Modification of Adenosine196 by Mettl3 Methyltransferase in the 5’-External Transcribed Spacer of 47S Pre-rRNA Affects rRNA Maturation

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. The Small Interfering RNAs (siRNA) Description and Lipid Nanoparticles (LNP) Formulation
2.2. Cell Culture and Transfection
2.3. RNA Isolation and RT-qPCR
2.4. Anti-m6A Immunoprecipitation
2.5. Formaldehyde-Crosslinked RNA-Immunoprecipitation (RIP)
2.6. Nascent RNA Purification and cDNA Preparation
2.7. Separation of Nuclear and Cytoplasmic Cell Fractions
2.8. Fluorescent in Situ Hybridization (FISH)
2.9. Estimation of rRNA Precursors’ Lifetime
2.10. Western Blotting
2.11. Ligation of DNA Probes Using RNA Template
2.12. Polysome Profiling
2.13. Bioinformatic Analysis
2.14. Statistical Analysis of the Experimental Data
3. Results
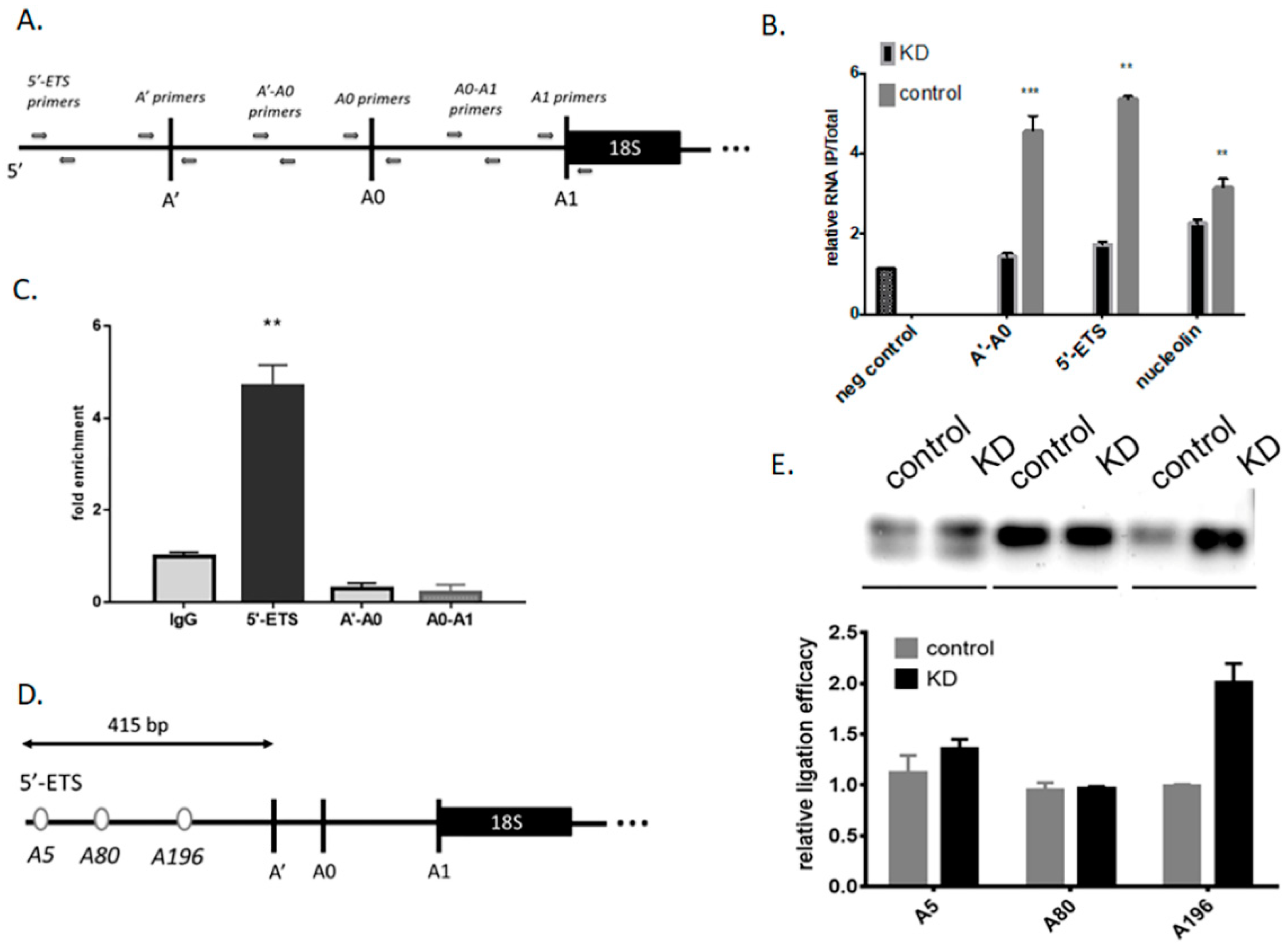
3.1. Mettl3 Methyltransferase Binds to 47S Pre-rRNA and Modifies Adenosine 196
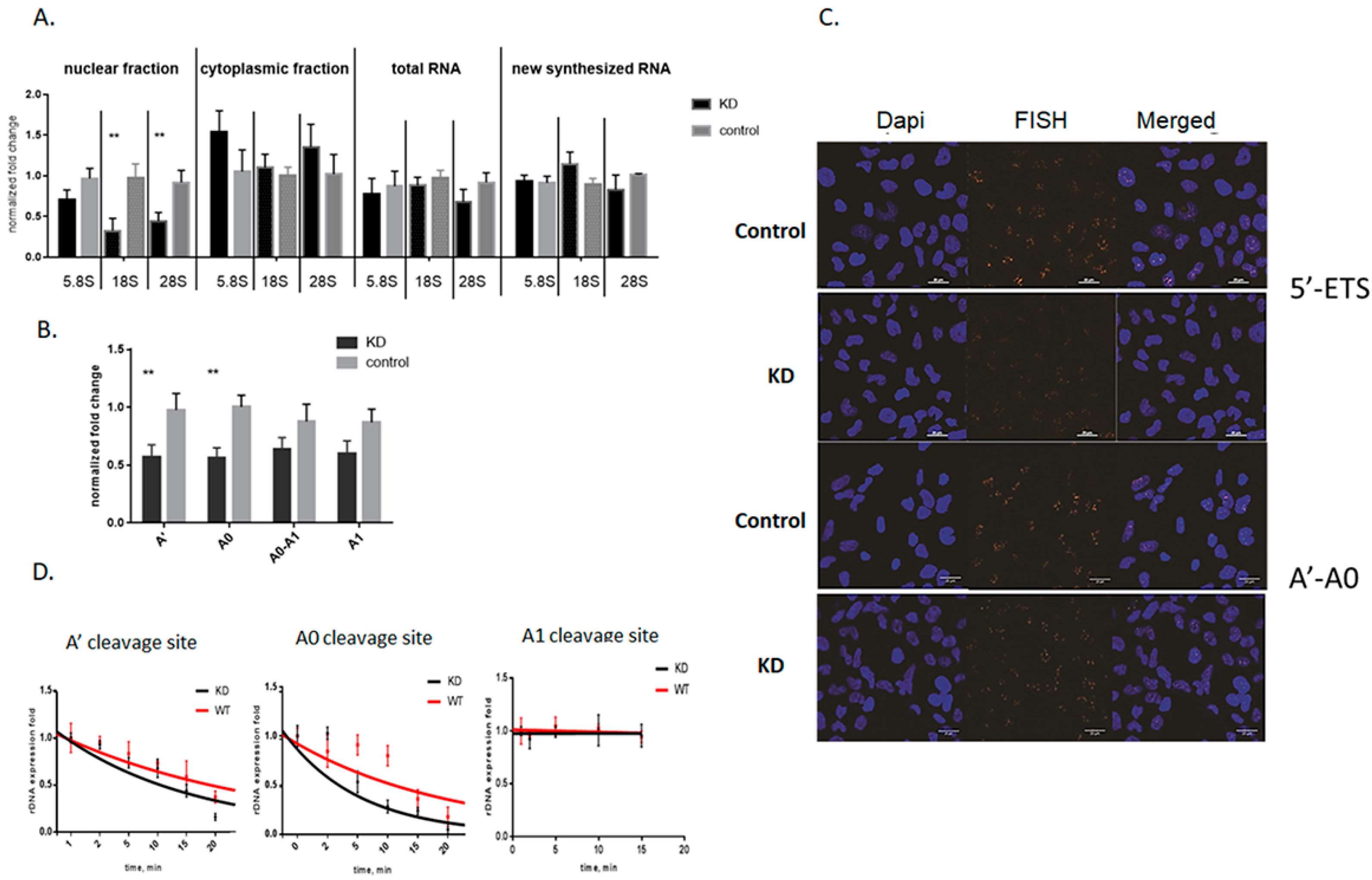
3.2. Knockdown of Mettl3 Methyltransferase Increases Rates of Pre-rRNA Maturation
3.3. Influence of Mettl3 Methyltransferase Knockdown on Pre-rRNA Processing Factors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sulima, S.O.; Hofman, I.J.; De Keersmaecker, K.; Dinman, J.D. How ribosomes translate cancer. Cancer Discov. 2017, 7, 1069–1087. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, J.; Thomas, G.; Volarević, S. Ribosome biogenesis in cancer: New players and therapeutic avenues. Nat. Rev. Cancer 2018, 18, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Boccaletto, P.; Machnicka, M.A.; Purta, E.; Piątkowski, P.; Bagiński, B.; Wirecki, T.K.; De Crécy-Lagard, V.; Ross, R.; Limbach, P.A.; Helm, M.; et al. MODOMICS: A database of RNA modification pathways. Nucleic Acids Res. 2018, 46, D303–D307. [Google Scholar] [CrossRef] [PubMed]
- Pinto, R.; Vågbø, C.B.; Jakobsson, M.E.; Kim, Y.; Baltissen, M.P.; O’Donohue, M.F.; Guzmán, U.H.; Małecki, J.M.; Wu, J.; Kirpekar, F.; et al. The human methyltransferase ZCCHC4 catalyses N6-methyladenosine modification of 28S ribosomal RNA. Nucleic Acids Res. 2020, 48, 830–846. [Google Scholar] [CrossRef]
