Lamin Mutations Cause Increased YAP Nuclear Entry in Muscle Stem Cells

 ,
, 

Abstract
1. Introduction
2. Materials and Methods
2.1. Human Cells and Cell Culture
2.2. Drug Treatments
2.3. Luciferase Reporter Assays
2.4. Immunocytochemistry and Image Analysis
2.5. SDS-PAGE and Protein Analysis
2.6. Quantification of Gene Expression
2.7. Statistical Analysis
3. Results
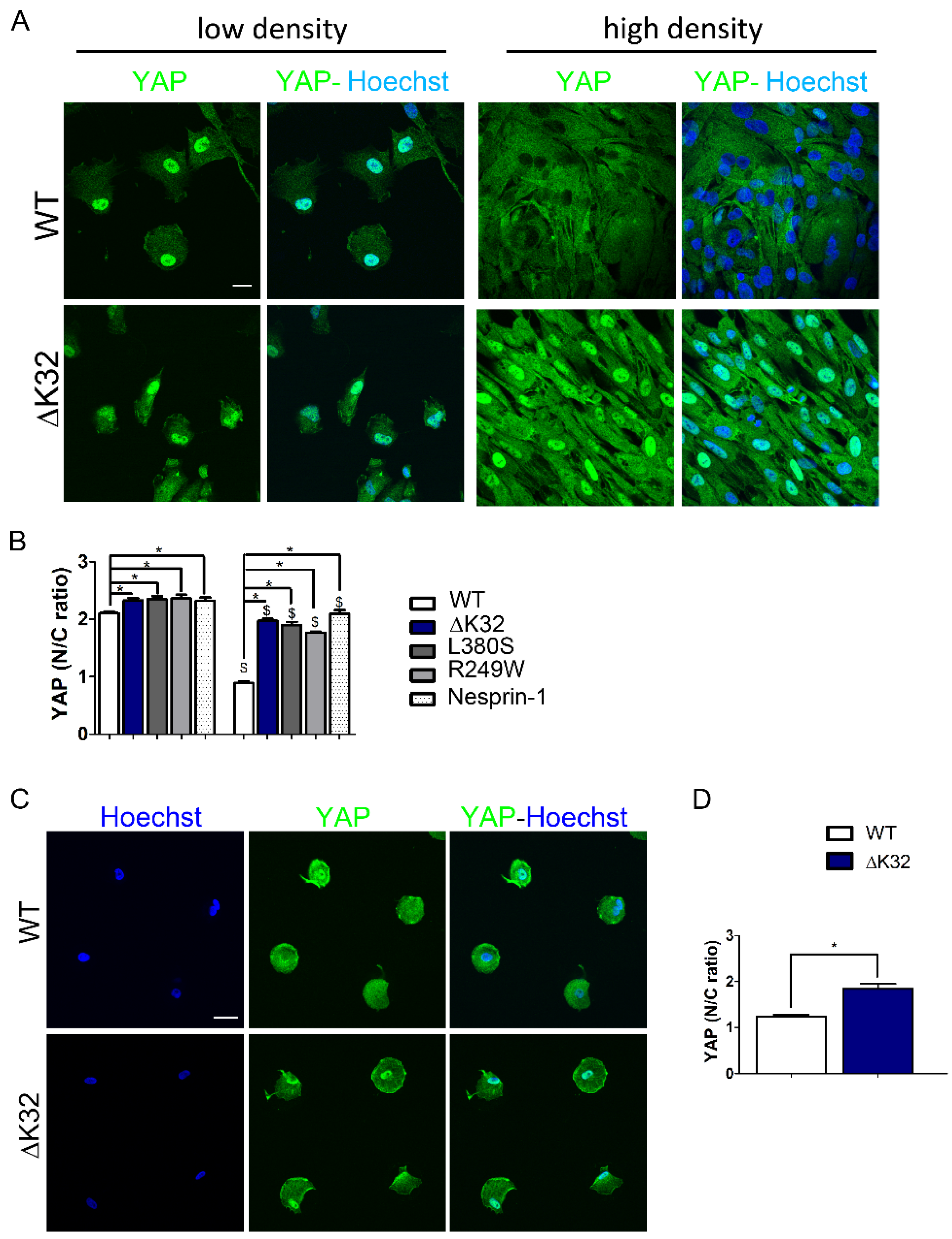
3.1. Impaired Yes-Associated Protein (YAP) Nuclear Exclusion in Confluent LMNA Mutant Muscle Stem Cells (MuSCs)
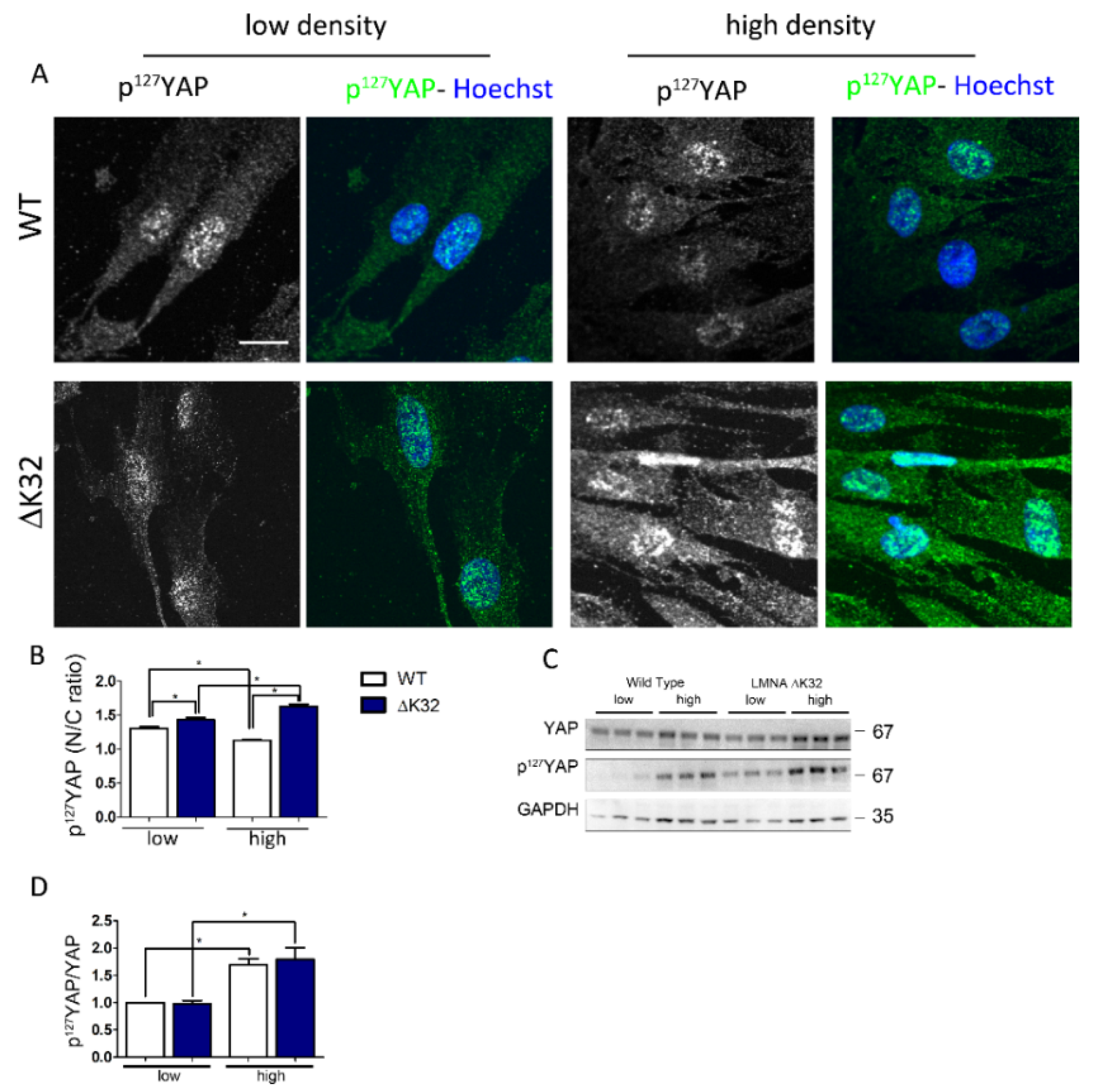
3.2. Phosphorylated Ser127-YAP Accumulates in the Nucleus of LMNA Mutated Muscle Stem Cells (MuSCs)
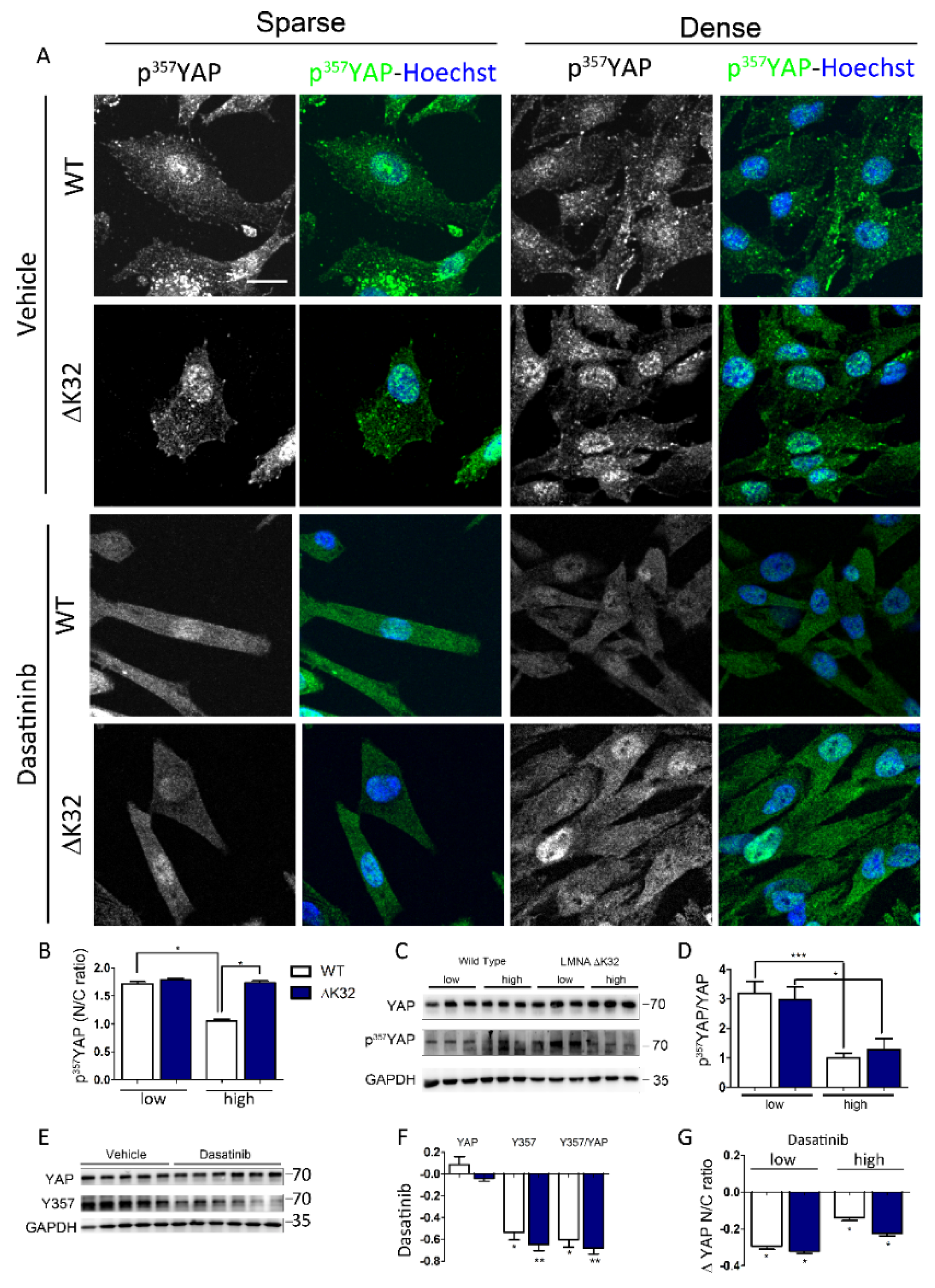
3.3. Src-Dependent Tyr Phosphorylation of YAP is Activated in LMNAΔK32 MuSCs
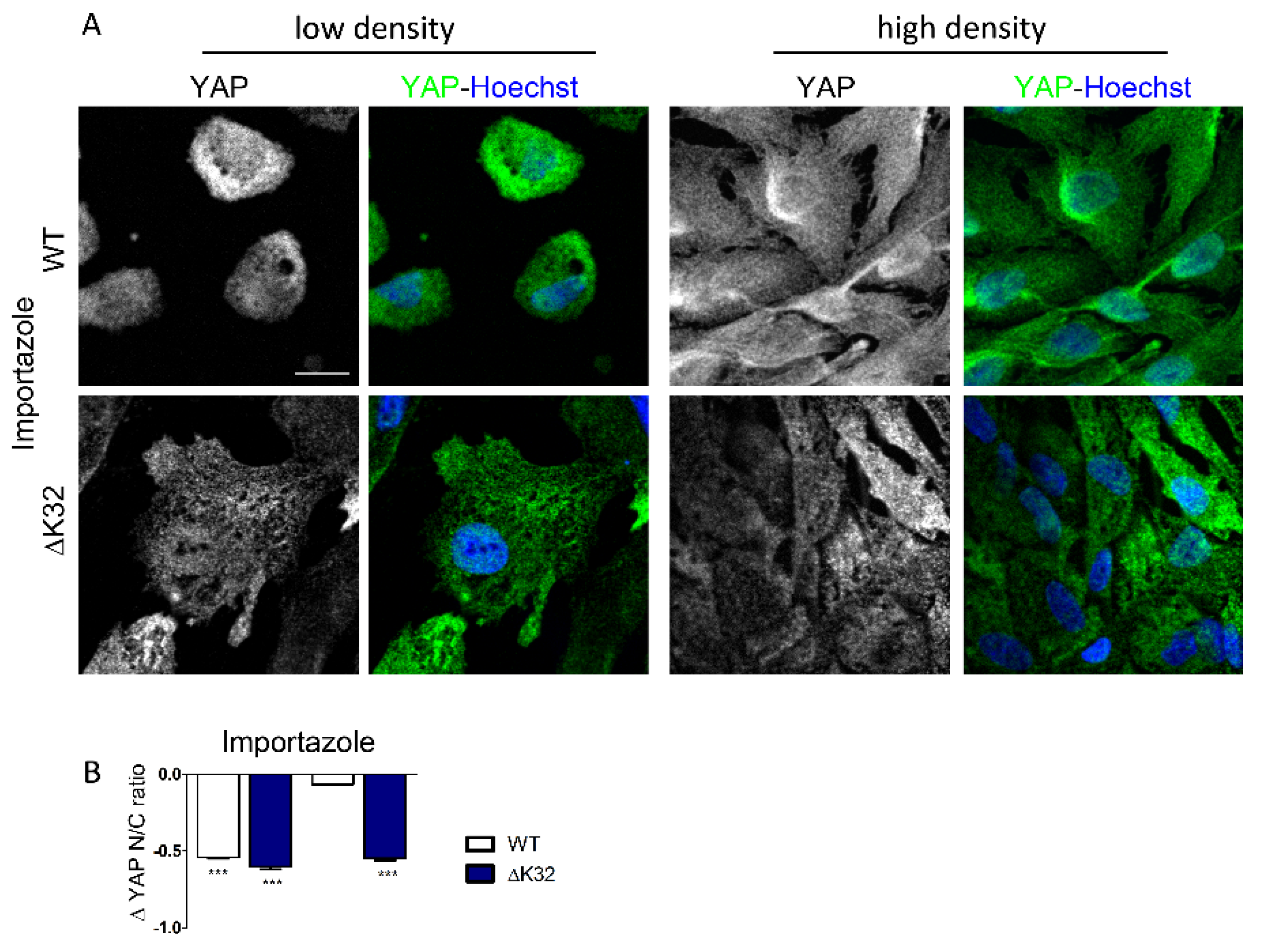
3.4. Blockade of Nuclear Import Inhibits Nuclear Accumulation of YAP in Confluent LMNAΔK32 MuSCs
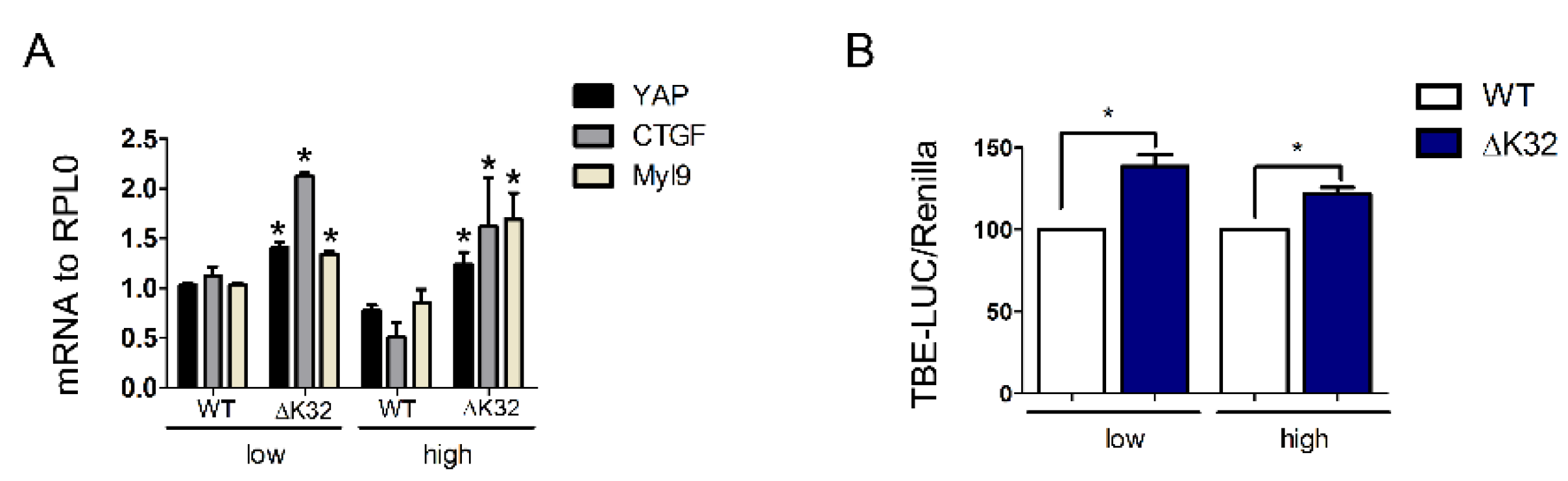
3.5. Functional Consequences of YAP Nuclear Accumulation in High Density LMNAΔK32 MuSCs
4. Discussion
4.1. Canonical Regulation of YAP Nucleo-Cytoplasmic Localization Via the Hippo Pathway
4.2. Nuclear Import of YAP in LMNAΔK32 Mutant Cells
4.3. Functional Consequence of YAP Deregulation in Myogenesis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Osmanagic-Myers, S.; Dechat, T.; Foisner, R. Lamins at the crossroads of mechanosignaling. Genes Dev. 2015, 29, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Worman, H.J.; Bonne, G. “Laminopathies”: A wide spectrum of human diseases. Exp. Cell Res. 2007, 313, 2121–2133. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, A.T.; Chikhaoui, K.; Yaou, R.B.; Bonne, G. Clinical and genetic heterogeneity in laminopathies. Biochem. Soc. Trans. 2011, 39, 1687–1692. [Google Scholar] [CrossRef] [PubMed]
