Redox Modifications of Proteins of the Mitochondrial Fusion and Fission Machinery

 , , , , ,
, , , , ,  ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. The Mitochondrial Fusion and Fission Machinery
3. Evolutionary Conservation of Proteins Involved in Mitochondrial Dynamics
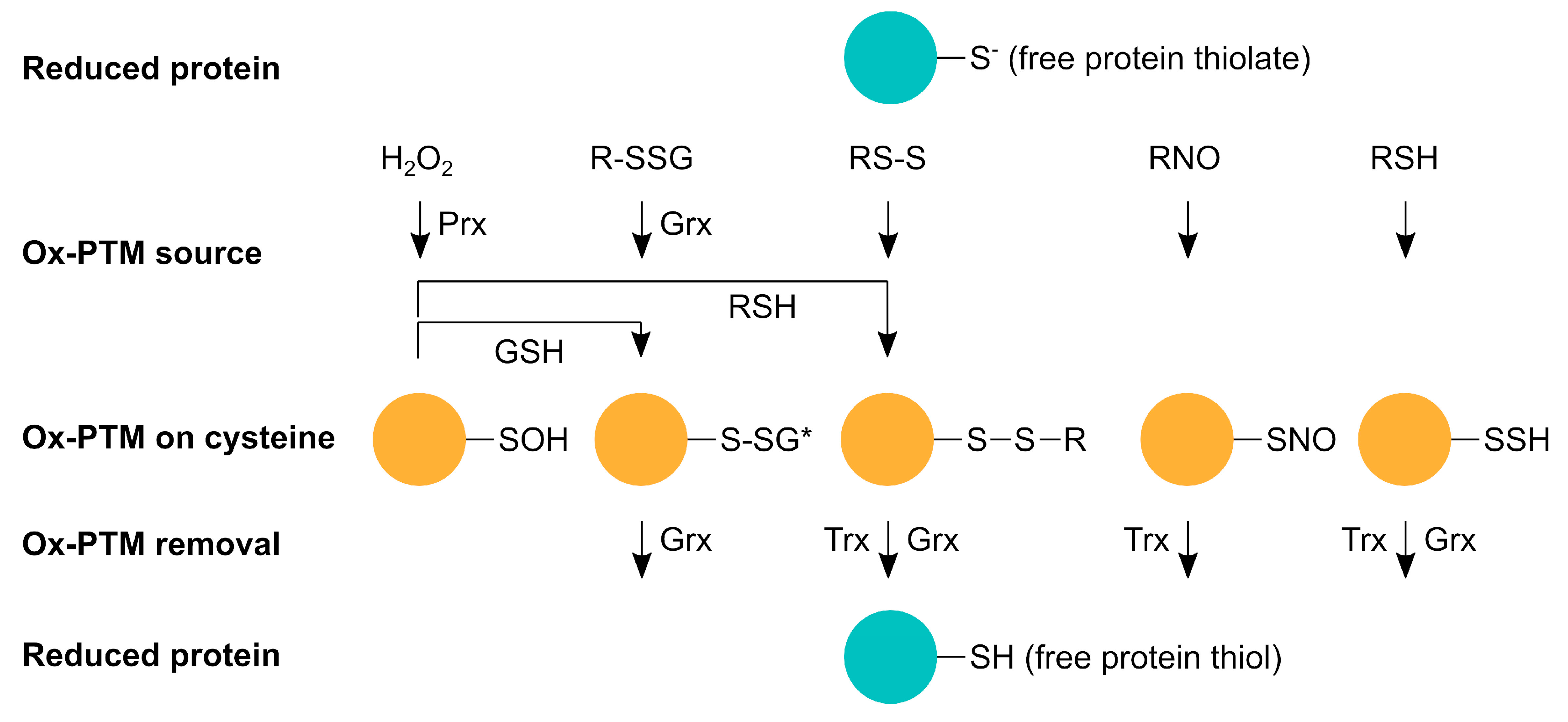
4. Redox Modifications of Proteins Involved in Mitochondrial Dynamics
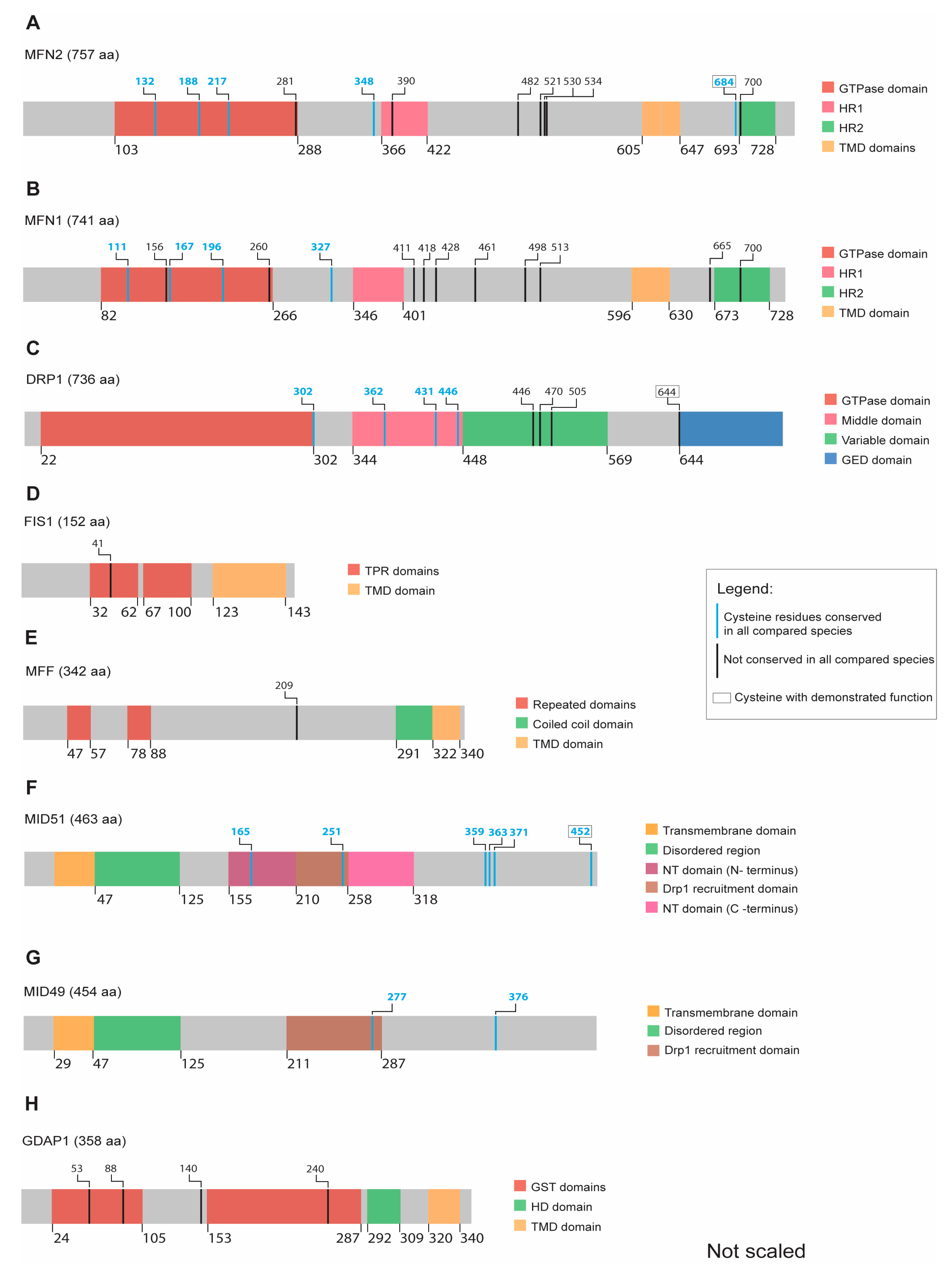
4.1. Mitofusins
4.2. DRP1
4.3. FIS1
4.4. MFF
4.5. MID49/51
4.6. GDAP1
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Miseta, A.; Csutora, P. Relationship between the occurrence of cysteine in proteins and the complexity of organisms. Mol. Biol. Evol. 2000, 17, 1232–1239. [Google Scholar] [CrossRef] [PubMed]
- Go, Y.-M.; Chandler, J.D.; Jones, D.P. The cysteine proteome. Free Radic. Biol. Med. 2015, 84, 227–245. [Google Scholar] [CrossRef] [PubMed]
- Gan, Z.R.; Wells, W.W. Identification and reactivity of the catalytic site of pig liver thioltransferase. J. Biol. Chem. 1987, 262, 6704–6707. [Google Scholar] [PubMed]
- Go, Y.-M.; Jones, D.P. The redox proteome. J. Biol. Chem. 2013, 288, 26512–26520. [Google Scholar] [CrossRef]
- Leichert, L.I.; Dick, T.P. Incidence and physiological relevance of protein thiol switches. Biol. Chem. 2015, 396, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Groitl, B.; Jakob, U. Thiol-based redox switches. Biochim. Biophys. Acta 2014, 1844, 1335–1343. [Google Scholar] [CrossRef]
- Poole, L.B.; Karplus, P.A.; Claiborne, A. Protein sulfenic acids in redox signaling. Annu. Rev. Pharmacol. Toxicol. 2004, 44, 325–347. [Google Scholar] [CrossRef]
- Fucci, L.; Oliver, C.N.; Coon, M.J.; Stadtman, E.R. Inactivation of key metabolic enzymes by mixed-function oxidation reactions: Possible implication in protein turnover and ageing. Proc. Natl. Acad. Sci. USA 1983, 80, 1521–1525. [Google Scholar] [CrossRef]
- Abe, K.; Kimura, H. The possible role of hydrogen sulfide as an endogenous neuromodulator. J. Neurosci. 1996, 16, 1066–1071. [Google Scholar] [CrossRef]
- Hosoki, R.; Matsuki, N.; Kimura, H. The possible role of hydrogen sulfide as an endogenous smooth muscle relaxant in synergy with nitric oxide. Biochem. Biophys. Res. Commun. 1997, 237, 527–531. [Google Scholar] [CrossRef]
- Hildebrandt, T.M.; Grieshaber, M.K. Three enzymatic activities catalyze the oxidation of sulfide to thiosulfate in mammalian and invertebrate mitochondria. FEBS J 2008, 275, 3352–3361. [Google Scholar] [CrossRef] [PubMed]
- Müller, F.H.; Bandeiras, T.M.; Urich, T.; Teixeira, M.; Gomes, C.M.; Kletzin, A. Coupling of the pathway of sulphur oxidation to dioxygen reduction: Characterization of a novel membrane-bound thiosulphate:quinone oxidoreductase. Mol. Microbiol. 2004, 53, 1147–1160. [Google Scholar] [CrossRef] [PubMed]
- Vitvitsky, V.; Yadav, P.K.; Kurthen, A.; Banerjee, R. Sulfide oxidation by a noncanonical pathway in red blood cells generates thiosulfate and polysulfides. J. Biol. Chem. 2015, 290, 8310–8320. [Google Scholar] [CrossRef] [PubMed]
- Kabil, O.; Banerjee, R. Redox biochemistry of hydrogen sulfide. J. Biol. Chem. 2010, 285, 21903–21907. [Google Scholar] [CrossRef]
- Dóka, É.; Ida, T.; Dagnell, M.; Abiko, Y.; Luong, N.C.; Balog, N.; Takata, T.; Espinosa, B.; Nishimura, A.; Cheng, Q.; et al. Control of protein function through oxidation and reduction of persulfidated states. Sci. Adv. 2020, 6, eaax8358. [Google Scholar] [CrossRef]
- Krishnan, N.; Fu, C.; Pappin, D.J.; Tonks, N.K. H2S-Induced sulfhydration of the phosphatase PTP1B and its role in the endoplasmic reticulum stress response. Sci. Signal. 2011, 4, ra86. [Google Scholar] [CrossRef]
- Sen, N.; Paul, B.D.; Gadalla, M.M.; Mustafa, A.K.; Sen, T.; Xu, R.; Kim, S.; Snyder, S.H. Hydrogen sulfide-linked sulfhydration of NF-κB mediates its antiapoptotic actions. Mol. Cell 2012, 45, 13–24. [Google Scholar] [CrossRef]
- Coletta, C.; Papapetropoulos, A.; Erdelyi, K.; Olah, G.; Módis, K.; Panopoulos, P.; Asimakopoulou, A.; Gerö, D.; Sharina, I.; Martin, E.; et al. Hydrogen sulfide and nitric oxide are mutually dependent in the regulation of angiogenesis and endothelium-dependent vasorelaxation. Proc. Natl. Acad. Sci. USA 2012, 109, 9161–9166. [Google Scholar] [CrossRef]
- Eberhardt, M.; Dux, M.; Namer, B.; Miljkovic, J.; Cordasic, N.; Will, C.; Kichko, T.I.; de la Roche, J.; Fischer, M.; Suárez, S.A.; et al. H2S and NO cooperatively regulate vascular tone by activating a neuroendocrine HNO-TRPA1-CGRP signalling pathway. Nat. Commun. 2014, 5, 4381. [Google Scholar] [CrossRef]
