Progenitor Cells Activated by Platelet Lysate in Human Articular Cartilage as a Tool for Future Cartilage Engineering and Reparative Strategies

 ,
,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Platelet Lysate (PL) Preparation
2.2. Cell Primary Cultures
2.2.1. Chondro-Progenitor Cells (CPCs)
2.2.2. Primary Articular Chondrocytes (ACs)
2.3. Growth Kinetics
2.4. Western Blot Analysis
2.5. Evaluation of Cell Senescence
2.6. Assay for In Vitro and In Vivo Neoplastic Transformation of CPCs
2.7. RNA Extraction and Real-Time Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
2.8. Immunofluorescence Staining and Immunophenotypic Characterization by Flow Cytometry
2.9. Colony-Forming Unit Fibroblast (CFU-F) Assay
2.10. In Vitro Multilineage Differentiation Potential
2.11. In Vivo Cartilage and Bone Formation
2.12. Histology and Immunohistochemistry
2.13. Production of Cell-Conditioned Media
2.14. Cytokine Identification in Cell Secretome
2.15. In Vitro Chemotaxis of CPCs
2.16. In Vitro Scratch Assay
2.17. Statistical Analysis
3. Results
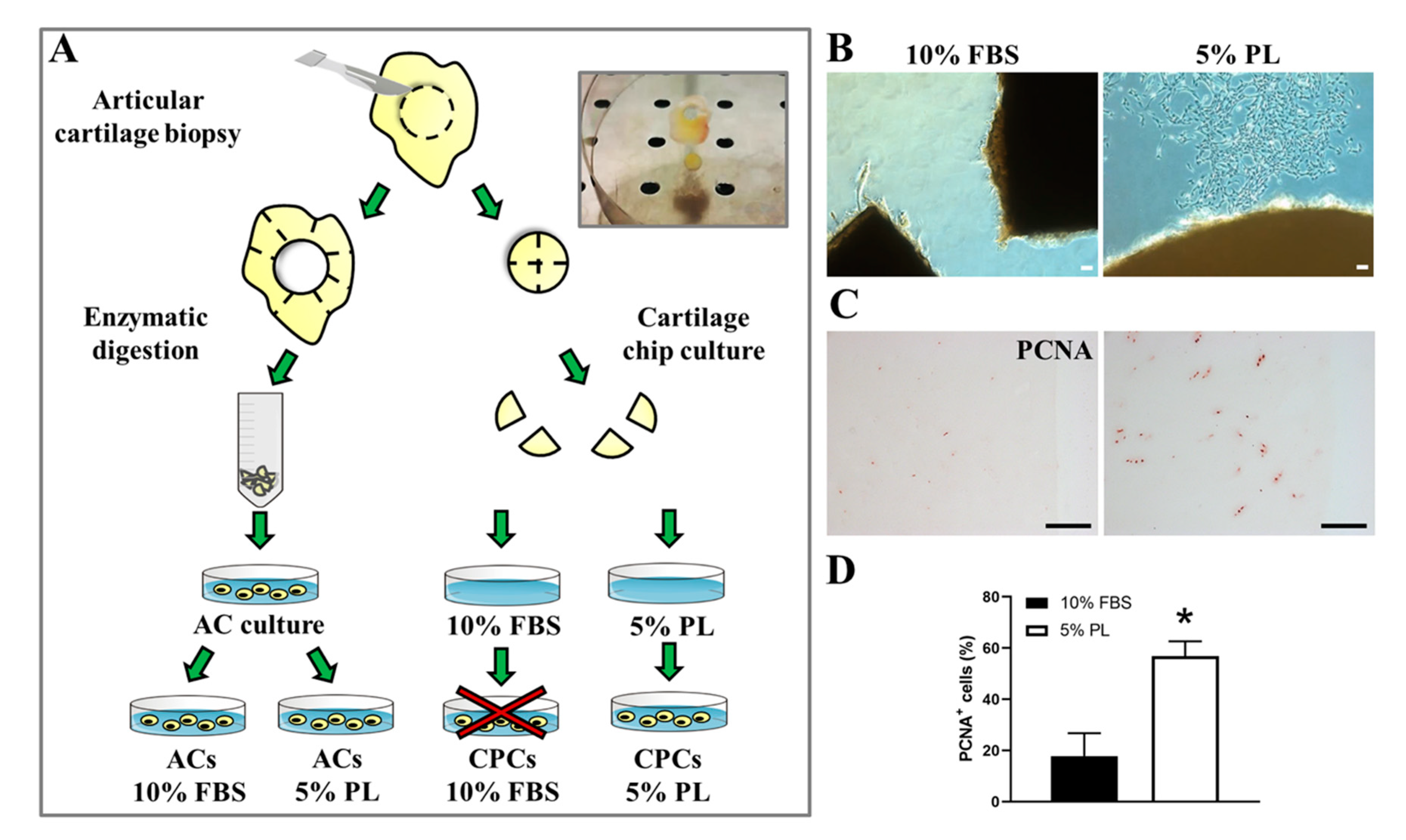
3.1. PL Induced Release of Cells with Fibroblastic-Like Phenotype from Ex Vivo Cultured Cartilage Chips and Promoted Their Proliferation
3.2. PL Increased the Proliferation of ACs and Reduced Their Senescence
3.3. Effect of PL on Gene Expression and Phenotype in Cartilage-Derived Cells
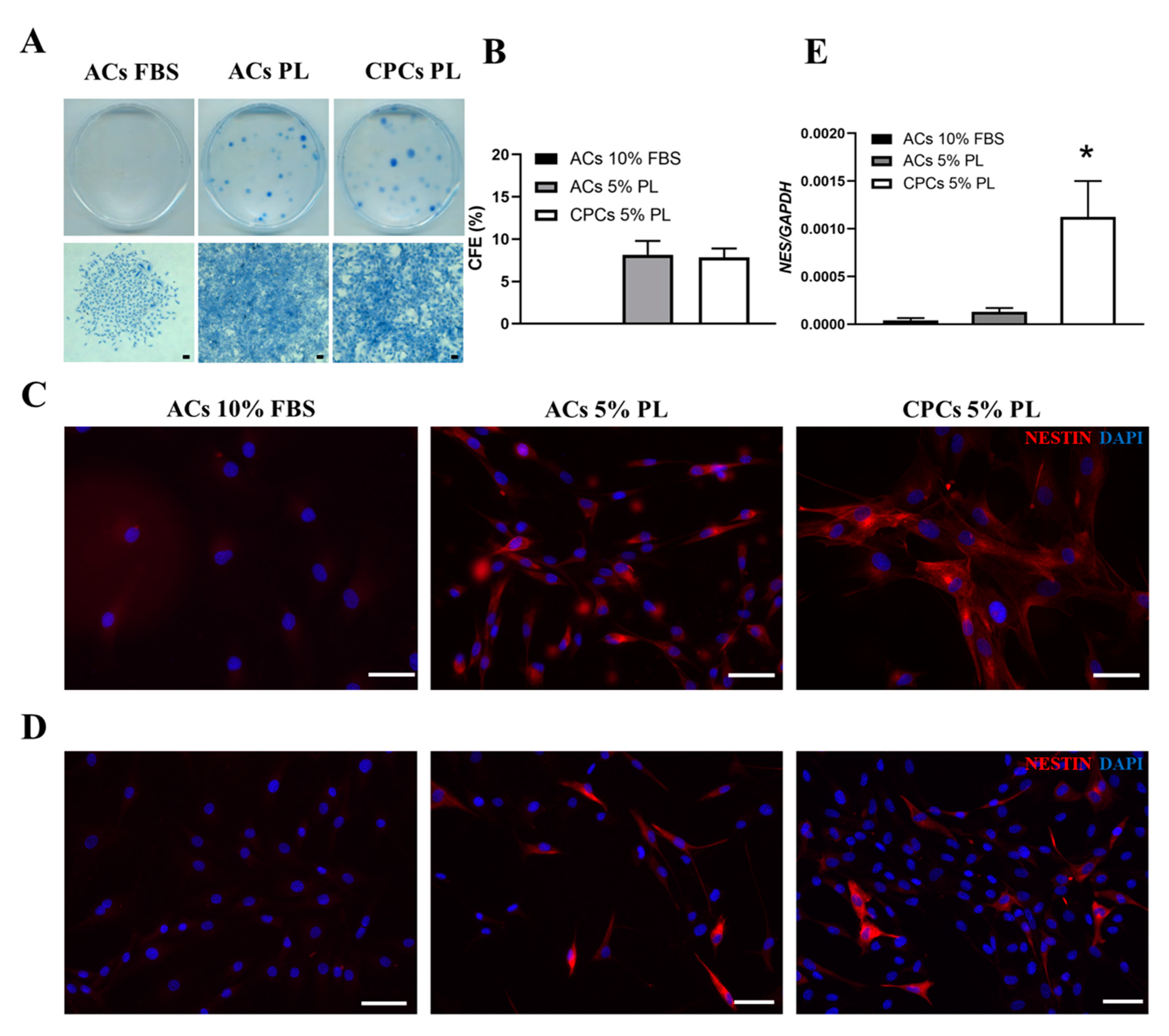
3.4. PL Modulated the Clonogenic Potential and Expression of Nestin Stem Marker in Cartilage-Derived Cells
3.5. Comparison of In Vitro Multilineage Differentiation Potential Between CPCs and ACs
3.6. CPCs-PL Produced Hyaline-Like Cartilage In Vivo Suitable for Tissue Engineering Strategies
3.7. Secretory Profile of CPCs and ACs Revealed an Intricate Scenario Including Tissue Turnover, Hypertrophy Counteraction, PL-Induced Pro-Inflammatory Effects and Chemoattractive Capability
3.8. PL-Recruited CPCs Showed Enhanced Motility Under Inflammatory Conditions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Jiang, Y.; Tuan, R.S. Origin and function of cartilage stem/progenitor cells in osteoarthritis. Nat. Rev. Rheumatol. 2015, 11, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Woolf, A.D. Global burden of osteoarthritis and musculoskeletal diseases. BMC Musculoskelet. Disord. 2015, 16, S3. [Google Scholar] [CrossRef][Green Version]
- Loeser, R.F.; Goldring, S.R.; Scanzello, C.R.; Goldring, M.B. Osteoarthritis: A disease of the joint as an organ. Arthritis Rheum. 2012, 64, 1697–1707. [Google Scholar] [CrossRef] [PubMed]
- Hunziker, E.B.; Lippuner, K.; Keel, M.J.B.; Shintani, N. An educational review of cartilage repair: Precepts & practice--myths & misconceptions--progress & prospects. Osteoarthr. Cartil. 2015, 23, 334–350. [Google Scholar]
- Huey, D.J.; Hu, J.C.; Athanasiou, K.A. Unlike bone, cartilage regeneration remains elusive. Science 2012, 338, 917–921. [Google Scholar] [CrossRef]
- Johnstone, B.; Hering, T.M.; Caplan, A.I.; Goldberg, V.M.; Yoo, J.U. In vitro chondrogenesis of bone marrow-derived mesenchymal progenitor cells. Exp. Cell Res. 1998, 238, 265–272. [Google Scholar] [CrossRef]