- Guilluy, C.; Osborne, L.D.; Van Landeghem, L.; Sharek, L.; Superfine, R.; Garcia-Mata, R.; Burridge, K. Isolated nuclei adapt to force and reveal a mechanotransduction pathway in the nucleus. Nat. Cell Biol. 2014, 16, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Swift, J.; Ivanovska, I.L.; Buxboim, A.; Harada, T.; Dingal, P.C.; Pinter, J.; Pajerowski, J.D.; Spinler, K.R.; Shin, J.W.; Tewari, M.; et al. Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science 2013, 341, 1240104. [Google Scholar] [CrossRef]
- Vaughan, A.; Alvarez-Reyes, M.; Bridger, J.M.; Broers, J.L.; Ramaekers, F.C.; Wehnert, M.; Morris, G.E.; Whitfield, W.G.F.; Hutchison, C.J. Both emerin and lamin C depend on lamin A for localization at the nuclear envelope. J. Cell Sci. 2001, 114, 2577–2590. [Google Scholar]
- Markiewicz, E.; Tilgner, K.; Barker, N.; van de Wetering, M.; Clevers, H.; Dorobek, M.; Hausmanowa-Petrusewicz, I.; Ramaekers, F.C.; Broers, J.L.; Blankesteijn, W.M.; et al. The inner nuclear membrane protein emerin regulates beta-catenin activity by restricting its accumulation in the nucleus. EMBO J. 2006, 25, 3275–3285. [Google Scholar] [CrossRef]
- Ho, C.Y.; Jaalouk, D.E.; Vartiainen, M.K.; Lammerding, J. Lamin A/C and emerin regulate MKL1-SRF activity by modulating actin dynamics. Nature 2013, 497, 507–511. [Google Scholar] [CrossRef]
- Schwartz, C.; Fischer, M.; Mamchaoui, K.; Bigot, A.; Lok, T.; Verdier, C.; Duperray, A.; Michel, R.; Holt, I.; Voit, T.; et al. Lamins and nesprin-1 mediate inside-out mechanical coupling in muscle cell precursors through FHOD1. Sci. Rep. 2017, 7, 1253. [Google Scholar] [CrossRef]
- Bertrand, A.T.; Ziaei, S.; Ehret, C.; Duchemin, H.; Mamchaoui, K.; Bigot, A.; Mayer, M.; Quijano-Roy, S.; Desguerre, I.; Laine, J.; et al. Cellular microenvironments reveal defective mechanosensing responses and elevated YAP signaling in LMNA-mutated muscle precursors. J. Cell Sci. 2014, 127, 2873–2884. [Google Scholar] [CrossRef]