- King, A.L.; Polhemus, D.J.; Bhushan, S.; Otsuka, H.; Kondo, K.; Nicholson, C.K.; Bradley, J.M.; Islam, K.N.; Calvert, J.W.; Tao, Y.-X.; et al. Hydrogen sulfide cytoprotective signaling is endothelial nitric oxide synthase-nitric oxide dependent. Proc. Natl. Acad. Sci. USA 2014, 111, 3182–3187. [Google Scholar] [CrossRef]
- Mustafa, A.K.; Sikka, G.; Gazi, S.K.; Steppan, J.; Jung, S.M.; Bhunia, A.K.; Barodka, V.M.; Gazi, F.K.; Barrow, R.K.; Wang, R.; et al. Hydrogen sulfide as endothelium-derived hyperpolarizing factor sulfhydrates potassium channels. Circ. Res. 2011, 109, 1259–1268. [Google Scholar] [CrossRef] [PubMed]
- Broniowska, K.A.; Keszler, A.; Basu, S.; Kim-Shapiro, D.B.; Hogg, N. Cytochrome c-mediated formation of S-nitrosothiol in cells. Biochem. J. 2012, 442, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Olson, N.; Kasahara, D.I.; Hristova, M.; Bernstein, R.; Janssen-Heininger, Y.; van der Vliet, A. Modulation of NF-κB and hypoxia-inducible factor--1 by S-nitrosoglutathione does not alter allergic airway inflammation in mice. Am. J. Respir. Cell Mol. Biol. 2011, 44, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Hara, M.R.; Agrawal, N.; Kim, S.F.; Cascio, M.B.; Fujimuro, M.; Ozeki, Y.; Takahashi, M.; Cheah, J.H.; Tankou, S.K.; Hester, L.D.; et al. S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat. Cell Biol. 2005, 7, 665–674. [Google Scholar] [CrossRef]
- Chang, A.H.K.; Sancheti, H.; Garcia, J.; Kaplowitz, N.; Cadenas, E.; Han, D. Respiratory substrates regulate S-nitrosylation of mitochondrial proteins through a thiol-dependent pathway. Chem. Res. Toxicol. 2014, 27, 794–804. [Google Scholar] [CrossRef]
- Piantadosi, C.A. Regulation of mitochondrial processes by protein S-nitrosylation. Biochim. Biophys. Acta 2012, 1820, 712–721. [Google Scholar] [CrossRef]
- Doulias, P.-T.; Tenopoulou, M.; Greene, J.L.; Raju, K.; Ischiropoulos, H. Nitric oxide regulates mitochondrial fatty acid metabolism through reversible protein S-nitrosylation. Sci. Signal. 2013, 6, rs1. [Google Scholar] [CrossRef]
- Bartesaghi, S.; Radi, R. Fundamentals on the biochemistry of peroxynitrite and protein tyrosine nitration. Redox Biol. 2018, 14, 618–625. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, C.-L.; Kang, P.T.; Garg, V.; Hu, K.; Green-Church, K.B.; Chen, Y.-R. Peroxynitrite-mediated oxidative modifications of complex II: Relevance in myocardial infarction. Biochemistry 2010, 49, 2529–2539. [Google Scholar] [CrossRef][Green Version]
- Kang, P.T.; Zhang, L.; Chen, C.-L.; Chen, J.; Green, K.B.; Chen, Y.-R. Protein thiyl radical mediates S-glutathionylation of complex I. Free Radic. Biol. Med. 2012, 53, 962–973. [Google Scholar] [CrossRef]
- Sykes, M.C.; Mowbray, A.L.; Jo, H. Reversible Glutathiolation of Caspase-3 by Glutaredoxin as a Novel Redox Signaling Mechanism in Tumor Necrosis Factor-α–Induced Cell Death. Circ. Res. 2007, 100, 152–154. [Google Scholar] [CrossRef]
- Klatt, P.; Lamas, S. Regulation of protein function by S-glutathiolation in response to oxidative and nitrosative stress. Eur. J. Biochem. 2000, 267, 4928–4944. [Google Scholar] [CrossRef] [PubMed]
- Pastore, A.; Piemonte, F. S-Glutathionylation signaling in cell biology: Progress and prospects. Eur. J. Pharm. Sci. 2012, 46, 279–292. [Google Scholar] [CrossRef] [PubMed]
- Beer, S.M.; Taylor, E.R.; Brown, S.E.; Dahm, C.C.; Costa, N.J.; Runswick, M.J.; Murphy, M.P. Glutaredoxin 2 catalyzes the reversible oxidation and glutathionylation of mitochondrial membrane thiol proteins: Implications for mitochondrial redox regulation and antioxidant DEFENSE. J. Biol. Chem. 2004, 279, 47939–47951. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef]
- Hanschmann, E.-M.; Godoy, J.R.; Berndt, C.; Hudemann, C.; Lillig, C.H. Thioredoxins, glutaredoxins, and peroxiredoxins--molecular mechanisms and health significance: From cofactors to antioxidants to redox signaling. Antioxid. Redox Signal. 2013, 19, 1539–1605. [Google Scholar] [CrossRef]
- Berndt, C.; Lillig, C.H.; Flohé, L. Redox regulation by glutathione needs enzymes. Front. Pharmacol. 2014, 5, 168. [Google Scholar] [CrossRef]
- Sengupta, R.; Holmgren, A. Thioredoxin and thioredoxin reductase in relation to reversible S-nitrosylation. Antioxid. Redox Signal. 2013, 18, 259–269. [Google Scholar] [CrossRef]