- Wakitani, S.; Imoto, K.; Yamamoto, T.; Saito, M.; Murata, N.; Yoneda, M. Human autologous culture expanded bone marrow mesenchymal cell transplantation for repair of cartilage defects in osteoarthritic knees. Osteoarthr. Cartil. 2002, 10, 199–206. [Google Scholar] [CrossRef]
- De Bari, C.; Dell’Accio, F.; Tylzanowski, P.; Luyten, F.P. Multipotent mesenchymal stem cells from adult human synovial membrane. Arthritis Rheum. 2001, 44, 1928–1942. [Google Scholar] [CrossRef]
- Dowthwaite, G.P.; Bishop, J.C.; Redman, S.N.; Khan, I.M.; Rooney, P.; Evans, D.J.R.; Haughton, L.; Bayram, Z.; Boyer, S.; Thomson, B.; et al. The surface of articular cartilage contains a progenitor cell populations. J. Cell Sci. 2004, 117, 889–897. [Google Scholar] [CrossRef]
- Pretzel, D.; Linss, S.; Rochler, S.; Endres, M.; Kaps, C.; Alsalameh, S.; Kinne, R.W. Relative percentage and zonal distribution of mesenchymal progenitor cells in human osteoarthritic and normal cartilage. Arthritis Res. Ther. 2011, 13, R64. [Google Scholar] [CrossRef]
- Jones, E.A.; Crawford, A.; English, A.; Henshaw, K.; Mundy, J.; Corscadden, D.; Chapman, T.; Emery, P.; Hatton, P.; McGonagle, D. Synovial fluid mesenchymal stem cells in health and early osteoarthritis: Detection and functional evaluation at the single-cell level. Arthritis Rheum. 2008, 58, 1731–1740. [Google Scholar] [CrossRef] [PubMed]
- Alsalameh, S.; Amin, R.; Gemba, T.; Lotz, M. Identification of Mesenchymal Progenitor Cells in Normal and Osteoarthritic Human Articular Cartilage. Arthritis Rheum. 2004, 50, 1522–1532. [Google Scholar] [CrossRef] [PubMed]
- Kon, E.; Filardo, G.; Di Martino, A.; Marcacci, M. Platelet-rich plasma (PRP) to treat sports injuries: Evidence to support its use. Knee Surg. Sports Traumatol. Arthrosc. 2011, 19, 516–527. [Google Scholar] [CrossRef] [PubMed]
- Pereira, R.C.; Scaranari, M.; Benelli, R.; Strada, P.; Reis, R.L.; Cancedda, R.; Gentili, C. Dual effect of platelet lysate on human articular cartilage: A maintenance of chondrogenic potential and a transient proinflammatory activity followed by an inflammation resolution. Tissue Eng. Part A 2013, 19, 1476–1488. [Google Scholar] [CrossRef] [PubMed]
- Romaldini, A.; Mastrogiacomo, M.; Cancedda, R.; Descalzi, F. Platelet Lysate Activates Human Subcutaneous Adipose Tissue Cells by Promoting Cell Proliferation and Their Paracrine Activity Toward Epidermal Keratinocytes. Front. Bioeng. Biotechnol. 2018, 6, 203. [Google Scholar] [CrossRef] [PubMed]
- Ruggiu, A.; Ulivi, V.; Sanguineti, F.; Cancedda, R.; Descalzi, F. The effect of platelet lysate on osteoblast proliferation associated with a transient increase of the inflammatory response in bone regeneration. Biomaterials 2013, 34, 9318–9330. [Google Scholar] [CrossRef]
- Krüger, J.P.; Hondke, S.; Endres, M.; Pruss, A.; Siclari, A.; Kaps, C. Human platelet-rich plasma stimulates migration and chondrogenic differentiation of human subchondral progenitor cells. J. Orthop. Res. 2012, 30, 845–852. [Google Scholar] [CrossRef]
- Cancedda, R.; Bollini, S.; Descalzi, F.; Mastrogiacomo, M.; Tasso, R. Learning from Mother Nature: Innovative Tools to Boost Endogenous Repair of Critical or Difficult-to-Heal Large Tissue Defects. Front. Bioeng. Biotechnol. 2017, 5, 1–13. [Google Scholar] [CrossRef]