- Bohnsack, K.E.; Höbartner, C.; Bohnsack, M.T. Eukaryotic 5-methylcytosine (m5C) RNA methyltransferases: Mechanisms, cellular functions, and links to disease. Genes 2019, 10, 102. [Google Scholar] [CrossRef]
- Wei, C.M.; Gershowitz, A.; Moss, B. Methylated nucleotides block 5′ terminus of HeLa cell messenger RNA. Cell 1975, 4, 379–386. [Google Scholar] [CrossRef]
- Tong, J.; Flavell, R.A.; Li, H.-B. RNA m6A modification and its function in diseases. Front. Med. 2018, 12, 481–489. [Google Scholar] [CrossRef]
- Shi, H.; Wei, J.; He, C. Where, when, and how: Context-dependent functions of RNA methylation writers, readers, and erasers. Mol. Cell 2019, 74, 640–650. [Google Scholar] [CrossRef]
- Aguilo, F.; Zhang, F.; Sancho, A.; Fidalgo, M.; Di Cecilia, S.; Vashisht, A.; Lee, D.-F.; Chen, C.-H.; Rengasamy, M.; Jahouh, F.; et al. Coordination of m6A mRNA methylation and gene transcription by ZFP217 regulates pluripotency and reprogramming. Cell Stem Cell 2015, 17, 689–704. [Google Scholar] [CrossRef]
- Meyer, K.D.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 2012, 149, 1635–1646. [Google Scholar] [CrossRef]
- Meyer, K.D.; Patil, D.P.; Zhou, J.; Zinoviev, A.; Skabkin, M.A.; Elemento, O.; Pestova, T.V.; Qian, S.-B.; Jaffrey, S.R.; Jaffrey, S.R.; et al. 5′ UTR m6A promotes cap-independent translation. Cell 2015, 163, 999–1010. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wan, J.; Gao, X.; Zhang, X.; Jaffrey, S.R.; Qian, S.-B. Dynamic m6A mRNA methylation directs translational control of heat shock response. Nature 2015, 526, 591–594. [Google Scholar] [CrossRef] [PubMed]
- Bushkin, G.G.; Pincus, D.; Morgan, J.T.; Richardson, K.; Lewis, C.; Chan, S.H.; Bartel, D.P.; Fink, G.R. m6A modification of a 3′ UTR site reduces RME1 mRNA levels to promote meiosis. Nat. Commun. 2019, 10, 3414. [Google Scholar] [CrossRef] [PubMed]