- Panciera, T.; Azzolin, L.; Cordenonsi, M.; Piccolo, S. Mechanobiology of YAP and TAZ in physiology and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; Rikeit, P.; Knaus, P.; Coirault, C. YAP-Mediated Mechanotransduction in Skeletal Muscle. Front. Physiol. 2016, 7, 41. [Google Scholar] [CrossRef] [PubMed]
- Irianto, J.; Pfeifer, C.R.; Ivanovska, I.L.; Swift, J.; Discher, D.E. Nuclear lamins in cancer. Cell. Mol. Bioeng. 2016, 9, 258–267. [Google Scholar] [CrossRef]
- Chow, K.H.; Factor, R.E.; Ullman, K.S. The nuclear envelope environment and its cancer connections. Nat. Rev. Cancer 2012, 12, 196–209. [Google Scholar] [CrossRef] [PubMed]
- Zink, D.; Fischer, A.H.; Nickerson, J.A. Nuclear structure in cancer cells. Nat. Rev. Cancer 2004, 4, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Dahl, K.N.; Ribeiro, A.J.; Lammerding, J. Nuclear shape, mechanics, and mechanotransduction. Circ. Res. 2008, 102, 1307–1318. [Google Scholar] [CrossRef] [PubMed]
- Elosegui-Artola, A.; Trepat, X.; Roca-Cusachs, P. Control of Mechanotransduction by Molecular Clutch Dynamics. Trends Cell Biol. 2018, 28, 356–367. [Google Scholar] [CrossRef]
- Aureille, J.; Buffiere-Ribot, V.; Harvey, B.E.; Boyault, C.; Pernet, L.; Andersen, T.; Bacola, G.; Balland, M.; Fraboulet, S.; Van Landeghem, L.; et al. Nuclear envelope deformation controls cell cycle progression in response to mechanical force. EMBO Rep. 2019, 20, e48084. [Google Scholar] [CrossRef]
- Zhou, C.; Rao, L.; Shanahan, C.M.; Zhang, Q. Nesprin-1/2: Roles in nuclear envelope organisation, myogenesis and muscle disease. Biochem. Soc. Trans. 2018, 46, 311–320. [Google Scholar] [CrossRef]
- Puckelwartz, M.J.; Kessler, E.J.; Kim, G.; Dewitt, M.M.; Zhang, Y.; Earley, J.U.; Depreux, F.F.; Holaska, J.; Mewborn, S.K.; Pytel, P.; et al. Nesprin-1 mutations in human and murine cardiomyopathy. J. Mol. Cell. Cardiol. 2010, 48, 600–608. [Google Scholar] [CrossRef]