- Cole, B.J.; Karas, V.; Hussey, K.; Pilz, K.; Fortier, L.A. Hyaluronic Acid Versus Platelet-Rich Plasma: A Prospective, Double-Blind Randomized Controlled Trial Comparing Clinical Outcomes and Effects on Intra-articular Biology for the Treatment of Knee Osteoarthritis. Am. J. Sports Med. 2017, 45, 339–346. [Google Scholar] [CrossRef]
- Siclari, A.; Mascaro, G.; Gentili, C.; Kaps, C.; Cancedda, R.; Boux, E. Cartilage repair in the knee with subchondral drilling augmented with a platelet-rich plasma-immersed polymer-based implant. Knee Surg. Sports Traumatol. Arthrosc. 2014, 22, 1225–1234. [Google Scholar] [CrossRef]
- Siclari, A.; Mascaro, G.; Gentili, C.; Cancedda, R.; Boux, E. A cell-free scaffold-based cartilage repair provides improved function hyaline-like repair at one year. Clin. Orthop. Relat. Res. 2012, 470, 910–919. [Google Scholar] [CrossRef]
- Akeda, K.; An, H.S.; Okuma, M.; Attawia, M.; Miyamoto, K.; Thonar, E.J.-M.A.; Lenz, M.E.; Sah, R.L.; Masuda, K. Platelet-rich plasma stimulates porcine articular chondrocyte proliferation and matrix biosynthesis. Osteoarthr. Cartil. 2006, 14, 1272–1280. [Google Scholar] [CrossRef] [PubMed]
- Van Buul, G.M.; Koevoet, W.L.M.; Kops, N.; Bos, P.K.; Verhaar, J.A.N.; Weinans, H.; Bernsen, M.R.; Van Osch, G.J.V.M. Platelet-rich plasma releasate inhibits inflammatory processes in osteoarthritic chondrocytes. Am. J. Sports Med. 2011, 39, 2362–2370. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Morgan, B.J.; Smith, R.; Fellows, C.R.; Thornton, C.; Snow, M.; Francis, L.W.; Khan, I.M. Platelet-rich plasma induces post-natal maturation of immature articular cartilage and correlates with LOXL1 activation. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Sellberg, F.; Berglund, E.; Ronaghi, M.; Strandberg, G.; Löf, H.; Sommar, P.; Lubenow, N.; Knutson, F.; Berglund, D. Composition of growth factors and cytokines in lysates obtained from fresh versus stored pathogen-inactivated platelet units. Transfus. Apher. Sci. 2016, 55, 333–337. [Google Scholar] [CrossRef]
- Wiese, C.; Rolletschek, A.; Kania, G.; Blyszczuk, P.; Tarasov, K.V.; Tarasova, Y.; Wersto, R.P.; Boheler, K.R.; Wobus, A.M. Nestin expression—A property of multi-lineage progenitor cells? Cell. Mol. Life Sci. 2004, 61, 2510–2522. [Google Scholar] [CrossRef]
- El Backly, R.; Ulivi, V.; Tonachini, L.; Cancedda, R.; Descalzi, F.; Mastrogiacomo, M. Platelet lysate induces in vitro wound healing of human keratinocytes associated with a strong proinflammatory response. Tissue Eng. Part A 2011, 17, 1787–1800. [Google Scholar] [CrossRef]
- Muraglia, A.; Ottonello, C.; Spanò, R.; Dozin, B.; Strada, P.; Grandizio, M.; Cancedda, R.; Mastrogiacomo, M. Biological activity of a standardized freeze-dried platelet derivative to be used as cell culture medium supplement. Platelets 2014, 25, 211–220. [Google Scholar] [CrossRef]
- Muraglia, A.; Nguyen, V.T.; Nardini, M.; Mogni, M.; Coviello, D.; Dozin, B.; Strada, P.; Baldelli, I.; Formica, M.; Cancedda, R.; et al. Culture medium supplements derived from human platelet and plasma: Cell commitment and proliferation support. Front. Bioeng. Biotechnol. 2017, 5, 66. [Google Scholar] [CrossRef]
