [Google Scholar] [CrossRef]
- Nagy, J.; Li, W.; Hertzberg, E.; Marotta, C. Elevated connexin43 immunoreactivity at sites of amyloid plaques in Alzheimer’s disease. Brain Res. 1996, 717, 173–178. [Google Scholar] [CrossRef]
- Richetin, K.; Roybon, L. Differential alteration of hippocampal function and plasticity in females and males of the APPxPS1 mouse model of Alzheimer’s disease. Neurobiol. Aging 2017, 220–231. [Google Scholar] [CrossRef]
- Liebmann, M.; Stahr, A.; Guenther, M.; Witte, O.W.; Frahm, C. Astrocytic Cx43 and Cx30 differentially modulate adult neurogenesis in mice. Neurosci. Lett. 2013, 545, 40–45. [Google Scholar] [CrossRef]
- Ransom, B.; Giaume, C. Gap junctions and hemichannels. Neuroglia 2013, 24, 292–305. [Google Scholar]
- Kalia, L.V.; Lang, A.E. Parkinson disease in 2015: evolving basic, pathological and clinical concepts in PD. Nat. Rev. Neurol. 2016, 12, 65. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.-E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef]
- Alexander, G.E. Biology of Parkinson’s disease: pathogenesis and pathophysiology of a multisystem neurodegenerative disorder. Dialogues Clin. Neurosci. 2004, 6, 259. [Google Scholar]
- Simon, D.K.; Tanner, C.M.; Brundin, P. Parkinson Disease Epidemiology, Pathology, Genetics, and Pathophysiology. Clin. Geriatr. Med. 2020, 36, 1–12. [Google Scholar] [CrossRef]
- Balestrino, R.; Schapira, A.H.V. Parkinson disease. Eur. J. Neurol. 2020, 27, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Elbaz, A.; Clavel, J.; Rathouz, P.J.; Moisan, F.; Galanaud, J.P.; Delemotte, B.; Alpérovitch, A.; Tzourio, C. Professional exposure to pesticides and Parkinson disease. Ann. Neurol. 2009, 66, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, O.F.; Lee, J.; Hing, N.Y.K.; Kim, S.-E.; Freeman, J.L.; Yuan, C. Lead (Pb) exposure reduces global DNA methylation level by non-competitive inhibition and alteration of dnmt expression. Metallomics 2017, 9, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, O.F.; Lin, L.; Bryan, C.J.; Xie, J.; Freeman, J.L.; Yuan, C. Profiling epigenetic changes in human cell line induced by atrazine exposure. Environ. Pollut. 2019, 258. [Google Scholar] [CrossRef]
- Tanner, C.M.; Kamel, F.; Ross, G.W.; Hoppin, J.A.; Goldman, S.M.; Korell, M.; Marras, C.; Bhudhikanok, G.S.; Kasten, M.; Chade, A.R. Rotenone, paraquat, and Parkinson’s disease. Environ. Health Perspect. 2011, 119, 866–872. [Google Scholar] [CrossRef] [Green Version]
- Jafari, S.; Etminan, M.; Aminzadeh, F.; Samii, A. Head injury and risk of Parkinson disease: a systematic review and meta-analysis. Mov. Disord. 2013, 28, 1222–1229. [Google Scholar] [CrossRef]
- Brück, D.; Wenning, G.K.; Stefanova, N.; Fellner, L. Glia and alpha-synuclein in neurodegeneration: a complex interaction. Neurobiol. Dis. 2016, 85, 262–274. [Google Scholar] [CrossRef] [Green Version]
- Rocha, E.M.; De Miranda, B.; Sanders, L.H. Alpha-synuclein: pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiol. Dis. 2018, 109, 249–257. [Google Scholar] [CrossRef]
- Joe, E.-H.; Choi, D.-J.; An, J.; Eun, J.-H.; Jou, I.; Park, S. Astrocytes, microglia, and Parkinson’s disease. Exp. Neurobiol. 2018, 27, 77–87. [Google Scholar] [CrossRef]