- Perovanovic, J.; Dell’Orso, S.; Gnochi, V.F.; Jaiswal, J.K.; Sartorelli, V.; Vigouroux, C.; Mamchaoui, K.; Mouly, V.; Bonne, G.; Hoffman, E.P. Laminopathies disrupt epigenomic developmental programs and cell fate. Sci. Transl. Med. 2016, 8, 335ra358. [Google Scholar] [CrossRef] [PubMed]
- Aure, K.; Mamchaoui, K.; Frachon, P.; Butler-Browne, G.S.; Lombes, A.; Mouly, V. Impact on oxidative phosphorylation of immortalization with the telomerase gene. Neuromuscul. Disord. 2007, 17, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Chaouch, S.; Mouly, V.; Goyenvalle, A.; Vulin, A.; Mamchaoui, K.; Negroni, E.; Di Santo, J.; Butler-Browne, G.; Torrente, Y.; Garcia, L.; et al. Immortalized skin fibroblasts expressing conditional MyoD as a renewable and reliable source of converted human muscle cells to assess therapeutic strategies for muscular dystrophies: Validation of an exon-skipping approach to restore dystrophin in Duchenne muscular dystrophy cells. Hum. Gene Ther. 2009, 20, 784–790. [Google Scholar] [PubMed]
- Soderholm, J.F.; Bird, S.L.; Kalab, P.; Sampathkumar, Y.; Hasegawa, K.; Uehara-Bingen, M.; Weis, K.; Heald, R. Importazole, a small molecule inhibitor of the transport receptor importin-beta. ACS Chem. Biol. 2011, 6, 700–708. [Google Scholar] [CrossRef] [PubMed]
- Kudo, N.; Wolff, B.; Sekimoto, T.; Schreiner, E.P.; Yoneda, Y.; Yanagida, M.; Horinouchi, S.; Yoshida, M. Leptomycin B inhibition of signal-mediated nuclear export by direct binding to CRM1. Exp. Cell Res. 1998, 242, 540–547. [Google Scholar] [CrossRef]
- Kim, N.G.; Koh, E.; Chen, X.; Gumbiner, B.M. E-cadherin mediates contact inhibition of proliferation through Hippo signaling-pathway components. Proc. Natl. Acad. Sci. USA 2011, 108, 11930–11935. [Google Scholar] [CrossRef]
- Zhao, B.; Wei, X.; Li, W.; Udan, R.S.; Yang, Q.; Kim, J.; Xie, J.; Ikenoue, T.; Yu, J.; Li, L.; et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007, 21, 2747–2761. [Google Scholar] [CrossRef]
