Charcot-Marie-Tooth Type 2B: A New Phenotype Associated with a Novel RAB7A Mutation and Inhibited EGFR Degradation

 , ,
, ,  ,
,  ,
,  ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Gene Sequence Analysis
2.3. Mutagenesis and Plasmid Construction
2.4. Antibodies
2.5. Cells and Transfection
2.6. Western Blotting and Co-Immunoprecipitation
2.7. Confocal Immunofluorescence Microscopy
2.8. Nucleotide Dissociation and GTPase Assay
2.9. EGFR Degradation Assay
2.10. Neurite Outgrowth Assay
2.11. Real-Time PCR
2.12. Molecular Modeling Studies
2.13. Statistical Analysis
3. Results
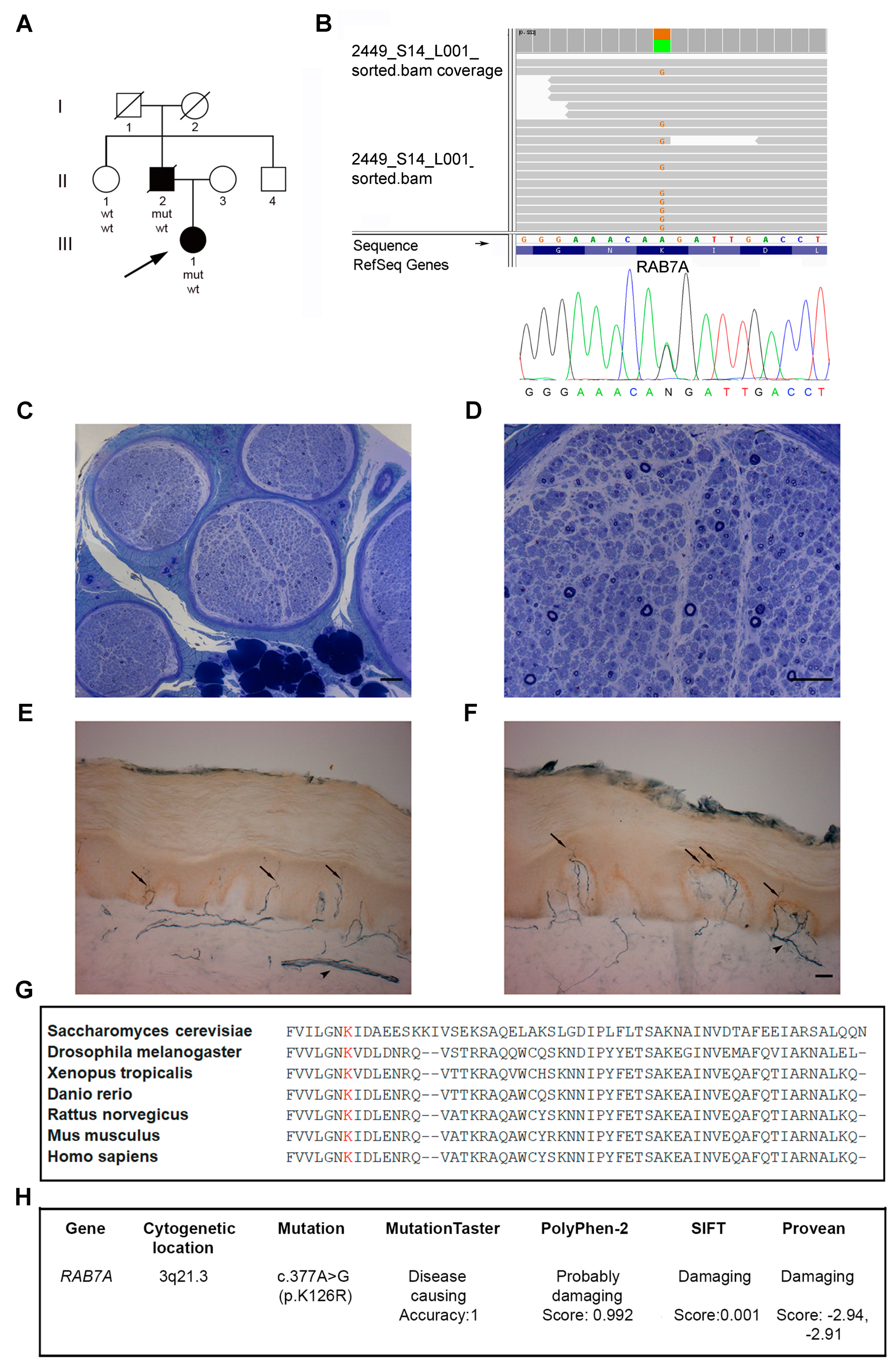
3.1. Patients
3.2. Identification of a Novel RAB7 Mutation Causing CMT2B
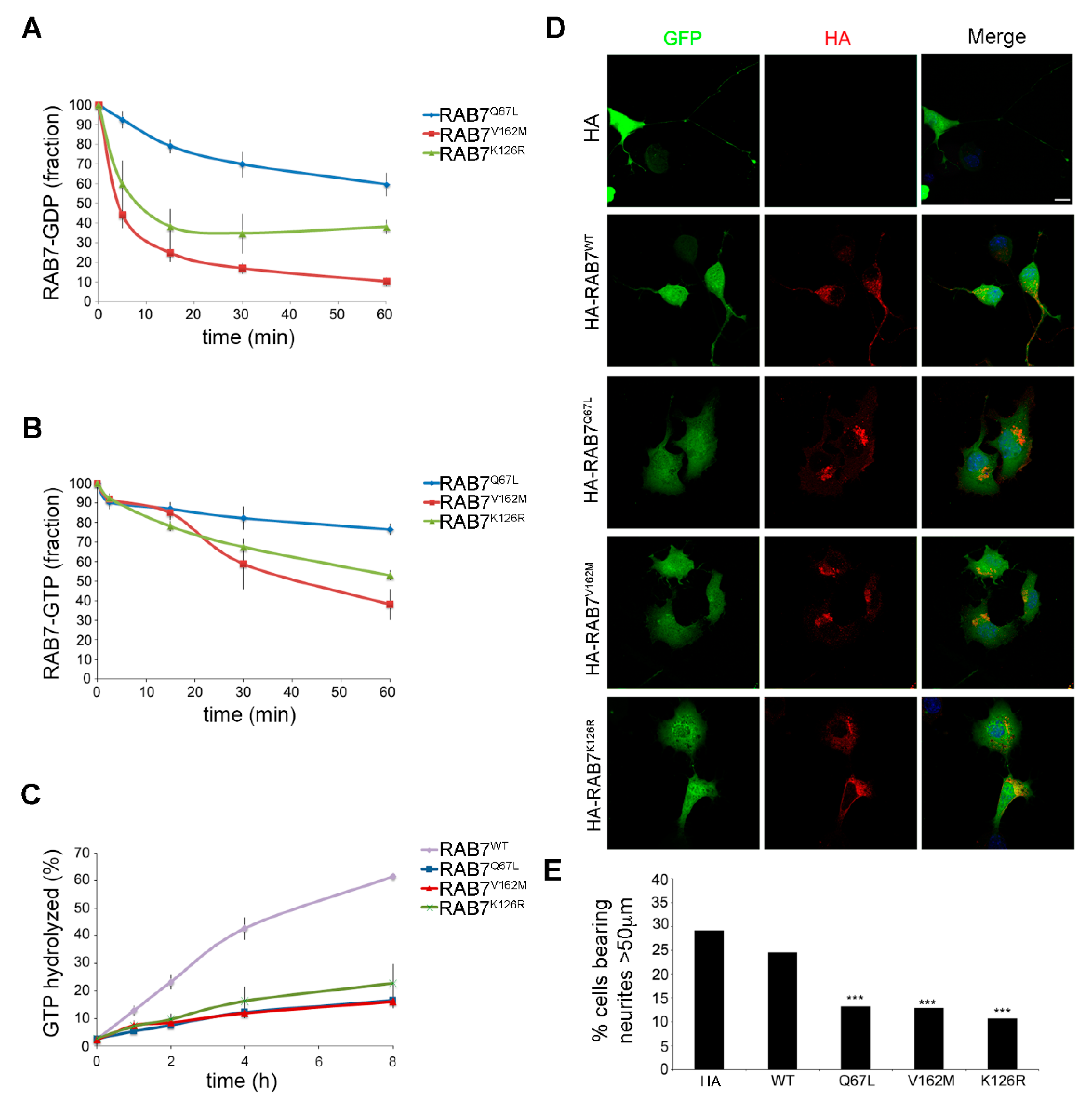
3.3. Characterization of the Biochemical Properties of the RAB7K126R Mutant Protein