- Mauer, J.; Luo, X.; Blanjoie, A.; Jiao, X.; Grozhik, A.V.; Patil, D.P.; Linder, B.; Pickering, B.F.; Vasseur, J.-J.; Gross, S.S.; et al. Reversible methylation of m6Am in the 5′ cap controls mRNA stability. Nature 2017, 541, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lu, Z.; Gomez, A.; Hon, G.C.; Yue, Y.; Han, D.; Fu, Y.; Parisien, M.; Dai, Q.; Ren, B.; et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 2014, 505, 117–120. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Sun, B.F.; Xiao, W.; Yang, X.; Sun, H.Y.; Zhao, Y.L.; Yang, Y.G. Dynamic m6A modification and its emerging regulatory role in mRNA splicing. Life Med. Sci. 2015, 60, 21–32. [Google Scholar]
- Ke, S.; Alemu, E.A.; Mertens, C.; Gantman, E.C.; Fak, J.J.; Mele, A.; Haripal, B.; Zucker-Scharff, I.; Moore, M.J.; Vågbø, C.B.; et al. A majority of m6A residues are in the last exons, allowing the potential for 3′ UTR regulation. Genes Dev. 2015, 29, 2037–2053. [Google Scholar] [CrossRef]
- Roundtree, I.A.; Luo, G.Z.; Zhang, Z.; Wang, X.; Zhou, T.; Cui, Y.; Sha, J.; Huang, X.; Guerrero, L.; He, E.; et al. YTHDC1 mediates nuclear export of N6-methyladenosine methylated mRNAs. Elife 2017, 6, e31311. [Google Scholar] [CrossRef]
- Liu, N.; Dai, Q.; Zheng, G.; He, C.; Parisien, M.; Pan, T. N6-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature 2015, 518, 560–564. [Google Scholar] [CrossRef]
- Zhou, K.I.; Parisien, M.; Dai, Q.; Liu, N.; Diatchenko, L.; Sachleben, J.R.; Pan, T. N6-methyladenosine modification in a long noncoding RNA hairpin predisposes its conformation to protein binding. J. Mol. Biol. 2016, 428, 822–833. [Google Scholar] [CrossRef]
- Akichika, S.; Hirano, S.; Shichino, Y.; Suzuki, T.; Nishimasu, H.; Ishitani, R.; Sugita, A.; Hirose, Y.; Iwasaki, S.; Suzuki, T.; et al. Cap-specific terminal N6-methylation of RNA by an RNA polymerase II-associated methyltransferase. Science 2019, 363, eaav0080. [Google Scholar] [CrossRef] [PubMed]
- Weisblum, B. Erythromycin resistance by ribosome modification. Antimicrob. Agents Chemother. 1995, 39, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Connolly, K.; Culver, G. Overexpression of RbfA in the absence of the KsgA checkpoint results in impaired translation initiation. Mol. Microbiol. 2013, 87, 968–981. [Google Scholar] [CrossRef] [PubMed]