- Van Nguyen, T.; Cancedda, R.; Descalzi, F. Platelet lysate activates quiescent cell proliferation and reprogramming in human articular cartilage: Involvement of hypoxia inducible factor 1. J. Tissue Eng. Regen. Med. 2018, 12, e1691–e1703. [Google Scholar] [CrossRef]
- Debacq-Chainiaux, F.; Erusalimsky, J.D.; Campisi, J.; Toussaint, O. Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat. Protoc. 2009, 4, 1798–1806. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.I.; Freedman, V.H.; Risser, R.; Pollack, R. Tumorigenicity of virus transformed cells in nude mice is correlated specifically with anchorage independent growth in vitro. Proc. Natl. Acad. Sci. USA 1975, 72, 4435–4439. [Google Scholar] [CrossRef] [PubMed]
- He, T.; Huang, Y.; Chak, J.C.; Klar, R.M. Recommendations for improving accuracy of gene expression data in bone and cartilage tissue engineering. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Fellows, C.R.; Williams, R.; Davies, I.R.; Gohil, K.; Baird, D.M.; Fairclough, J.; Rooney, P.; Archer, C.W.; Khan, I.M. Characterisation of a divergent progenitor cell sub-populations in human osteoarthritic cartilage: The role of telomere erosion and replicative senescence. Sci. Rep. 2017, 7, 41421. [Google Scholar] [CrossRef]
- Mankin, H.J.; Dorfman, H.; Lippiello, L.; Zarin, A. Biochemical and metabolic abnormalities in articular cartilage from arthritic human hips. II. Correlation of morphology with biochemical and metabolic data. J. Bone Joint Surg. Am. 1971, 53, 523–537. [Google Scholar] [CrossRef]
- Williams, R.; Khan, I.M.; Richardson, K.; Nelson, L.; McCarthy, H.E.; Analbelsi, T.; Singhrao, S.K.; Dowthwaite, G.P.; Jones, R.E.; Baird, D.M.; et al. Identification and clonal characterisation of a progenitor cell sub-population in normal human articular cartilage. PLoS ONE 2010, 5, e13246. [Google Scholar] [CrossRef]
- Sherr, C.J. Growth factor-regulated G1 cyclins. Stem Cells 1994, 12, 47–55. [Google Scholar]
- Moussa, M.; Lajeunesse, D.; Hilal, G.; El Atat, O.; Haykal, G.; Serhal, R.; Chalhoub, A.; Khalil, C.; Alaaeddine, N. Platelet rich plasma (PRP) induces chondroprotection via increasing autophagy, anti-inflammatory markers, and decreasing apoptosis in human osteoarthritic cartilage. Exp. Cell Res. 2017, 352, 146–156. [Google Scholar] [CrossRef]
- Porter, A.G.; Jänicke, R.U. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999, 6, 99–104. [Google Scholar] [CrossRef]
- Hamada, T.; Sakai, T.; Hiraiwa, H.; Nakashima, M.; Ono, Y.; Mitsuyama, H.; Ishiguro, N. Surface markers and gene expression to characterize the differentiation of monolayer expanded human articular chondrocytes. Nagoya J. Med. Sci. 2013, 75, 101–111. [Google Scholar]
- Diaz-Romero, J.; Gaillard, J.P.; Grogan, S.P.; Nesic, D.; Trub, T.; Mainil-Varlet, P. Immunophenotypic analysis of human articular chondrocytes: Changes in surface markers associated with cell expansion in monolayer culture. J. Cell. Physiol. 2005, 202, 731–742. [Google Scholar] [CrossRef] [PubMed]