3.4. Expression of the RAB7K126R Mutant Inhibits Neurite outgrowth in Neuro2A Cells
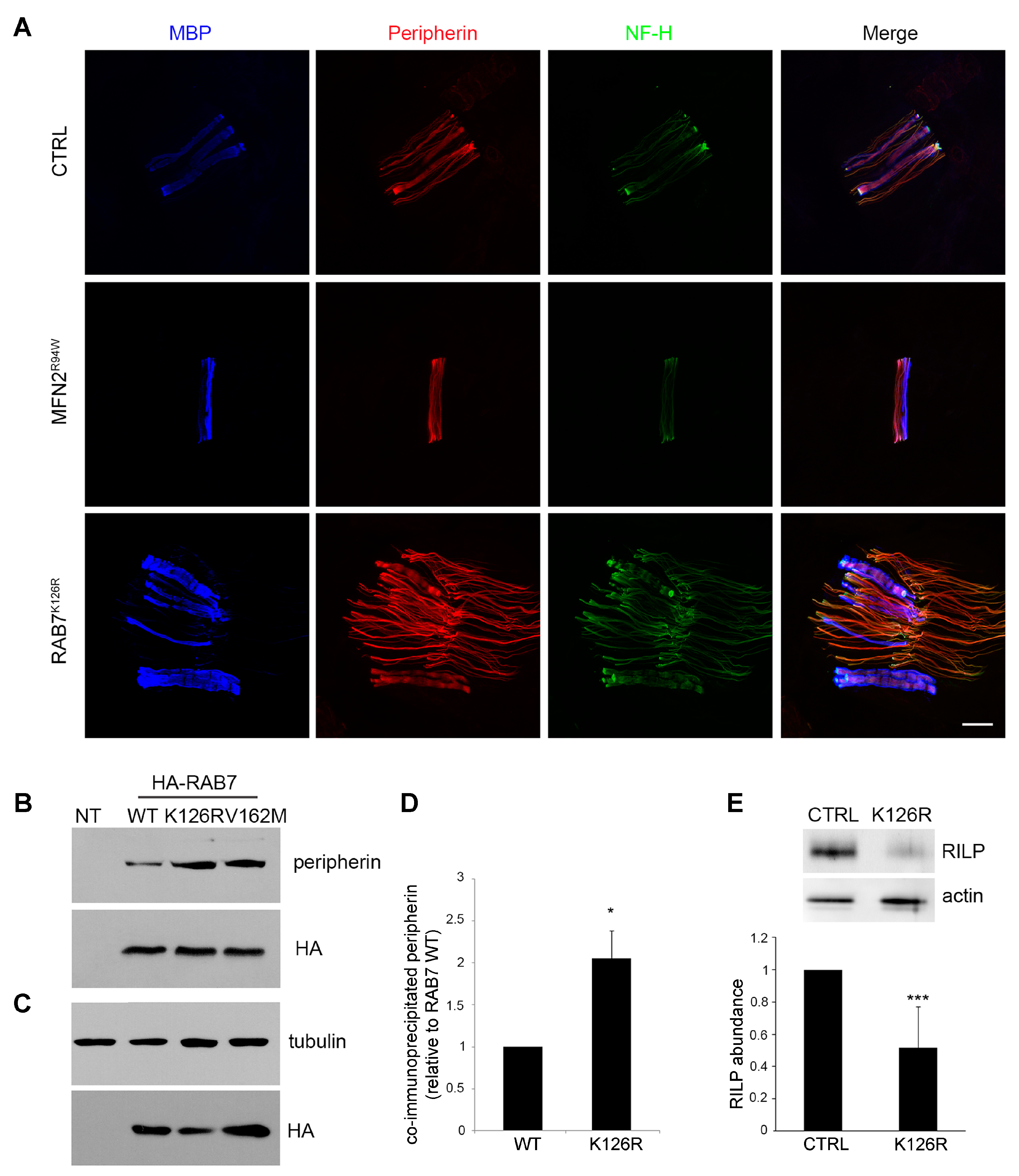
3.5. Analysis of Peripherin and RILP, two known Downstream RAB7 Effectors
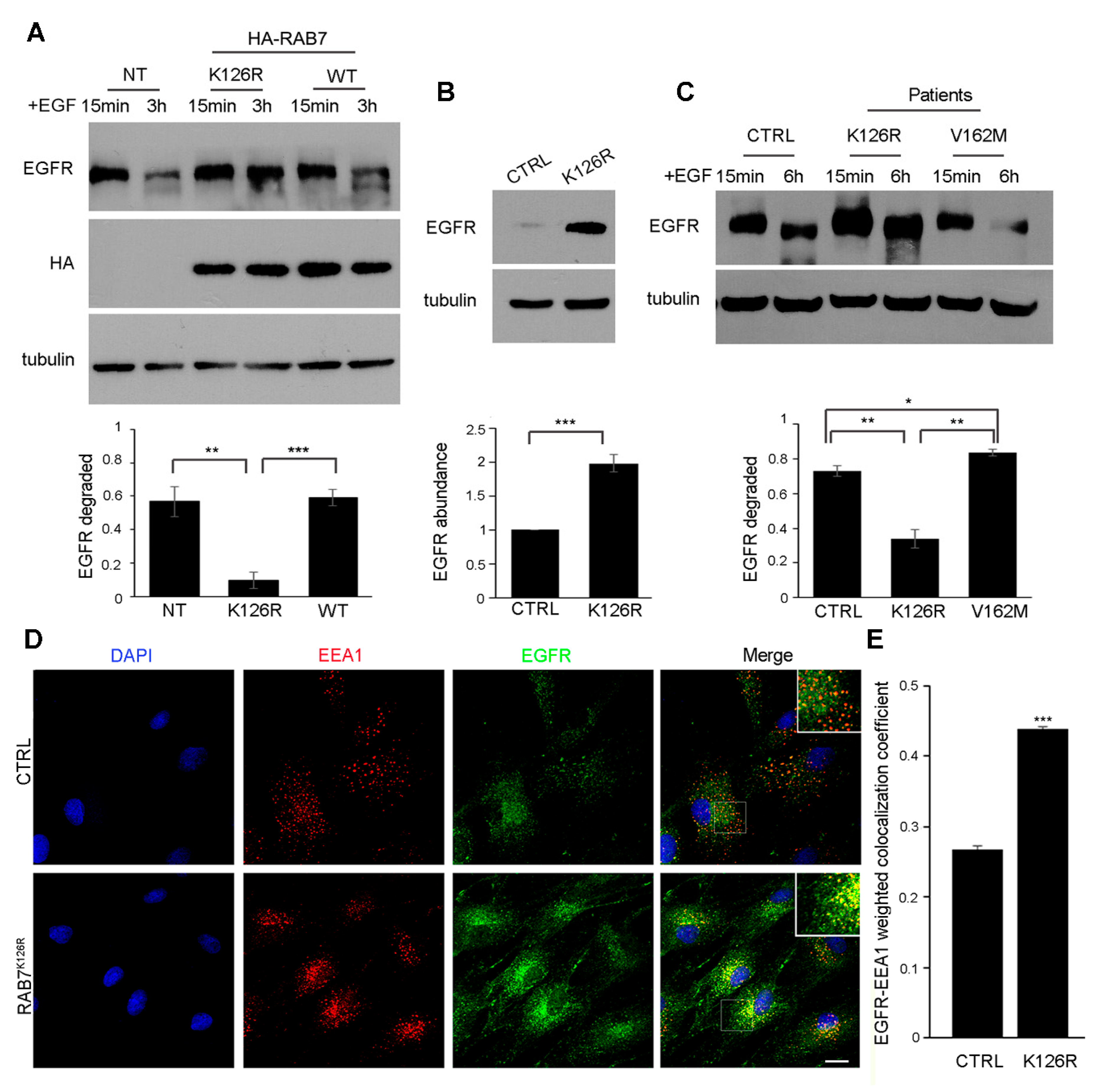