- Sergiev, P.V.; Golovina, A.Y.; Osterman, I.A.; Nesterchuk, M.V.; Sergeeva, O.V.; Chugunova, A.A.; Evfratov, S.A.; Andreianova, E.S.; Pletnev, P.I.; Petriukov, K.S.; et al. N6-methylated adenosine in RNA: From bacteria to humans. J. Mol. Biol. 2016, 428, 2134–2145. [Google Scholar] [CrossRef]
- Ma, H.; Wang, X.; Cai, J.; Dai, Q.; Natchiar, S.K.; Lv, R.; Chen, K.; Lu, Z.; Chen, H.; Shi, Y.G.; et al. N6-Methyladenosine methyltransferase ZCCHC4 mediates ribosomal RNA methylation. Nat. Chem. Biol. 2019, 15, 88–94. [Google Scholar] [CrossRef]
- Van Tran, N.; Ernst, F.G.M.; Hawley, B.R.; Zorbas, C.; Ulryck, N.; Hackert, P.; Bohnsack, K.E.; Bohnsack, M.T.; Jaffrey, S.R.; Lafontaine, D.L.J.; et al. The human 18S rRNA m6A methyltransferase METTL5 is stabilized by TRMT112. Nucleic Acids Res. 2019, 47, 7719–7733. [Google Scholar] [CrossRef]
- Reynolds, A.; Leake, D.; Boese, Q.; Scaringe, S.; Marshall, W.S.; Khvorova, A. Rational siRNA design for RNA interference. Nat. Biotechnol. 2004, 22, 326–330. [Google Scholar] [CrossRef]
- Pei, Y.; Tuschl, T. On the art of identifying effective and specific siRNAs. Nat. Methods 2006, 3, 670–676. [Google Scholar] [CrossRef]
- Anderson, E.M.; Birmingham, A.; Baskerville, S.; Reynolds, A.; Maksimova, E.; Leake, D.; Fedorov, Y.; Karpilow, J.; Khvorova, A. Experimental validation of the importance of seed complement frequency to siRNA specificity. RNA 2008, 14, 853–861. [Google Scholar] [CrossRef]
- Jackson, A.L.; Burchard, J.; Leake, D.; Reynolds, A.; Schelter, J.; Guo, J.; Johnson, J.M.; Lim, L.; Karpilow, J.; Marshall, W.; et al. Position-specific chemical modification of siRNAs reduces “off-target” transcript silencing. RNA 2006, 12, 1197–1205. [Google Scholar] [CrossRef]
- Jayaraman, M.; Ansell, S.M.; Mui, B.L.; Tam, Y.K.; Chen, J.; Du, X.; Butler, D.; Eltepu, L.; Matsuda, S.; Rajeev, K.G.; et al. Maximizing the potency of siRNA lipid nanoparticles for hepatic gene silencing in vivo. Angew. Chem. Int. Ed. Engl. 2012, 51, 8529–8533. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C.; Ou, K.; Belliveau, N.M.; Leaver, T.J.; Wild, A.W.; Huft, J.; Lin, P.J.; Chen, S.; Leung, A.K.; Hansen, C.L.; et al. Microfluidic-based manufacture of siRNA-lipid nanoparticles for therapeutic applications. Methods Mol. Biol. 2014, 1141, 109–120. [Google Scholar] [PubMed]