- Aragona, M.; Panciera, T.; Manfrin, A.; Giulitti, S.; Michielin, F.; Elvassore, N.; Dupont, S.; Piccolo, S. A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell 2013, 154, 1047–1059. [Google Scholar] [CrossRef]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef]
- Calvo, F.; Ege, N.; Grande-Garcia, A.; Hooper, S.; Jenkins, R.P.; Chaudhry, S.I.; Harrington, K.; Williamson, P.; Moeendarbary, E.; Charras, G.; et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 2013, 15, 637–646. [Google Scholar] [CrossRef]
- Judson, R.N.; Tremblay, A.M.; Knopp, P.; White, R.B.; Urcia, R.; De Bari, C.; Zammit, P.S.; Camargo, F.D.; Wackerhage, H. The Hippo pathway member Yap plays a key role in influencing fate decisions in muscle satellite cells. J. Cell Sci. 2012, 125, 6009–6019. [Google Scholar] [CrossRef] [PubMed]
- Wackerhage, H.; Del Re, D.P.; Judson, R.N.; Sudol, M.; Sadoshima, J. The Hippo signal transduction network in skeletal and cardiac muscle. Sci. Signal 2014, 7, re4. [Google Scholar] [CrossRef] [PubMed]
- Goodman, C.A.; Dietz, J.M.; Jacobs, B.L.; McNally, R.M.; You, J.S.; Hornberger, T.A. Yes-Associated Protein is up-regulated by mechanical overload and is sufficient to induce skeletal muscle hypertrophy. FEBS Lett. 2015, 589, 1491–1497. [Google Scholar] [CrossRef] [PubMed]
- Dupont, S. Role of YAP/TAZ in cell-matrix adhesion-mediated signalling and mechanotransduction. Exp. Cell Res. 2016, 343, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.; Kim, W.; Kim, S.; Kim, Y.; Song, Y.; Bilousov, O.; Kim, J.; Lee, T.; Cha, B.; Kim, M.; et al. Phosphorylation by NLK inhibits YAP-14-3-3-interactions and induces its nuclear localization. EMBO Rep. 2017, 18, 61–71. [Google Scholar] [CrossRef]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the Roots of Cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef]
- Yu, F.X.; Zhao, B.; Guan, K.L. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 2015, 163, 811–828. [Google Scholar] [CrossRef]