- Kienzle, G.; Von Kempis, J. Vascular cell adhesion molecule 1 (CD106) on primary human articular chondrocytes: Functional regulation of expression by cytokines and comparison with intercellular adhesion molecule 1 (CD54) and very late activation antigen 2. Arthritis Rheum. 1998, 41, 1296–1305. [Google Scholar] [CrossRef]
- Bernal, A.; Arranz, L. Nestin-expressing progenitor cells: Function, identity and therapeutic implications. Cell. Mol. Life Sci. 2018, 75, 2177–2195. [Google Scholar] [CrossRef] [PubMed]
- Mignone, J.L.; Kukekov, V.; Chiang, A.-S.; Steindler, D.; Enikolopov, G. Neural stem and progenitor cells in nestin-GFP transgenic mice. J. Comp. Neurol. 2004, 469, 311–324. [Google Scholar] [CrossRef] [PubMed]
- Pereira, R.C.; Benelli, R.; Canciani, B.; Scaranari, M.; Daculsi, G.; Cancedda, R.; Gentili, C. Beta-tricalcium phosphate ceramic triggers fast and robust bone formation by human mesenchymal stem cells. J. Tissue Eng. Regen. Med. 2019, 13, 1007–1018. [Google Scholar] [CrossRef]
- Mueller, M.B.; Tuan, R.S. Functional characterization of hypertrophy in chondrogenesis of human mesenchymal stem cells. Arthritis Rheum. 2008, 58, 1377–1388. [Google Scholar] [CrossRef]
- Sanchez, C.; Bay-Jensen, A.C.; Pap, T.; Dvir-Ginzberg, M.; Quasnichka, H.; Barrett-Jolley, R.; Mobasheri, A.; Henrotin, Y. Chondrocyte secretome: A source of novel insights and exploratory biomarkers of osteoarthritis. Osteoarthr. Cartil. 2017, 25, 1199–1209. [Google Scholar] [CrossRef]
- Schwab, W.; Gavlik, J.M.; Beichler, T.; Funk, R.H.W.; Albrecht, S.; Magdolen, V.; Luther, T.; Kasper, M.; Shakibaei, M. Expression of the urokinase-type plasminogen activator receptor in human articular chondrocytes: Association with caveolin and β 1-integrin. Histochem. Cell. Biol. 2001, 115, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.; Chun, C.-H.; Chun, J.-S. Dkk-1 expression in chondrocytes inhibits experimental osteoarthritic cartilage destruction in mice. Arthritis Rheum. 2012, 64, 2568–2578. [Google Scholar] [CrossRef]
- Bonyadi Rad, E.; Musumeci, G.; Pichler, K.; Heidary, M.; Szychlinska, M.A.; Castrogiovanni, P.; Marth, E.; Böhm, C.; Srinivasaiah, S.; Krönke, G.; et al. Runx2 mediated Induction of Novel Targets ST2 and Runx3 Leads to Cooperative Regulation of Hypertrophic Differentiation in ATDC5 Chondrocytes. Sci. Rep. 2017, 7, 17947. [Google Scholar] [CrossRef]
- Hatfield, J.T.; Anderson, P.J.; Powell, B.C. Retinol-binding protein 4 is expressed in chondrocytes of developing mouse long bones: Implications for a local role in formation of the secondary ossification center. Histochem. Cell Biol. 2013, 139, 727–734. [Google Scholar] [CrossRef] [PubMed]
- Scotece, M.; Koskinen-Kolasa, A.; Moilanen, T.; Moilanen, E.; Vuolteenaho, K. Novel adipokine associated with osteoarthritis: Retinol binding protein 4 is produced by cartilage and correlates with matrix metalloproteinasesin osteoarthritis patients. Osteoarthr. Cartil. 2018, 26, S126–S127. [Google Scholar] [CrossRef]
- Sandell, L.J.; Xing, X.; Franz, C.; Davies, S.; Chang, L.W.; Patra, D. Exuberant expression of chemokine genes by adult human articular chondrocytes in response to IL-1β. Osteoarthr. Cartil. 2008, 16, 1560–1571. [Google Scholar] [CrossRef] [PubMed]