- Love, K.T.; Mahon, K.P.; Levins, C.G.; Whitehead, K.A.; Querbes, W.; Dorkin, J.R.; Qin, J.; Cantley, W.; Qin, L.L.; Frank-Kamenetsky, M.; et al. Lipid-like materials for low-dose, in vivo gene silencing. Proc. Natl. Acad. Sci. USA 2010, 107, 1864–1869. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wang, H.; Wei, Z.; Zhang, S.; Hua, G.; Zhang, S.W.; Zhang, L.; Gao, S.-J.; Meng, J.; Huang, Y.; et al. MeT-DB V2.0: Elucidating context-specific functions of N6-methyl-adenosine methyltranscriptome. Nucleic Acids Res. 2018, 46, D281–D287. [Google Scholar] [CrossRef] [PubMed]
- Geula, S.; Moshitch-Moshkovitz, S.; Dominissini, D.; Mansour, A.A.; Kol, N.; Salmon-Divon, M.; Hershkovitz, V.; Peer, E.; Mor, N.; Ben-Haim, M.S.; et al. m6A mRNA methylation facilitates resolution of naïve pluripotency toward differentiation. Science 2015, 347, 1002–1006. [Google Scholar] [CrossRef]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Sorek, R.; et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012, 485, 201–206. [Google Scholar] [CrossRef]
- Liu, W.; Yan, J.; Zhang, Z.; Pian, H.; Liu, C.; Li, Z. Identification of a selective DNA ligase for accurate recognition and ultrasensitive quantification of N6-methyladenosine in RNA at one-nucleotide resolution. Chem. Sci. 2018, 9, 3354–3359. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, B.S.; Roundtree, I.A.; Lu, Z.; Han, D.; Ma, H.; Weng, X.; Chen, K.; Shi, H.; He, C.; et al. N6-methyladenosine modulates messenger RNA translation efficiency. Cell 2015, 161, 1388–1399. [Google Scholar] [CrossRef]
- Sloan, K.E.; Warda, A.S.; Sharma, S.; Entian, K.-D.; Lafontaine, D.L.J.; Bohnsack, M.T. Tuning the ribosome: The influence of rRNA modification on eukaryotic ribosome biogenesis and function. RNA Biol. 2017, 14, 1138–1152. [Google Scholar] [CrossRef]
- Puvion-Dutilleul, F.; Mazan, S.; Nicoloso, M.; Pichard, E.; Bachellerie, J.P.; Puvion, E. Alterations of nucleolar ultrastructure and ribosome biogenesis by actinomycin D. Implications for U3 snRNP function. Eur. J. Cell Biol. 1992, 58, 149–162. [Google Scholar]