- Dóka, É.; Pader, I.; Bíró, A.; Johansson, K.; Cheng, Q.; Ballagó, K.; Prigge, J.R.; Pastor-Flores, D.; Dick, T.P.; Schmidt, E.E.; et al. A novel persulfide detection method reveals protein persulfide- and polysulfide-reducing functions of thioredoxin and glutathione systems. Sci. Adv. 2016, 2, e1500968. [Google Scholar] [CrossRef]
- Lillig, C.H.; Berndt, C. Glutaredoxins in thiol/disulfide exchange. Antioxid. Redox Signal. 2013, 18, 1654–1665. [Google Scholar] [CrossRef]
- Chang, T.-S.; Jeong, W.; Woo, H.A.; Lee, S.M.; Park, S.; Rhee, S.G. Characterization of Mammalian Sulfiredoxin and Its Reactivation of Hyperoxidized Peroxiredoxin through Reduction of Cysteine Sulfinic Acid in the Active Site to Cysteine. J. Biol. Chem. 2004, 279, 50994–51001. [Google Scholar] [CrossRef] [PubMed]
- Biteau, B.; Labarre, J.; Toledano, M.B. ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae sulphiredoxin. Nature 2003, 425, 980–984. [Google Scholar] [CrossRef] [PubMed]
- Akter, S.; Fu, L.; Jung, Y.; Conte, M.L.; Lawson, J.R.; Lowther, W.T.; Sun, R.; Liu, K.; Yang, J.; Carroll, K.S. Chemical proteomics reveals new targets of cysteine sulfinic acid reductase. Nat. Chem. Biol. 2018, 14, 995–1004. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C.; Hampton, M.B. Redox biology: Signaling via a peroxiredoxin sensor. Nat. Chem. Biol. 2015, 11, 5–6. [Google Scholar] [CrossRef]
- Stöcker, S.; Maurer, M.; Ruppert, T.; Dick, T.P. A role for 2-Cys peroxiredoxins in facilitating cytosolic protein thiol oxidation. Nat. Chem. Biol. 2018, 14, 148–155. [Google Scholar] [CrossRef]
- Soares Moretti, A.I.; Martins Laurindo, F.R. Protein disulfide isomerases: Redox connections in and out of the endoplasmic reticulum. Arch. Biochem. Biophys. 2017, 617, 106–119. [Google Scholar] [CrossRef]
- Tian, C.; Alomar, F.; Moore, C.J.; Shao, C.H.; Kutty, S.; Singh, J.; Bidasee, K.R. Reactive carbonyl species and their roles in sarcoplasmic reticulum Ca2+ cycling defect in the diabetic heart. Heart Fail. Rev. 2014, 19, 101–112. [Google Scholar] [CrossRef]
- Watanabe, Y.; Cohen, R.A.; Matsui, R. Redox Regulation of Ischemic Angiogenesis Another Aspect of Reactive Oxygen Species. Circ. J. 2016, 80, 1278–1284. [Google Scholar] [CrossRef]
- Venditti, P.; Di Stefano, L.; Di Meo, S. Mitochondrial metabolism of reactive oxygen species. Mitochondrion 2013, 13, 71–82. [Google Scholar] [CrossRef]
- Hamanaka, R.B.; Chandel, N.S. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem. Sci. 2010, 35, 505–513. [Google Scholar] [CrossRef]
- Kussmaul, L.; Hirst, J. The mechanism of superoxide production by NADH:ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Proc. Natl. Acad. Sci. USA 2006, 103, 7607–7612. [Google Scholar] [CrossRef] [PubMed]
- Pryde, K.R.; Hirst, J. Superoxide is produced by the reduced flavin in mitochondrial complex I: A single, unified mechanism that applies during both forward and reverse electron transfer. J. Biol. Chem. 2011, 286, 18056–18065. [Google Scholar] [CrossRef] [PubMed]
- Kudin, A.P.; Bimpong-Buta, N.Y.-B.; Vielhaber, S.; Elger, C.E.; Kunz, W.S. Characterization of superoxide-producing sites in isolated brain mitochondria. J. Biol. Chem. 2004, 279, 4127–4135. [Google Scholar] [CrossRef]
- Votyakova, T.V.; Reynolds, I.J. DeltaPsi(m)-Dependent and -independent production of reactive oxygen species by rat brain mitochondria. J. Neurochem. 2001, 79, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Fiskum, G.; Schubert, D. Generation of reactive oxygen species by the mitochondrial electron transport chain. J. Neurochem. 2002, 80, 780–787. [Google Scholar] [CrossRef] [PubMed]
- Chouchani, E.T.; Methner, C.; Nadtochiy, S.M.; Logan, A.; Pell, V.R.; Ding, S.; James, A.M.; Cochemé, H.M.; Reinhold, J.; Lilley, K.S.; et al. Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I. Nat. Med. 2013, 19, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Bleier, L.; Wittig, I.; Heide, H.; Steger, M.; Brandt, U.; Dröse, S. Generator-specific targets of mitochondrial reactive oxygen species. Free Radic. Biol. Med. 2015, 78, 1–10. [Google Scholar] [CrossRef]
- Turrens, J.F.; Alexandre, A.; Lehninger, A.L. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch. Biochem. Biophys. 1985, 237, 408–414. [Google Scholar] [CrossRef]
- Muller, F.L.; Liu, Y.; Van Remmen, H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. J. Biol. Chem. 2004, 279, 49064–49073. [Google Scholar] [CrossRef]