- Pattappa, G.; Peroglio, M.; Sakai, D.; Mochida, J.; Benneker, L.M.; Alini, M.; Grad, S. CCL5/RANTES is a key chemoattractant released by degenerative intervertebral discs in organ culture. Eur. Cell. Mater. 2014, 27, 124–136. [Google Scholar] [CrossRef]
- Mazzetti, I.; Magagnoli, G.; Paoletti, S.; Uguccioni, M.; Olivotto, E.; Vitellozzi, R.; Cattini, L.; Facchini, A.; Borzì, R.M. A Role for Chemokines in the Induction of Chondrocyte Phenotype Modulation. Arthritis Rheum. 2004, 50, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Koelling, S.; Kruegel, J.; Irmer, M.; Path, J.R.; Sadowski, B.; Miro, X.; Miosge, N. Migratory Chondrogenic Progenitor Cells from Repair Tissue during the Later Stages of Human Osteoarthritis. Cell Stem Cell 2009, 4, 324–335. [Google Scholar] [CrossRef]
- Seol, D.; McCabe, D.J.; Choe, H.; Zheng, H.; Yu, Y.; Jang, K.; Walter, M.W.; Lehman, A.D.; Ding, L.; Buckwalter, J.A.; et al. Chondrogenic progenitor cells respond to cartilage injury. Arthritis Rheum. 2012, 64, 3626–3637. [Google Scholar] [CrossRef]
- Joos, H.; Wildner, A.; Hogrefe, C.; Reichel, H.; Brenner, R.E. Interleukin-1 beta and tumor necrosis factor alpha inhibit migration activity of chondrogenic progenitor cells from non-fibrillated osteoarthritic cartilage. Arthritis Res. Ther. 2013, 15, R119. [Google Scholar] [CrossRef]
- Fiedler, J.; Röderer, G.; Günther, K.P.; Brenner, R.E. BMP-2, BMP-4, and PDGF-bb stimulate chemotactic migration of primary human mesenchymal progenitor cells. J. Cell. Biochem. 2002, 87, 305–312. [Google Scholar] [CrossRef]
- Fiedler, J.; Brill, C.; Blum, W.F.; Brenner, R.E. IGF-I and IGF-II stimulate directed cell migration of bone-marrow-derived human mesenchymal progenitor cells. Biochem. Biophys. Res. Commun. 2006, 345, 1177–1183. [Google Scholar] [CrossRef]
- Chatterjee, M.; Gawaz, M. Platelet-derived CXCL12 (SDF-1α): Basic mechanisms and clinical implications. J. Thromb. Haemost. 2013, 11, 1954–1967. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.H.; Ozanne, S.E.; Hales, C.N. Analysis of expression of growth factor receptors in replicatively and oxidatively senescent human fibroblasts. FEBS Lett. 2005, 579, 6388–6394. [Google Scholar] [CrossRef] [PubMed]
- Itahana, K.; Dimri, G.P.; Hara, E.; Itahana, Y.; Zou, Y.; Desprez, P.Y.; Campisi, J. A role for p53 in maintaining and establishing the quiescence growth arrest in human cells. J. Biol. Chem. 2002, 277, 18206–18214. [Google Scholar] [CrossRef]
- Lee, S.; Schmitt, C.A. The dynamic nature of senescence in cancer. Nat. Cell Biol. 2019, 21, 94–101. [Google Scholar] [CrossRef]
- Liu, H.Y.; Huang, C.F.; Lin, T.C.; Tsai, C.Y.; Tina Chen, S.Y.; Liu, A.; Chen, W.H.; Wei, H.J.; Wang, M.F.; Williams, D.F.; et al. Delayed animal aging through the recovery of stem cell senescence by platelet rich plasma. Biomaterials 2014, 35, 9767–9776. [Google Scholar] [CrossRef]
- Jeon, O.H.; Kim, C.; Laberge, R.-M.; Demaria, M.; Rathod, S.; Vasserot, A.P.; Chung, J.W.; Kim, D.H.; Poon, Y.; David, N.; et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat. Med. 2017, 23, 775–781. [Google Scholar] [CrossRef]