- Kovacs, G.G.; Breydo, L.; Green, R.; Kis, V.; Puska, G.; Lőrincz, P.; Perju-Dumbrava, L.; Giera, R.; Pirker, W.; Lutz, M. Intracellular processing of disease-associated α-synuclein in the human brain suggests prion-like cell-to-cell spread. Neurobiol. Dis. 2014, 69, 76–92. [Google Scholar] [CrossRef]
- Damier, P.; Hirsch, E.; Zhang, P.; Agid, Y.; Javoy-Agid, F. Glutathione peroxidase, glial cells and Parkinson’s disease. Neuroscience 1993, 52, 1–6. [Google Scholar] [CrossRef]
- Díaz, E.F.; Labra, V.C.; Alvear, T.F.; Mellado, L.A.; Inostroza, C.A.; Oyarzún, J.E.; Salgado, N.; Quintanilla, R.A.; Orellana, J.A. Connexin 43 hemichannels and pannexin-1 channels contribute to the α-synuclein-induced dysfunction and death of astrocytes. Glia 2019, 67, 1598–1619. [Google Scholar] [CrossRef]
- Konnova, E.A.; Swanberg, M. Animal Models of Parkinson’s Disease. In Parkinson’s Disease: Pathogenesis and Clinical Aspects [Internet]; Stoker, T.B., Greenland, J.C., Eds.; Codon Publications: Brisbane, Australia, 2018; pp. 83–106. [Google Scholar] [CrossRef]
- Rufer, M.; Wirth, S.; Hofer, A.; Dermietzel, R.; Pastor, A.; Kettenmann, H.; Unsicker, K. Regulation of connexin-43, GFAP, and FGF-2 is not accompanied by changes in astroglial coupling in MPTP-lesioned, FGF-2-treated Parkisonian mice. J. Neurosci. Res. 1996, 46, 606–617. [Google Scholar] [CrossRef]
- Kawasaki, A.; Hayashi, T.; Nakachi, K.; Trosko, J.E.; Sugihara, K.; Kotake, Y.; Ohta, S. Modulation of connexin 43 in rotenone-induced model of Parkinson’s disease. Neuroscience 2009, 160, 61–68. [Google Scholar] [CrossRef]
- Zhang, S.; Liang, R.; Zhou, F.; Huang, X.; Ding, J.-H.; Hu, G. Reversal of rotenone-induced dysfunction of astrocytic connexin43 by opening mitochondrial ATP-sensitive potassium channels. Cell. Mol. Neurobiol. 2011, 31, 111–117. [Google Scholar] [CrossRef]
- Rimkute, L.; Kraujalis, T.; Snipas, M.; Palacios-Prado, N.; Jotautis, V.; Skeberdis, V.A.; Bukauskas, F.F. Modulation of Connexin-36 Gap Junction Channels by Intracellular pH and Magnesium Ions. Front. Physiol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Teubner, B.; Degen, J.; Söhl, G.; Güldenagel, M.; Bukauskas, F.; Trexler, E.; Verselis, V.; De Zeeuw, C.; Lee, C.; Kozak, C. Functional expression of the murine connexin 36 gene coding for a neuron-specific gap junctional protein. J. Membr. Biol. 2000, 176, 249–262. [Google Scholar] [CrossRef] [Green Version]
- Srinivas, M.; Rozental, R.; Kojima, T.; Dermietzel, R.; Mehler, M.; Condorelli, D.F.; Kessler, J.A.; Spray, D.C. Functional properties of channels formed by the neuronal gap junction protein connexin36. J. Neurosci. 1999, 19, 9848–9855. [Google Scholar] [CrossRef]
- Wichmann, T.; DeLong, M. Basal ganglia discharge abnormalities in Parkinson’s disease. In Parkinson’s Disease and Related Disorders; Springer: New York, NY, USA, 2006; pp. 21–25. [Google Scholar]
- Traub, R.D.; Whittington, M.A. Cortical Oscillations in Health and Disease; Oxford University Press: New York, NY, USA, 2010. [Google Scholar]
- Schwab, B.C.; Heida, T.; Zhao, Y.; van Gils, S.A.; van Wezel, R.J. Pallidal gap junctions-triggers of synchrony in Parkinson’s disease? Mov Disord 2014, 29, 1486–1494. [Google Scholar] [CrossRef] [Green Version]
- Phookan, S.; Sutton, A.C.; Walling, I.; Smith, A.; O’Connor, K.A.; Campbell, J.C.; Calos, M.; Yu, W.; Pilitsis, J.G.; Brotchie, J.M.; et al. Gap junction blockers attenuate beta oscillations and improve forelimb function in hemiparkinsonian rats. Exp. Neurol 2015, 265, 160–170. [Google Scholar] [CrossRef]