- Han, D.; Antunes, F.; Canali, R.; Rettori, D.; Cadenas, E. Voltage-dependent anion channels control the release of the superoxide anion from mitochondria to cytosol. J. Biol. Chem. 2003, 278, 5557–5563. [Google Scholar] [CrossRef]
- Lillig, C.H.; Berndt, C.; Holmgren, A. Glutaredoxin systems. Biochim. Biophys. Acta 2008, 1780, 1304–1317. [Google Scholar] [CrossRef] [PubMed]
- Pai, H.V.; Starke, D.W.; Lesnefsky, E.J.; Hoppel, C.L.; Mieyal, J.J. What is the functional significance of the unique location of glutaredoxin 1 (GRx1) in the intermembrane space of mitochondria? Antioxid. Redox Signal. 2007, 9, 2027–2033. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Chan, D.C. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 634–646. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Chen, H.; Fiket, M.; Alexander, C.; Chan, D.C. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J. Cell Biol. 2007, 178, 749–755. [Google Scholar] [CrossRef] [PubMed]
- Palmer, C.S.; Osellame, L.D.; Laine, D.; Koutsopoulos, O.S.; Frazier, A.E.; Ryan, M.T. MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep. 2011, 12, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liu, L.; Wu, S.; Xing, D. Drp1, Mff, Fis1, and MiD51 are coordinated to mediate mitochondrial fission during UV irradiation-induced apoptosis. FASEB J. 2016, 30, 466–476. [Google Scholar] [CrossRef]
- Niemann, A.; Wagner, K.M.; Ruegg, M.; Suter, U. GDAP1 mutations differ in their effects on mitochondrial dynamics and apoptosis depending on the mode of inheritance. Neurobiol. Dis. 2009, 36, 509–520. [Google Scholar] [CrossRef]
- Huber, N.; Bieniossek, C.; Wagner, K.M.; Elsässer, H.-P.; Suter, U.; Berger, I.; Niemann, A. Glutathione-conjugating and membrane-remodeling activity of GDAP1 relies on amphipathic C-terminal domain. Sci. Rep. 2016, 6, 36930. [Google Scholar] [CrossRef]
- González-Sánchez, P.; Pla-Martín, D.; Martínez-Valero, P.; Rueda, C.B.; Calpena, E.; del Arco, A.; Palau, F.; Satrústegui, J. CMT-linked loss-of-function mutations in GDAP1 impair store-operated Ca2+ entry-stimulated respiration. Sci. Rep. 2017, 7, 42993. [Google Scholar] [CrossRef]
- Kawalec, M.; Zabłocka, B.; Kabzińska, D.; Neska, J.; Beręsewicz, M. Mitofusin 2 expression dominates over mitofusin 1 exclusively in mouse dorsal root ganglia—A possible explanation for peripheral nervous system involvement in Charcot-Marie-Tooth 2A. Folia Neuropathol. 2014, 52, 436–442. [Google Scholar] [CrossRef]
- Detmer, S.A.; Chan, D.C. Complementation between mouse Mfn1 and Mfn2 protects mitochondrial fusion defects caused by CMT2A disease mutations. J. Cell Biol. 2007, 176, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-J.; Cao, Y.-L.; Feng, J.-X.; Qi, Y.; Meng, S.; Yang, J.-F.; Zhong, Y.-T.; Kang, S.; Chen, X.; Lan, L.; et al. Structural insights of human mitofusin-2 into mitochondrial fusion and CMT2A onset. Nat. Commun. 2019, 10, 4914. [Google Scholar] [CrossRef] [PubMed]
- Pyakurel, A.; Savoia, C.; Hess, D.; Scorrano, L. Extracellular regulated kinase phosphorylates mitofusin 1 to control mitochondrial morphology and apoptosis. Mol. Cell 2015, 58, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Schrepfer, E.; Scorrano, L. Mitofusins, from Mitochondria to Metabolism. Mol. Cell 2016, 61, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Koshiba, T.; Detmer, S.A.; Kaiser, J.T.; Chen, H.; McCaffery, J.M.; Chan, D.C. Structural basis of mitochondrial tethering by mitofusin complexes. Science 2004, 305, 858–862. [Google Scholar] [CrossRef]
- Mattie, S.; Riemer, J.; Wideman, J.G.; McBride, H.M. A new mitofusin topology places the redox-regulated C terminus in the mitochondrial intermembrane space. J. Cell Biol. 2018, 217, 507–515. [Google Scholar] [CrossRef]
- Narendra, D.; Tanaka, A.; Suen, D.-F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef]
- Gegg, M.E.; Cooper, J.M.; Chau, K.-Y.; Rojo, M.; Schapira, A.H.V.; Taanman, J.-W. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum. Mol. Genet. 2010, 19, 4861–4870. [Google Scholar] [CrossRef]
- Chen, Y.; Dorn, G.W. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 2013, 340, 471–475. [Google Scholar] [CrossRef]