- Fickert, S.; Fiedler, J.; Brenner, R.E. Identification of subpopulations with characteristics of mesenchymal progenitor cells from human osteoarthritic cartilage using triple staining for cell surface markers. Arthritis. Res. Ther. 2004, 6, R422. [Google Scholar] [CrossRef]
- Jayasuriya, C.T.; Hu, N.; Li, J.; Lemme, N.; Terek, R.; Ehrlich, M.G.; Chen, Q. Molecular characterization of mesenchymal stem cells in human osteoarthritis cartilage reveals contribution to the OA phenotype. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef]
- Jiang, Y.; Cai, Y.; Zhang, W.; Yin, Z.; Hu, C.; Tong, T.; Lu, P.; Zhang, S.; Neculai, D.; Tuan, R.S.; et al. Human Cartilage-Derived Progenitor Cells From Committed Chondrocytes for Efficient Cartilage Repair and Regeneration. Stem Cells Transl. Med. 2016, 5, 733–744. [Google Scholar] [CrossRef]
- Méndez-Ferrer, S.; Michurina, T.V.; Ferraro, F.; Mazloom, A.R.; MacArthur, B.D.; Lira, S.A.; Scadden, D.T.; Ma’ayan, A.; Enikolopov, G.N.; Frenette, P.S. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 2010, 466, 829–834. [Google Scholar] [CrossRef]
- Fellows, C.R.; Khan, I.M.; Archer, C.W. Articular cartilage contains a nestin positive stem cell population. Eur. Cell. Mater. 2014, 28, 55. [Google Scholar]
- Dell’Accio, F.; De Bari, C.; Luyten, F.P. Molecular markers predictive of the capacity of expanded human articular chondrocytes to form stable cartilage in vivo. Arthritis Rheum. 2001, 44, 1608–1619. [Google Scholar] [CrossRef]
- Healy, C.; Uwanogho, D.; Sharpe, P.T. Expression of the chicken Sox9 gene marks the onset of cartilage differentiation. Ann. N. Y. Acad. Sci. 1996, 785, 261–262. [Google Scholar] [CrossRef]
- Khan, I.M.; Bishop, J.C.; Gilbert, S.; Archer, C.W. Clonal chondroprogenitors maintain telomerase activity and Sox9 expression during extended monolayer culture and retain chondrogenic potential. Osteoarthr. Cartil. 2009, 17, 518–528. [Google Scholar] [CrossRef]
- Grogan, S.P.; Miyaki, S.; Asahara, H.; D’Lima, D.D.; Lotz, M.K. Mesenchymal progenitor cell markers in human articular cartilage: Normal distribution and changes in osteoarthritis. Arthritis Res. Ther. 2009, 11, 1–13. [Google Scholar] [CrossRef]
- Boswell, S.G.; Cole, B.J.; Sundman, E.A.; Karas, V.; Fortier, L.A. Platelet-rich plasma: A milieu of bioactive factors. Arthroscopy. 2012, 28, 429–439. [Google Scholar] [CrossRef]
- Freson, K. The platelet proteome. In Platelets, 4th ed.; Michelson, A.D., Ed.; Elsevier/Academic Press: San Diego, CA, USA, 2019; pp. 155–167. [Google Scholar]
- Kabiri, A.; Hashemibeni, B.; Pourazar, A.; Mardani, M.; Esfandiari, E.; Esmaeili, A. Platelet-rich plasma application in chondrogenesis. Adv. Biomed. Res. 2014, 3, 138. [Google Scholar] [CrossRef]
- Wang, K.; Li, J.; Li, Z.; Wang, B.; Qin, Y.; Zhang, N.; Zhang, H.; Su, X.; Wang, Y.; Zhu, H. Chondrogenic Progenitor Cells Exhibit Superiority Over Mesenchymal Stem Cells and Chondrocytes in Platelet-Rich Plasma Scaffold-Based Cartilage Regeneration. Am. J. Sports Med. 2019, 47, 2200–2215. [Google Scholar] [CrossRef]