- Bates, G.P.; Dorsey, R.; Gusella, J.F.; Hayden, M.R.; Kay, C.; Leavitt, B.R.; Nance, M.; Ross, C.A.; Scahill, R.I.; Wetzel, R. Huntington disease. Nat. Rev. Dis. Primers 2015, 1, 15005. [Google Scholar] [CrossRef]
- Waldvogel, H.J.; Kim, E.H.; Tippett, L.J.; Vonsattel, J.P.; Faull, R.L. The Neuropathology of Huntington’s Disease. Curr Top. Behav Neurosci 2015, 22, 33–80. [Google Scholar] [PubMed]
- MacDonald, M.E.; Ambrose, C.M.; Duyao, M.P.; Myers, R.H.; Lin, C.; Srinidhi, L.; Barnes, G.; Taylor, S.A.; James, M.; Groot, N. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- Chen, H.; Chan, D.C. Mitochondrial dynamics–fusion, fission, movement, and mitophagy–in neurodegenerative diseases. Hum. Mol. Genet. 2009, 18, R169–R176. [Google Scholar] [CrossRef] [PubMed]
- Shirendeb, U.; Reddy, A.P.; Manczak, M.; Calkins, M.J.; Mao, P.; Tagle, D.A.; Hemachandra Reddy, P. Abnormal mitochondrial dynamics, mitochondrial loss and mutant huntingtin oligomers in Huntington’s disease: implications for selective neuronal damage. Hum. Mol. Genet. 2011, 20, 1438–1455. [Google Scholar] [CrossRef] [Green Version]
- Manczak, M.; Reddy, P.H. Mitochondrial division inhibitor 1 protects against mutant huntingtin-induced abnormal mitochondrial dynamics and neuronal damage in Huntington’s disease. Hum. Mol. Genet. 2015, 24, 7308–7325. [Google Scholar] [CrossRef] [Green Version]
- Squitieri, F.; Falleni, A.; Cannella, M.; Orobello, S.; Fulceri, F.; Lenzi, P.; Fornai, F. Abnormal morphology of peripheral cell tissues from patients with Huntington disease. J. Neural Transm. 2010, 117, 77. [Google Scholar] [CrossRef]
- Khakh, B.S.; Beaumont, V.; Cachope, R.; Munoz-Sanjuan, I.; Goldman, S.A.; Grantyn, R. Unravelling and Exploiting Astrocyte Dysfunction in Huntington’s Disease. Trends Neurosci 2017, 40, 422–437. [Google Scholar] [CrossRef]
- Zhou, B.; Zuo, Y.X.; Jiang, R.T. Astrocyte morphology: Diversity, plasticity, and role in neurological diseases. Cns Neurosci 2019, 25, 665–673. [Google Scholar] [CrossRef]
- Dvorzhak, A.; Vagner, T.; Kirmse, K.; Grantyn, R. Functional Indicators of Glutamate Transport in Single Striatal Astrocytes and the Influence of Kir4.1 in Normal and Huntington Mice. J. Neurosci 2016, 36, 4959–4975. [Google Scholar] [CrossRef] [Green Version]
- Jiang, R.; Diaz-Castro, B.; Looger, L.L.; Khakh, B.S. Dysfunctional Calcium and Glutamate Signaling in Striatal Astrocytes from Huntington’s Disease Model Mice. J. Neurosci 2016, 36, 3453–3470. [Google Scholar] [CrossRef] [Green Version]
- Tong, X.; Ao, Y.; Faas, G.C.; Nwaobi, S.E.; Xu, J.; Haustein, M.D.; Anderson, M.A.; Mody, I.; Olsen, M.L.; Sofroniew, M.V.; et al. Astrocyte Kir4.1 ion channel deficits contribute to neuronal dysfunction in Huntington’s disease model mice. Nat. Neurosci 2014, 17, 694–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramaswamy, S.; McBride, J.L.; Kordower, J.H. Animal models of Huntington’s disease. Ilar J. 2007, 48, 356–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrasch-Parwez, E.; Habbes, H.W.; Weickert, S.; Löbbecke-Schumacher, M.; Striedinger, K.; Wieczorek, S.; Dermietzel, R.; Epplen, J.T. Fine-structural analysis and connexin expression in the retina of a transgenic model of Huntington’s disease. J. Comp. Neurol 2004, 479, 181–197. [Google Scholar] [CrossRef] [PubMed]