- Mukherjee, R.; Chakrabarti, O. Regulation of Mitofusin1 by Mahogunin Ring Finger-1 and the proteasome modulates mitochondrial fusion. Biochim. Biophys. Acta 2016, 1863, 3065–3083. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.T.C.; Garcin, P.O.; Fu, M.; Masoudi, M.; St-Pierre, P.; Panté, N.; Nabi, I.R. Distinct mechanisms controlling rough and smooth endoplasmic reticulum contacts with mitochondria. J. Cell Sci. 2015, 128, 2759–2765. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; St-Pierre, P.; Shankar, J.; Wang, P.T.C.; Joshi, B.; Nabi, I.R. Regulation of mitophagy by the Gp78 E3 ubiquitin ligase. Mol. Biol. Cell 2013, 24, 1153–1162. [Google Scholar] [CrossRef] [PubMed]
- Barsoum, M.J.; Yuan, H.; Gerencser, A.A.; Liot, G.; Kushnareva, Y.; Gräber, S.; Kovacs, I.; Lee, W.D.; Waggoner, J.; Cui, J.; et al. Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J. 2006, 25, 3900–3911. [Google Scholar] [CrossRef] [PubMed]
- Shutt, T.; Geoffrion, M.; Milne, R.; McBride, H.M. The intracellular redox state is a core determinant of mitochondrial fusion. EMBO Rep. 2012, 13, 909–915. [Google Scholar] [CrossRef] [PubMed]
- Thaher, O.; Wolf, C.; Dey, P.N.; Pouya, A.; Wüllner, V.; Tenzer, S.; Methner, A. The thiol switch C684 in Mitofusin-2 mediates redox-induced alterations of mitochondrial shape and respiration. Neurochem. Int. 2018, 117, 167–173. [Google Scholar] [CrossRef]
- Bleazard, W.; McCaffery, J.M.; King, E.J.; Bale, S.; Mozdy, A.; Tieu, Q.; Nunnari, J.; Shaw, J.M. The dynamin-related GTPase Dnm1 regulates mitochondrial fission in yeast. Nat. Cell Biol. 1999, 1, 298–304. [Google Scholar] [CrossRef]
- Smirnova, E.; Griparic, L.; Shurland, D.L.; van der Bliek, A.M. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 2001, 12, 2245–2256. [Google Scholar] [CrossRef]
- Reddy, P.H.; Reddy, T.P.; Manczak, M.; Calkins, M.J.; Shirendeb, U.; Mao, P. Dynamin-related protein 1 and mitochondrial fragmentation in neurodegenerative diseases. Brain Res. Rev. 2011, 67, 103–118. [Google Scholar] [CrossRef]
- Oliver, D.; Reddy, P.H. Dynamics of Dynamin-Related Protein 1 in Alzheimer’s Disease and Other Neurodegenerative Diseases. Cells 2019, 8, 961. [Google Scholar] [CrossRef]
- Shin, H.W.; Shinotsuka, C.; Torii, S.; Murakami, K.; Nakayama, K. Identification and subcellular localization of a novel mammalian dynamin-related protein homologous to yeast Vps1p and Dnm1p. J. Biochem. 1997, 122, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Imoto, M.; Tachibana, I.; Urrutia, R. Identification and functional characterization of a novel human protein highly related to the yeast dynamin-like GTPase Vps1p. J. Cell Sci. 1998, 111, 1341–1349. [Google Scholar] [PubMed]
- Yoon, Y.; Pitts, K.R.; Dahan, S.; McNiven, M.A. A novel dynamin-like protein associates with cytoplasmic vesicles and tubules of the endoplasmic reticulum in mammalian cells. J. Cell Biol. 1998, 140, 779–793. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-R.; Manlandro, C.M.; Arnoult, D.; Stadler, J.; Posey, A.E.; Hill, R.B.; Blackstone, C. A lethal de novo mutation in the middle domain of the dynamin-related GTPase Drp1 impairs higher order assembly and mitochondrial division. J. Biol. Chem. 2010, 285, 32494–32503. [Google Scholar] [CrossRef]
- Ingerman, E.; Perkins, E.M.; Marino, M.; Mears, J.A.; McCaffery, J.M.; Hinshaw, J.E.; Nunnari, J. Dnm1 forms spirals that are structurally tailored to fit mitochondria. J. Cell Biol. 2005, 170, 1021–1027. [Google Scholar] [CrossRef]
- Michalska, B.M.; Kwapiszewska, K.; Szczepanowska, J.; Kalwarczyk, T.; Patalas-Krawczyk, P.; Szczepański, K.; Hołyst, R.; Duszynski, J.; Szymański, J. Insight into the fission mechanism by quantitative characterization of Drp1 protein distribution in the living cell. Sci. Rep. 2018, 8, 8122. [Google Scholar] [CrossRef]
- Kwapiszewska, K.; Kalwarczyk, T.; Michalska, B.; Szczepański, K.; Szymański, J.; Patalas-Krawczyk, P.; Andryszewski, T.; Iwan, M.; Duszynski, J.; Hołyst, R. Determination of oligomerization state of Drp1 protein in living cells at nanomolar concentrations. Sci. Rep. 2019, 9, 5906. [Google Scholar] [CrossRef]
- Qi, Z.; Huang, Z.; Xie, F.; Chen, L. Dynamin-related protein 1: A critical protein in the pathogenesis of neural system dysfunctions and neurodegenerative diseases. J. Cell. Physiol. 2019, 234, 10032–10046. [Google Scholar] [CrossRef]
- Cho, D.-H.; Nakamura, T.; Fang, J.; Cieplak, P.; Godzik, A.; Gu, Z.; Lipton, S.A. S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science 2009, 324, 102–105. [Google Scholar] [CrossRef]