- Popov, A.; Smirnov, E.; Kováčik, L.; Raška, O.; Hagen, G.; Stixová, L.; Raška, I. Duration of the first steps of the human rRNA processing. Nucleus 2013, 4, 134–141. [Google Scholar] [CrossRef]
- Lin, S.; Choe, J.; Du, P.; Triboulet, R.; Gregory, R.I. The m6A methyltransferase METTL3 promotes translation in human cancer cells. Mol. Cell 2016, 62, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Ignatova, V.V.; Jansen, P.W.T.C.; Baltissen, M.P.; Vermeulen, M.; Schneider, R. The interactome of a family of potential methyltransferases in HeLa cells. Sci. Rep. 2019, 9, 6584. [Google Scholar] [CrossRef] [PubMed]
- Henras, A.K.; Plisson-Chastang, C.; O’Donohue, M.-F.; Chakraborty, A.; Gleizes, P.-E. An overview of pre-ribosomal RNA processing in eukaryotes. Wiley Interdiscip. Rev. RNA 2015, 6, 225–242. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Nazar, R.N. Modification of rRNA as a “quality control mechanism” in ribosome biogenesis. FEBS Lett. 2002, 523, 182–186. [Google Scholar] [CrossRef]
- Li, F.; Kennedy, S.; Hajian, T.; Gibson, E.; Seitova, A.; Xu, C.; Arrowsmith, C.; Vedadi, M. A Radioactivity-Based Assay for Screening Human m6 A-RNA Methyltransferase, METTL3-METTL14 Complex, and Demethylase ALKBH5. J. Biomol. Screen. 2016, 213, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Choe, J.; Lin, S.; Zhang, W.; Liu, Q.; Wang, L.; Ramirez-Moya, J.; Du, P.; Kim, W.; Tang, S.; Santisteban, P.; et al. mRNA circularization by METTL3-eIF3h enhances translation and promotes oncogenesis. Nature 2018, 561, 556–560. [Google Scholar] [CrossRef]
- Barbieri, I.; Tzelepis, K.; Pandolfini, L.; Shi, J.; Millán-Zambrano, G.; Robson, S.C.; Aspris, D.; Migliori, V.; Bannister, A.J.; De Braekeleer, E.; et al. Promoter-bound METTL3 maintains myeloid leukaemia by m6A-dependent translation control. Nature 2017, 552, 126–131. [Google Scholar] [CrossRef]
- Li, X.; Tang, J.; Huang, W.; Wang, F.; Li, P.; Qin, C.; Qin, Z.; Zou, Q.; Wei, J.; Yang, H.; et al. The M6A methyltransferase METTL3: Acting as a tumor suppressor in renal cell carcinoma. Oncotarget 2017, 8, 96103–96116. [Google Scholar] [CrossRef]