- Wolf, R.C.; Sambataro, F.; Vasic, N.; Baldas, E.M.; Ratheiser, I.; Bernhard Landwehrmeyer, G.; Depping, M.S.; Thomann, P.A.; Sprengelmeyer, R.; Süssmuth, S.D. Visual system integrity and cognition in early Huntington’s disease. Eur. J. Neurosci. 2014, 40, 2417–2426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.J.; Han, J.W.; Kim, H.J.; Kim, I.B.; Lee, M.Y.; Oh, S.J.; Chung, J.W.; Chun, M.H. The immunocytochemical localization of connexin 36 at rod and cone gap junctions in the guinea pig retina. Eur. J. Neurosci. 2003, 18, 2925–2934. [Google Scholar] [CrossRef] [PubMed]
- Cummings, D.M.; Yamazaki, I.; Cepeda, C.; Paul, D.L.; Levine, M.S. Neuronal coupling via connexin36 contributes to spontaneous synaptic currents of striatal medium-sized spiny neurons. J. Neurosci. Res. 2008, 86, 2147–2158. [Google Scholar] [CrossRef] [PubMed]
- Cummings, D.M.; Cepeda, C.; Levine, M.S. Alterations in striatal synaptic transmission are consistent across genetic mouse models of Huntington’s disease. Asn Neuro 2010, 2, AN20100007. [Google Scholar] [CrossRef] [Green Version]
- Vis, J.C.; Nicholson, L.F.; Faull, R.L.; Evans, W.H.; Severs, N.J.; Green, C.R. Connexin expression in Huntington’s diseased human brain. Cell Biol Int 1998, 22, 837–847. [Google Scholar] [CrossRef]
- Nakase, T.; Fushiki, S.; Naus, C.C. Astrocytic gap junctions composed of connexin 43 reduce apoptotic neuronal damage in cerebral ischemia. Stroke 2003, 34, 1987–1993. [Google Scholar] [CrossRef] [Green Version]
- Nakase, T.; Söhl, G.; Theis, M.; Willecke, K.; Naus, C.C. Increased apoptosis and inflammation after focal brain ischemia in mice lacking connexin43 in astrocytes. Am. J. Pathol 2004, 164, 2067–2075. [Google Scholar] [CrossRef] [Green Version]
- Siushansian, R.; Bechberger, J.F.; Cechetto, D.F.; Hachinski, V.C.; Naus, C.C. Connexin43 null mutation increases infarct size after stroke. J. Comp. Neurol 2001, 440, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Paschon, V.; Higa, G.S.; Resende, R.R.; Britto, L.R.; Kihara, A.H. Blocking of connexin-mediated communication promotes neuroprotection during acute degeneration induced by mechanical trauma. Plos One 2012, 7, e45449. [Google Scholar] [CrossRef] [PubMed]
- Akopian, A.; Kumar, S.; Ramakrishnan, H.; Roy, K.; Viswanathan, S.; Bloomfield, S.A. Targeting neuronal gap junctions in mouse retina offers neuroprotection in glaucoma. J. Clin. Invest. 2017, 127, 2647–2661. [Google Scholar] [CrossRef]
- Akopian, A.; Atlasz, T.; Pan, F.; Wong, S.; Zhang, Y.; Völgyi, B.; Paul, D.L.; Bloomfield, S.A. Gap junction-mediated death of retinal neurons is connexin and insult specific: a potential target for neuroprotection. J. Neurosci 2014, 34, 10582–10591. [Google Scholar] [CrossRef] [Green Version]
- Chakravarthy, B.R.; Wang, J.; Tremblay, R.; Atkinson, T.G.; Wang, F.; Li, H.; Buchan, A.M.; Durkin, J.P. Comparison of the changes in protein kinase C induced by glutamate in primary cortical neurons and by in vivo cerebral ischaemia. Cell Signal. 1998, 10, 291–295. [Google Scholar] [CrossRef]
- Naus, C.C.; Bechberger, J.F.; Zhang, Y.; Venance, L.; Yamasaki, H.; Juneja, S.C.; Kidder, G.M.; Giaume, C. Altered gap junctional communication, intercellular signaling, and growth in cultured astrocytes deficient in connexin43. J. Neurosci Res. 1997, 49, 528–540. [Google Scholar] [CrossRef]