- Bossy, B.; Petrilli, A.; Klinglmayr, E.; Chen, J.; Lütz-Meindl, U.; Knott, A.B.; Masliah, E.; Schwarzenbacher, R.; Bossy-Wetzel, E. S-Nitrosylation of DRP1 does not affect enzymatic activity and is not specific to Alzheimer’s disease. J. Alzheimers Dis. 2010, 20, S513–S526. [Google Scholar] [CrossRef]
- Nakamura, T.; Lipton, S.A. Redox modulation by S-nitrosylation contributes to protein misfolding, mitochondrial dynamics, and neuronal synaptic damage in neurodegenerative diseases. Cell Death Differ. 2011, 18, 1478–1486. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.; Nakamura, T.; Holland, E.A.; McKercher, S.R.; Lipton, S.A. S-nitrosylation of Cdk5: Potential implications in amyloid-β-related neurotoxicity in Alzheimer disease. Prion 2012, 6, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Rizza, S.; Cardaci, S.; Montagna, C.; Di Giacomo, G.; De Zio, D.; Bordi, M.; Maiani, E.; Campello, S.; Borreca, A.; Puca, A.A.; et al. S-nitrosylation drives cell senescence and aging in mammals by controlling mitochondrial dynamics and mitophagy. Proc. Natl. Acad. Sci. USA 2018, 115, E3388–E3397. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-M.; Youn, S.-W.; Sudhahar, V.; Das, A.; Chandhri, R.; Cuervo Grajal, H.; Kweon, J.; Leanhart, S.; He, L.; Toth, P.T.; et al. Redox Regulation of Mitochondrial Fission Protein Drp1 by Protein Disulfide Isomerase Limits Endothelial Senescence. Cell Rep. 2018, 23, 3565–3578. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Zhang, Q.; Zhang, P.; Sun, L.; Peng, C.; Yuan, Z.; Cheng, J. c-Abl-mediated Drp1 phosphorylation promotes oxidative stress-induced mitochondrial fragmentation and neuronal cell death. Cell Death Dis. 2017, 8, e3117. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.-S.; Kim, J.-E. PDI-mediated S-nitrosylation of DRP1 facilitates DRP1-S616 phosphorylation and mitochondrial fission in CA1 neurons. Cell Death Dis. 2018, 9, 869. [Google Scholar] [CrossRef]
- Tsushima, K.; Bugger, H.; Wende, A.R.; Soto, J.; Jenson, G.A.; Tor, A.R.; McGlauflin, R.; Kenny, H.C.; Zhang, Y.; Souvenir, R.; et al. Mitochondrial Reactive Oxygen Species in Lipotoxic Hearts Induce Post-Translational Modifications of AKAP121, DRP1, and OPA1 That Promote Mitochondrial Fission. Circ. Res. 2018, 122, 58–73. [Google Scholar] [CrossRef]
- Mozdy, A.D.; McCaffery, J.M.; Shaw, J.M. Dnm1p GTPase-mediated mitochondrial fission is a multi-step process requiring the novel integral membrane component Fis1p. J. Cell Biol. 2000, 151, 367–380. [Google Scholar] [CrossRef]
- Otera, H.; Wang, C.; Cleland, M.M.; Setoguchi, K.; Yokota, S.; Youle, R.J.; Mihara, K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J. Cell Biol. 2010, 191, 1141–1158. [Google Scholar] [CrossRef]
- Losón, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 2013, 24, 659–667. [Google Scholar] [CrossRef]
- Jofuku, A.; Ishihara, N.; Mihara, K. Analysis of functional domains of rat mitochondrial Fis1, the mitochondrial fission-stimulating protein. Biochem. Biophys. Res. Commun. 2005, 333, 650–659. [Google Scholar] [CrossRef] [PubMed]
- Dohm, J.A.; Lee, S.J.; Hardwick, J.M.; Hill, R.B.; Gittis, A.G. Cytosolic domain of the human mitochondrial fission protein fis1 adopts a TPR fold. Proteins 2004, 54, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Fox, R.J.; Burwell, L.S.; Yoon, Y. Regulation of mitochondrial fission and apoptosis by the mitochondrial outer membrane protein hFis1. J. Cell Sci. 2005, 118, 4141–4151. [Google Scholar] [CrossRef] [PubMed]
- Wells, R.C.; Picton, L.K.; Williams, S.C.P.; Tan, F.J.; Hill, R.B. Direct binding of the dynamin-like GTPase, Dnm1, to mitochondrial dynamics protein Fis1 is negatively regulated by the Fis1 N-terminal arm. J. Biol. Chem. 2007, 282, 33769–33775. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Mehta, S.L.; Li, P.A. Glutamate induces mitochondrial dynamic imbalance and autophagy activation: Preventive effects of selenium. PLoS ONE 2012, 7, e39382. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wu, J.; Wu, R.; Ma, J.; Du, G.; Jiao, R.; Tian, Y.; Zheng, Z.; Yuan, Z. DJ-1 promotes the proteasomal degradation of Fis1: Implications of DJ-1 in neuronal protection. Biochem. J. 2012, 447, 261–269. [Google Scholar] [CrossRef]
- Liu, H.; Mao, P.; Wang, J.; Wang, T.; Xie, C.-H. Allicin Protects PC12 Cells Against 6-OHDA-Induced Oxidative Stress and Mitochondrial Dysfunction via Regulating Mitochondrial Dynamics. Cell. Physiol. Biochem. 2015, 36, 966–979. [Google Scholar] [CrossRef]