- Oh, H.; Irvine, K.D. In vivo regulation of Yorkie phosphorylation and localization. Development 2008, 135, 1081–1088. [Google Scholar] [CrossRef]
- Zhao, B.; Lei, Q.Y.; Guan, K.L. The Hippo-YAP pathway: New connections between regulation of organ size and cancer. Curr. Opin. Cell Biol. 2008, 20, 638–646. [Google Scholar] [CrossRef]
- Chan, E.H.; Nousiainen, M.; Chalamalasetty, R.B.; Schafer, A.; Nigg, E.A.; Sillje, H.H. The Ste20-like kinase Mst2 activates the human large tumor suppressor kinase Lats1. Oncogene 2005, 24, 2076–2086. [Google Scholar] [CrossRef]
- Mo, J.S.; Park, H.W.; Guan, K.L. The Hippo signaling pathway in stem cell biology and cancer. EMBO Rep. 2014, 15, 642–656. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Chun, A.; Cheung, K.; Rashidi, B.; Yang, X. Tumor suppressor LATS1 is a negative regulator of oncogene YAP. J. Biol. Chem. 2008, 283, 5496–5509. [Google Scholar] [CrossRef] [PubMed]
- Lei, Q.Y.; Zhang, H.; Zhao, B.; Zha, Z.Y.; Bai, F.; Pei, X.H.; Zhao, S.; Xiong, Y.; Guan, K.L. TAZ promotes cell proliferation and epithelial-mesenchymal transition and is inhibited by the hippo pathway. Mol. Cell. Biol. 2008, 28, 2426–2436. [Google Scholar] [CrossRef] [PubMed]
- Oka, T.; Mazack, V.; Sudol, M. Mst2 and Lats kinases regulate apoptotic function of Yes kinase-associated protein (YAP). J. Biol. Chem. 2008, 283, 27534–27546. [Google Scholar] [CrossRef] [PubMed]
- Wada, K.; Itoga, K.; Okano, T.; Yonemura, S.; Sasaki, H. Hippo pathway regulation by cell morphology and stress fibers. Development 2011, 138, 3907–3914. [Google Scholar] [CrossRef]
- Das, A.; Fischer, R.S.; Pan, D.; Waterman, C.M. YAP Nuclear Localization in the Absence of Cell-Cell Contact Is Mediated by a Filamentous Actin-dependent, Myosin II- and Phospho-YAP-independent Pathway during Extracellular Matrix Mechanosensing. J. Biol. Chem. 2016, 291, 6096–6110. [Google Scholar] [CrossRef]