- Scemes, E.; Dermietzel, R.; Spray, D.C. Calcium waves between astrocytes from Cx43 knockout mice. Glia 1998, 24, 65–73. [Google Scholar] [CrossRef]
- Li, W.E.; Ochalski, P.A.; Hertzberg, E.L.; Nagy, J.I. Immunorecognition, ultrastructure and phosphorylation status of astrocytic gap junctions and connexin43 in rat brain after cerebral focal ischaemia. Eur J. Neurosci 1998, 10, 2444–2463. [Google Scholar] [CrossRef]
- de Pina-Benabou, M.H.; Szostak, V.; Kyrozis, A.; Rempe, D.; Uziel, D.; Urban-Maldonado, M.; Benabou, S.; Spray, D.C.; Federoff, H.J.; Stanton, P.K.; et al. Blockade of gap junctions in vivo provides neuroprotection after perinatal global ischemia. Stroke 2005, 36, 2232–2237. [Google Scholar] [CrossRef] [Green Version]
- Farahani, R.; Pina-Benabou, M.H.; Kyrozis, A.; Siddiq, A.; Barradas, P.C.; Chiu, F.C.; Cavalcante, L.A.; Lai, J.C.; Stanton, P.K.; Rozental, R. Alterations in metabolism and gap junction expression may determine the role of astrocytes as "good samaritans" or executioners. Glia 2005, 50, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Davidson, J.O.; Drury, P.P.; Green, C.R.; Nicholson, L.F.; Bennet, L.; Gunn, A.J. Connexin hemichannel blockade is neuroprotective after asphyxia in preterm fetal sheep. Plos One 2014, 9, e96558. [Google Scholar] [CrossRef] [PubMed]
- Frantseva, M.V.; Kokarovtseva, L.; Perez Velazquez, J.L. Ischemia-induced brain damage depends on specific gap-junctional coupling. J. Cereb Blood Flow Metab 2002, 22, 453–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frantseva, M.V.; Kokarovtseva, L.; Naus, C.G.; Carlen, P.L.; MacFabe, D.; Perez Velazquez, J.L. Specific gap junctions enhance the neuronal vulnerability to brain traumatic injury. J. Neurosci 2002, 22, 644–653. [Google Scholar] [CrossRef] [PubMed]
- Voytenko, L.P.; Lushnikova, I.V.; Savotchenko, A.V.; Isaeva, E.V.; Skok, M.V.; Lykhmus, O.Y.; Patseva, M.A.; Skibo, G.G. Hippocampal GABAergic interneurons coexpressing alpha7-nicotinic receptors and connexin-36 are able to improve neuronal viability under oxygen-glucose deprivation. Brain Res. 2015, 1616, 134–145. [Google Scholar] [CrossRef]
- Yin, X.; Feng, L.; Ma, D.; Yin, P.; Wang, X.; Hou, S.; Hao, Y.; Zhang, J.; Xin, M.; Feng, J. Roles of astrocytic connexin-43, hemichannels, and gap junctions in oxygen-glucose deprivation/reperfusion injury induced neuroinflammation and the possible regulatory mechanisms of salvianolic acid B and carbenoxolone. J. Neuroinflammation 2018, 15, 97. [Google Scholar] [CrossRef]
- Almad, A.A.; Doreswamy, A.; Gross, S.K.; Richard, J.P.; Huo, Y.; Haughey, N.; Maragakis, N.J. Connexin 43 in astrocytes contributes to motor neuron toxicity in amyotrophic lateral sclerosis. Glia 2016, 64, 1154–1169. [Google Scholar] [CrossRef] [Green Version]
- Ek-Vitorin, J.F.; King, T.J.; Heyman, N.S.; Lampe, P.D.; Burt, J.M. Selectivity of connexin 43 channels is regulated through protein kinase C-dependent phosphorylation. Circ. Res. 2006, 98, 1498–1505. [Google Scholar] [CrossRef] [Green Version]
- Rivedal, E.; Leithe, E. Connexin43 synthesis, phosphorylation, and degradation in regulation of transient inhibition of gap junction intercellular communication by the phorbol ester TPA in rat liver epithelial cells. Exp. Cell Res. 2005, 302, 143–152. [Google Scholar] [CrossRef]