- Tangmansakulchai, K.; Abubakar, Z.; Kitiyanant, N.; Suwanjang, W.; Leepiyasakulchai, C.; Govitrapong, P.; Chetsawang, B. Calpastatin overexpression reduces oxidative stress-induced mitochondrial impairment and cell death in human neuroblastoma SH-SY5Y cells by decreasing calpain and calcineurin activation, induction of mitochondrial fission and destruction of mitochondrial fusion. Mitochondrion 2016, 30, 151–161. [Google Scholar]
- Gandre-Babbe, S.; van der Bliek, A.M. The novel tail-anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol. Biol. Cell 2008, 19, 2402–2412. [Google Scholar] [CrossRef]
- Willems, P.H.G.M.; Rossignol, R.; Dieteren, C.E.J.; Murphy, M.P.; Koopman, W.J.H. Redox Homeostasis and Mitochondrial Dynamics. Cell Metab. 2015, 22, 207–218. [Google Scholar] [CrossRef]
- Toyama, E.Q.; Herzig, S.; Courchet, J.; Lewis, T.L.; Losón, O.C.; Hellberg, K.; Young, N.P.; Chen, H.; Polleux, F.; Chan, D.C.; et al. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 2016, 351, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Ducommun, S.; Deak, M.; Sumpton, D.; Ford, R.J.; Núñez Galindo, A.; Kussmann, M.; Viollet, B.; Steinberg, G.R.; Foretz, M.; Dayon, L.; et al. Motif affinity and mass spectrometry proteomic approach for the discovery of cellular AMPK targets: Identification of mitochondrial fission factor as a new AMPK substrate. Cell Signal. 2015, 27, 978–988. [Google Scholar] [CrossRef] [PubMed]
- Sarraf, S.A.; Raman, M.; Guarani-Pereira, V.; Sowa, M.E.; Huttlin, E.L.; Gygi, S.P.; Harper, J.W. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 2013, 496, 372–376. [Google Scholar] [CrossRef]
- Lee, L.; Seager, R.; Nakamura, Y.; Wilkinson, K.A.; Henley, J.M. Parkin-mediated ubiquitination contributes to the constitutive turnover of mitochondrial fission factor (Mff). PLoS ONE 2019, 14, e0213116. [Google Scholar] [CrossRef]
- Bak, D.W.; Pizzagalli, M.D.; Weerapana, E. Identifying Functional Cysteine Residues in the Mitochondria. ACS Chem. Biol. 2017, 12, 947–957. [Google Scholar] [CrossRef]
- Simpson, J.C.; Wellenreuther, R.; Poustka, A.; Pepperkok, R.; Wiemann, S. Systematic subcellular localization of novel proteins identified by large-scale cDNA sequencing. EMBO Rep. 2000, 1, 287–292. [Google Scholar] [CrossRef]
- Bartsakoulia, M.; Pyle, A.; Troncoso-Chandía, D.; Vial-Brizzi, J.; Paz-Fiblas, M.V.; Duff, J.; Griffin, H.; Boczonadi, V.; Lochmüller, H.; Kleinle, S.; et al. A novel mechanism causing imbalance of mitochondrial fusion and fission in human myopathies. Hum. Mol. Genet. 2018, 27, 1186–1195. [Google Scholar] [CrossRef]
- Samangouei, P.; Crespo-Avilan, G.E.; Cabrera-Fuentes, H.; Hernández-Reséndiz, S.; Ismail, N.I.; Katwadi, K.B.; Boisvert, W.A.; Hausenloy, D.J. MiD49 and MiD51: New mediators of mitochondrial fission and novel targets for cardioprotection. Cond. Med. 2018, 1, 239–246. [Google Scholar]
- Losón, O.C.; Meng, S.; Ngo, H.; Liu, R.; Kaiser, J.T.; Chan, D.C. Crystal structure and functional analysis of MiD49, a receptor for the mitochondrial fission protein Drp1. Protein Sci. 2015, 24, 386–394. [Google Scholar] [CrossRef]
- Losón, O.C.; Liu, R.; Rome, M.E.; Meng, S.; Kaiser, J.T.; Shan, S.-O.; Chan, D.C. The mitochondrial fission receptor MiD51 requires ADP as a cofactor. Structure 2014, 22, 367–377. [Google Scholar] [CrossRef]
- Richter, V.; Palmer, C.S.; Osellame, L.D.; Singh, A.P.; Elgass, K.; Stroud, D.A.; Sesaki, H.; Kvansakul, M.; Ryan, M.T. Structural and functional analysis of MiD51, a dynamin receptor required for mitochondrial fission. J. Cell Biol. 2014, 204, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Kalia, R.; Wang, R.Y.-R.; Yusuf, A.; Thomas, P.V.; Agard, D.A.; Shaw, J.M.; Frost, A. Structural basis of mitochondrial receptor binding and constriction by DRP1. Nature 2018, 558, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Palmer, C.S.; Elgass, K.D.; Parton, R.G.; Osellame, L.D.; Stojanovski, D.; Ryan, M.T. Adaptor proteins MiD49 and MiD51 can act independently of Mff and Fis1 in Drp1 recruitment and are specific for mitochondrial fission. J. Biol. Chem. 2013, 288, 27584–27593. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Zhai, Y.; Chen, M.; Zhang, K.; Chen, Q.; Pang, X.; Sun, F. New interfaces on MiD51 for Drp1 recruitment and regulation. PLoS ONE 2019, 14, e0211459. [Google Scholar] [CrossRef]