- Ege, N.; Dowbaj, A.M.; Jiang, M.; Howell, M.; Hooper, S.; Foster, C.; Jenkins, R.P.; Sahai, E. Quantitative Analysis Reveals that Actin and Src-Family Kinases Regulate Nuclear YAP1 and Its Export. Cell Syst. 2018, 6, 692.e13–708.e13. [Google Scholar] [CrossRef]
- Kofler, M.; Speight, P.; Little, D.; Di Ciano-Oliveira, C.; Szaszi, K.; Kapus, A. Mediated nuclear import and export of TAZ and the underlying molecular requirements. Nat. Commun. 2018, 9, 4966. [Google Scholar] [CrossRef]
- Levy, D.; Adamovich, Y.; Reuven, N.; Shaul, Y. Yap1 phosphorylation by c-Abl is a critical step in selective activation of proapoptotic genes in response to DNA damage. Mol. Cell 2008, 29, 350–361. [Google Scholar] [CrossRef]
- Wang, S.; Li, H.; Wang, G.; Zhang, T.; Fu, B.; Ma, M.; Quan, Z.; Chen, G. Yes-associated protein (YAP) expression is involved in epithelial-mesenchymal transition in hepatocellular carcinoma. Clin. Transl. Oncol. 2016, 18, 172–177. [Google Scholar] [CrossRef]
- Zwerger, M.; Jaalouk, D.E.; Lombardi, M.L.; Isermann, P.; Mauermann, M.; Dialynas, G.; Herrmann, H.; Wallrath, L.L.; Lammerding, J. Myopathic lamin mutations impair nuclear stability in cells and tissue and disrupt nucleo-cytoskeletal coupling. Hum. Mol. Genet. 2013, 22, 2335–2349. [Google Scholar] [CrossRef] [PubMed]
- Earle, A.J.; Kirby, T.J.; Fedorchak, G.R.; Isermann, P.; Patel, J.; Iruvanti, S.; Moore, S.A.; Bonne, G.; Wallrath, L.L.; Lammerding, J. Mutant lamins cause nuclear envelope rupture and DNA damage in skeletal muscle cells. Nat. Mater. 2019. [Google Scholar] [CrossRef] [PubMed]
- Jahed, Z.; Mofrad, M.R. The nucleus feels the force, LINCed in or not! Curr. Opin. Cell Biol. 2019, 58, 114–119. [Google Scholar] [CrossRef]
- Ohgushi, M.; Minaguchi, M.; Sasai, Y. Rho-Signaling-Directed YAP/TAZ Activity Underlies the Long-Term Survival and Expansion of Human Embryonic Stem Cells. Cell Stem Cell 2015, 17, 448–461. [Google Scholar] [CrossRef]