- Bohnsack, K.E.; Bohnsack, M.T. Uncovering the assembly pathway of human ribosomes and its emerging links to disease. EMBO J. 2019, 38, e100278. [Google Scholar] [CrossRef]
- Liang, Y.; Zhan, G.; Chang, K.J.; Yang, Y.P.; Wang, L.; Lin, J.; Hsu, C.H. The roles of m6A RNA modifiers in human cancer. J. Chin. Med. Assoc. 2020, 83, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Schöller, E.; Weichmann, F.; Treiber, T.; Ringle, S.; Treiber, N.; Flatley, A.; Feederle, R.; Bruckmann, A.; Meister, G. Interactions, localization, and phosphorylation of the m6A generating METTL3-METTL14-WTAP complex. RNA 2018, 24, 499–512. [Google Scholar] [CrossRef] [PubMed]
- Tomecki, R.; Sikorski, P.J.; Zakrzewska-Placzek, M. Comparison of preribosomal RNA processing pathways in yeast, plant and human cells—Focus on coordinated action of endo-and exoribonucleases. FEBS Lett. 2017, 591, 1801–1850. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sergeeva, O.; Sergeev, P.; Melnikov, P.; Prikazchikova, T.; Dontsova, O.; Zatsepin, T. Modification of Adenosine196 by Mettl3 Methyltransferase in the 5’-External Transcribed Spacer of 47S Pre-rRNA Affects rRNA Maturation. Cells 2020, 9, 1061. https://doi.org/10.3390/cells9041061
Sergeeva O, Sergeev P, Melnikov P, Prikazchikova T, Dontsova O, Zatsepin T. Modification of Adenosine196 by Mettl3 Methyltransferase in the 5’-External Transcribed Spacer of 47S Pre-rRNA Affects rRNA Maturation. Cells. 2020; 9(4):1061. https://doi.org/10.3390/cells9041061
Chicago/Turabian StyleSergeeva, Olga, Philipp Sergeev, Pavel Melnikov, Tatiana Prikazchikova, Olga Dontsova, and Timofei Zatsepin. 2020. "Modification of Adenosine196 by Mettl3 Methyltransferase in the 5’-External Transcribed Spacer of 47S Pre-rRNA Affects rRNA Maturation" Cells 9, no. 4: 1061. https://doi.org/10.3390/cells9041061
APA StyleSergeeva, O., Sergeev, P., Melnikov, P., Prikazchikova, T., Dontsova, O., & Zatsepin, T. (2020). Modification of Adenosine196 by Mettl3 Methyltransferase in the 5’-External Transcribed Spacer of 47S Pre-rRNA Affects rRNA Maturation. Cells, 9(4), 1061. https://doi.org/10.3390/cells9041061