- Hanstein, R.; Trotter, J.; Behl, C.; Clement, A.B. Increased connexin 43 expression as a potential mediator of the neuroprotective activity of the corticotropin-releasing hormone. Mol. Endocrinol 2009, 23, 1479–1493. [Google Scholar] [CrossRef] [Green Version]
- Deng, Z.H.; Liao, J.; Zhang, J.Y.; Liang, C.; Song, C.H.; Han, M.; Wang, L.H.; Xue, H.; Zhang, K.; Zabeau, L.; et al. Inhibition of the connexin 43 elevation may be involved in the neuroprotective activity of leptin against brain ischemic injury. Cell Mol. Neurobiol 2014, 34, 871–879. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Guo, Y.; Yang, W.; Zheng, P.; Zeng, J.; Tong, W. Involvement of Connexin40 in the Protective Effects of Ginsenoside Rb1 Against Traumatic Brain Injury. Cell Mol. Neurobiol 2016, 36, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Hou, Z.; Sun, J.; Huang, Y.; Wang, L.; Zhou, Z.; Zhou, L.H.; Cai, Y. Protective effects of Tongxinluo on cerebral ischemia/reperfusion injury related to Connexin 43/Calpain II/Bax/Caspase-3 pathway in rat. J. Ethnopharmacol 2017, 198, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, C.J.; Lu, Y.; Zong, X.G.; Luo, C.; Sun, J.; Guo, L.J. Baclofen mediates neuroprotection on hippocampal CA1 pyramidal cells through the regulation of autophagy under chronic cerebral hypoperfusion. Sci Rep. 2015, 5, 14474. [Google Scholar] [CrossRef] [Green Version]
- Kozoriz, M.G.; Bechberger, J.F.; Bechberger, G.R.; Suen, M.W.; Moreno, A.P.; Maass, K.; Willecke, K.; Naus, C.C. The connexin43 C-terminal region mediates neuroprotection during stroke. J. Neuropathol Exp. Neurol 2010, 69, 196–206. [Google Scholar] [CrossRef] [Green Version]
- Hugyecz, M.; Mracskó, É.; Hertelendy, P.; Farkas, E.; Domoki, F.; Bari, F. Hydrogen supplemented air inhalation reduces changes of prooxidant enzyme and gap junction protein levels after transient global cerebral ischemia in the rat hippocampus. Brain Res. 2011, 1404, 31–38. [Google Scholar] [CrossRef]
- Li, H.; Luo, Y.; Yang, P.; Liu, J. Hydrogen as a complementary therapy against ischemic stroke: A review of the evidence. J. Neurol. Sci. 2019, 396, 240–246. [Google Scholar] [CrossRef]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez, O.F.; Rodríguez, A.V.; Velasco-España, J.M.; Murillo, L.C.; Sutachan, J.-J.; Albarracin, S.-L. Role of Connexins 30, 36, and 43 in Brain Tumors, Neurodegenerative Diseases, and Neuroprotection. Cells 2020, 9, 846. https://doi.org/10.3390/cells9040846
Sánchez OF, Rodríguez AV, Velasco-España JM, Murillo LC, Sutachan J-J, Albarracin S-L. Role of Connexins 30, 36, and 43 in Brain Tumors, Neurodegenerative Diseases, and Neuroprotection. Cells. 2020; 9(4):846. https://doi.org/10.3390/cells9040846
Chicago/Turabian StyleSánchez, Oscar F., Andrea V. Rodríguez, José M. Velasco-España, Laura C. Murillo, Jhon-Jairo Sutachan, and Sonia-Luz Albarracin. 2020. "Role of Connexins 30, 36, and 43 in Brain Tumors, Neurodegenerative Diseases, and Neuroprotection" Cells 9, no. 4: 846. https://doi.org/10.3390/cells9040846
APA StyleSánchez, O. F., Rodríguez, A. V., Velasco-España, J. M., Murillo, L. C., Sutachan, J.-J., & Albarracin, S.-L. (2020). Role of Connexins 30, 36, and 43 in Brain Tumors, Neurodegenerative Diseases, and Neuroprotection. Cells, 9(4), 846. https://doi.org/10.3390/cells9040846