- Zhao, J.; Liu, T.; Jin, S.; Wang, X.; Qu, M.; Uhlén, P.; Tomilin, N.; Shupliakov, O.; Lendahl, U.; Nistér, M. Human MIEF1 recruits Drp1 to mitochondrial outer membranes and promotes mitochondrial fusion rather than fission. EMBO J. 2011, 30, 2762–2778. [Google Scholar] [CrossRef]
- Liu, H.; Nakagawa, T.; Kanematsu, T.; Uchida, T.; Tsuji, S. Isolation of 10 differentially expressed cDNAs in differentiated Neuro2a cells induced through controlled expression of the GD3 synthase gene. J. Neurochem. 1999, 72, 1781–1790. [Google Scholar] [CrossRef]
- Baxter, R.V.; Ben Othmane, K.; Rochelle, J.M.; Stajich, J.E.; Hulette, C.; Dew-Knight, S.; Hentati, F.; Ben Hamida, M.; Bel, S.; Stenger, J.E.; et al. Ganglioside-induced differentiation-associated protein-1 is mutant in Charcot-Marie-Tooth disease type 4A/8q21. Nat. Genet. 2002, 30, 21–22. [Google Scholar] [CrossRef]
- Cuesta, A.; Pedrola, L.; Sevilla, T.; García-Planells, J.; Chumillas, M.J.; Mayordomo, F.; LeGuern, E.; Marín, I.; Vílchez, J.J.; Palau, F. The gene encoding ganglioside-induced differentiation-associated protein 1 is mutated in axonal Charcot-Marie-Tooth type 4A disease. Nat. Genet. 2002, 30, 22–25. [Google Scholar] [CrossRef]
- Niemann, A.; Ruegg, M.; La Padula, V.; Schenone, A.; Suter, U. Ganglioside-induced differentiation associated protein 1 is a regulator of the mitochondrial network: New implications for Charcot-Marie-Tooth disease. J. Cell Biol. 2005, 170, 1067–1078. [Google Scholar] [CrossRef]
- Pedrola, L.; Espert, A.; Wu, X.; Claramunt, R.; Shy, M.E.; Palau, F. GDAP1, the protein causing Charcot-Marie-Tooth disease type 4A, is expressed in neurons and is associated with mitochondria. Hum. Mol. Genet. 2005, 14, 1087–1094. [Google Scholar] [CrossRef] [PubMed]
- Shield, A.J.; Murray, T.P.; Board, P.G. Functional characterisation of ganglioside-induced differentiation-associated protein 1 as a glutathione transferase. Biochem. Biophys. Res. Commun. 2006, 347, 859–866. [Google Scholar] [CrossRef]
- Noack, R.; Frede, S.; Albrecht, P.; Henke, N.; Pfeiffer, A.; Knoll, K.; Dehmel, T.; Meyer Zu Hörste, G.; Stettner, M.; Kieseier, B.C.; et al. Charcot-Marie-Tooth disease CMT4A: GDAP1 increases cellular glutathione and the mitochondrial membrane potential. Hum. Mol. Genet. 2012, 21, 150–162. [Google Scholar] [CrossRef] [PubMed]
- López Del Amo, V.; Seco-Cervera, M.; García-Giménez, J.L.; Whitworth, A.J.; Pallardó, F.V.; Galindo, M.I. Mitochondrial defects and neuromuscular degeneration caused by altered expression of Drosophila Gdap1: Implications for the Charcot-Marie-Tooth neuropathy. Hum. Mol. Genet. 2015, 24, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Niemann, A.; Huber, N.; Wagner, K.M.; Somandin, C.; Horn, M.; Lebrun-Julien, F.; Angst, B.; Pereira, J.A.; Halfter, H.; Welzl, H.; et al. The Gdap1 knockout mouse mechanistically links redox control to Charcot-Marie-Tooth disease. Brain 2014, 137, 668–682. [Google Scholar] [CrossRef] [PubMed]
- Marco, A.; Cuesta, A.; Pedrola, L.; Palau, F.; Marín, I. Evolutionary and structural analyses of GDAP1, involved in Charcot-Marie-Tooth disease, characterize a novel class of glutathione transferase-related genes. Mol. Biol. Evol. 2004, 21, 176–187. [Google Scholar] [CrossRef] [PubMed]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wolf, C.; López del Amo, V.; Arndt, S.; Bueno, D.; Tenzer, S.; Hanschmann, E.-M.; Berndt, C.; Methner, A. Redox Modifications of Proteins of the Mitochondrial Fusion and Fission Machinery. Cells 2020, 9, 815. https://doi.org/10.3390/cells9040815
Wolf C, López del Amo V, Arndt S, Bueno D, Tenzer S, Hanschmann E-M, Berndt C, Methner A. Redox Modifications of Proteins of the Mitochondrial Fusion and Fission Machinery. Cells. 2020; 9(4):815. https://doi.org/10.3390/cells9040815
Chicago/Turabian StyleWolf, Christina, Víctor López del Amo, Sabine Arndt, Diones Bueno, Stefan Tenzer, Eva-Maria Hanschmann, Carsten Berndt, and Axel Methner. 2020. "Redox Modifications of Proteins of the Mitochondrial Fusion and Fission Machinery" Cells 9, no. 4: 815. https://doi.org/10.3390/cells9040815
APA StyleWolf, C., López del Amo, V., Arndt, S., Bueno, D., Tenzer, S., Hanschmann, E.-M., Berndt, C., & Methner, A. (2020). Redox Modifications of Proteins of the Mitochondrial Fusion and Fission Machinery. Cells, 9(4), 815. https://doi.org/10.3390/cells9040815