- Zhong, L.; Huang, X.; Rodrigues, E.D.; Leijten, J.C.H.; Verrips, T.; El Khattabi, M.; Karperien, M.; Post, J.N. Endogenous DKK1 and FRZB Regulate Chondrogenesis and Hypertrophy in Three-Dimensional Cultures of Human Chondrocytes and Human Mesenchymal Stem Cells. Stem Cells Dev. 2016, 25, 1808–1817. [Google Scholar] [CrossRef]
- Lian, J.B.; McKee, M.D.; Todd, A.M.; Gerstenfeld, L.C. Induction of bone-related proteins, osteocalcin and osteopontin, and their matrix ultrastructural localization with development of chondrocyte hypertrophy in vitro. J. Cell. Biochem. 1993, 52, 206–219. [Google Scholar] [CrossRef]
- Martel-Pelletier, J.; Cloutier, J.M.; Pelletier, J.P. Cathepsin B and cysteine protease inhibitors in human osteoarthritis. J. Orthop. Res. 1990, 8, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Herlihy, S.E.; Brown, M.L.; Pilling, D.; Weeks, B.R.; Myers, L.K.; Gomer, R.H. Role of the Neutrophil Chemorepellent Soluble Dipeptidyl Peptidase IV in Decreasing Inflammation in a Murine Model of Arthritis. Arthritis Rheumatol. 2015, 67, 2634–2638. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Liu, D.; Zheng, J.; Shi, P.; Chou, P.-H.; Oh, C.; Chen, D.; An, H.S.; Chee, A. Annulus fibrosus cells express and utilize C-C chemokine receptor 5 (CCR5) for migration. Spine J. 2017, 17, 720–726. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Zheng, H.; Buckwalter, J.A.; Martin, J.A. Single cell sorting identifies progenitor cell population from full thickness bovine articular cartilage. Osteoarthr. Cartil. 2014, 22, 1318–1326. [Google Scholar] [CrossRef] [PubMed]
- Jayasuriya, C.T.; Twomey-Kozak, J.; Newberry, J.; Desai, S.; Feltman, P.; Franco, J.R.; Li, N.; Terek, R.; Ehrlich, M.G.; Owens, B.D. Human Cartilage-Derived Progenitors Resist Terminal Differentiation and Require CXCR4 Activation to Successfully Bridge Meniscus Tissue Tears. Stem Cells 2019, 37, 102–114. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carluccio, S.; Martinelli, D.; Palamà, M.E.F.; Pereira, R.C.; Benelli, R.; Guijarro, A.; Cancedda, R.; Gentili, C. Progenitor Cells Activated by Platelet Lysate in Human Articular Cartilage as a Tool for Future Cartilage Engineering and Reparative Strategies. Cells 2020, 9, 1052. https://doi.org/10.3390/cells9041052
Carluccio S, Martinelli D, Palamà MEF, Pereira RC, Benelli R, Guijarro A, Cancedda R, Gentili C. Progenitor Cells Activated by Platelet Lysate in Human Articular Cartilage as a Tool for Future Cartilage Engineering and Reparative Strategies. Cells. 2020; 9(4):1052. https://doi.org/10.3390/cells9041052
Chicago/Turabian StyleCarluccio, Simonetta, Daniela Martinelli, Maria Elisabetta Federica Palamà, Rui Cruz Pereira, Roberto Benelli, Ana Guijarro, Ranieri Cancedda, and Chiara Gentili. 2020. "Progenitor Cells Activated by Platelet Lysate in Human Articular Cartilage as a Tool for Future Cartilage Engineering and Reparative Strategies" Cells 9, no. 4: 1052. https://doi.org/10.3390/cells9041052
APA StyleCarluccio, S., Martinelli, D., Palamà, M. E. F., Pereira, R. C., Benelli, R., Guijarro, A., Cancedda, R., & Gentili, C. (2020). Progenitor Cells Activated by Platelet Lysate in Human Articular Cartilage as a Tool for Future Cartilage Engineering and Reparative Strategies. Cells, 9(4), 1052. https://doi.org/10.3390/cells9041052