- Musah, S.; Wrighton, P.J.; Zaltsman, Y.; Zhong, X.; Zorn, S.; Parlato, M.B.; Hsiao, C.; Palecek, S.P.; Chang, Q.; Murphy, W.L.; et al. Substratum-induced differentiation of human pluripotent stem cells reveals the coactivator YAP is a potent regulator of neuronal specification. Proc. Natl. Acad. Sci. USA 2014, 111, 13805–13810. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene name | Abbreviation | Forward/Reverse | Sequence |
|---|---|---|---|
| Human Yes-associated protein 1 | hYAP1 | fw | GCTACAGTGTCCCTCGAACC |
| rev | CCGGTGCATGTGTCTCCTTA | ||
| Human Connective tissue growth factor | h-CTGF | fw | ACCGACTGGAAGACACGTTTG |
| rev | CCAGGTCAGCTTCGCAAGG | ||
| Human Connective tissue growth factor | h-RPLPO | fw | CTCCAAGCAGATGCAGCAGA |
| rev | ATAGCCTTGCGCATCATGGT | ||
| Human Glyceraldehyde 3-phosphate dehydrogenase | h-GAPDH | fw | TGC-CAT-GTA-GAC-CCC-TTG-AA |
| rev | TGG-TTG-AGC-ACA-GGG-TAG-TT | ||
| Human Myosin light chain 9 | h-Myl9 | fw | CGA-ATA-CCT-GGA-GGG-CAT-GAT |
| rev | AAA-CCT-GAG-GCT-TCC-TCG-TC |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Owens, D.J.; Fischer, M.; Jabre, S.; Moog, S.; Mamchaoui, K.; Butler-Browne, G.; Coirault, C. Lamin Mutations Cause Increased YAP Nuclear Entry in Muscle Stem Cells. Cells 2020, 9, 816. https://doi.org/10.3390/cells9040816
Owens DJ, Fischer M, Jabre S, Moog S, Mamchaoui K, Butler-Browne G, Coirault C. Lamin Mutations Cause Increased YAP Nuclear Entry in Muscle Stem Cells. Cells. 2020; 9(4):816. https://doi.org/10.3390/cells9040816
Chicago/Turabian StyleOwens, Daniel J., Martina Fischer, Saline Jabre, Sophie Moog, Kamel Mamchaoui, Gillian Butler-Browne, and Catherine Coirault. 2020. "Lamin Mutations Cause Increased YAP Nuclear Entry in Muscle Stem Cells" Cells 9, no. 4: 816. https://doi.org/10.3390/cells9040816
APA StyleOwens, D. J., Fischer, M., Jabre, S., Moog, S., Mamchaoui, K., Butler-Browne, G., & Coirault, C. (2020). Lamin Mutations Cause Increased YAP Nuclear Entry in Muscle Stem Cells. Cells, 9(4), 816. https://doi.org/10.3390/cells9040816