3.6. Expression of the RAB7K126R Mutant Inhibits Ligand-Induced EGFR Degradation
3.7. EGFR Amount, Degradation and Intracellular Distribution are Altered in Patient Fibroblasts Carrying the RAB7K126R Mutation
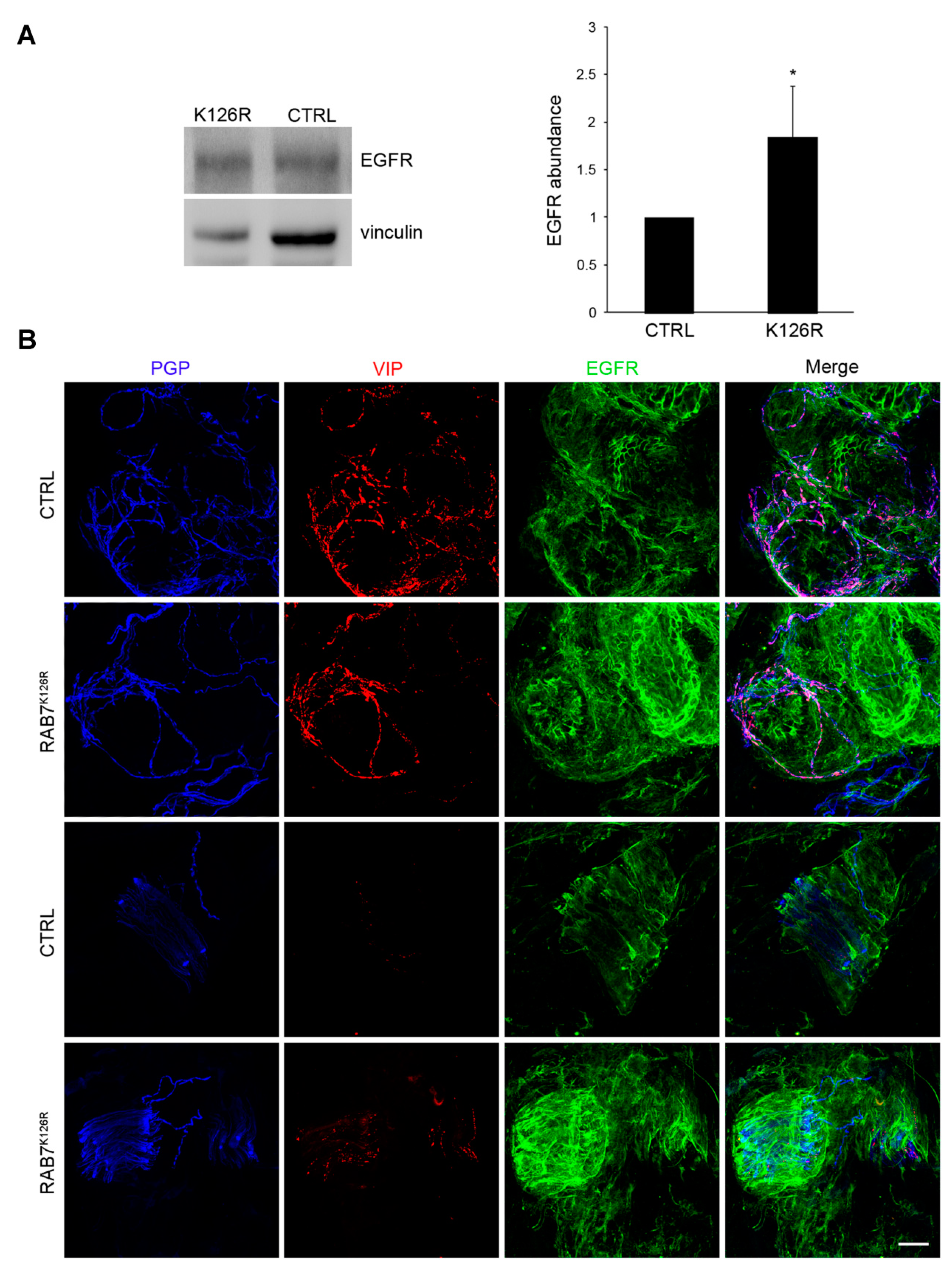
3.8. EGFR Amount is Altered in Patient Tissues
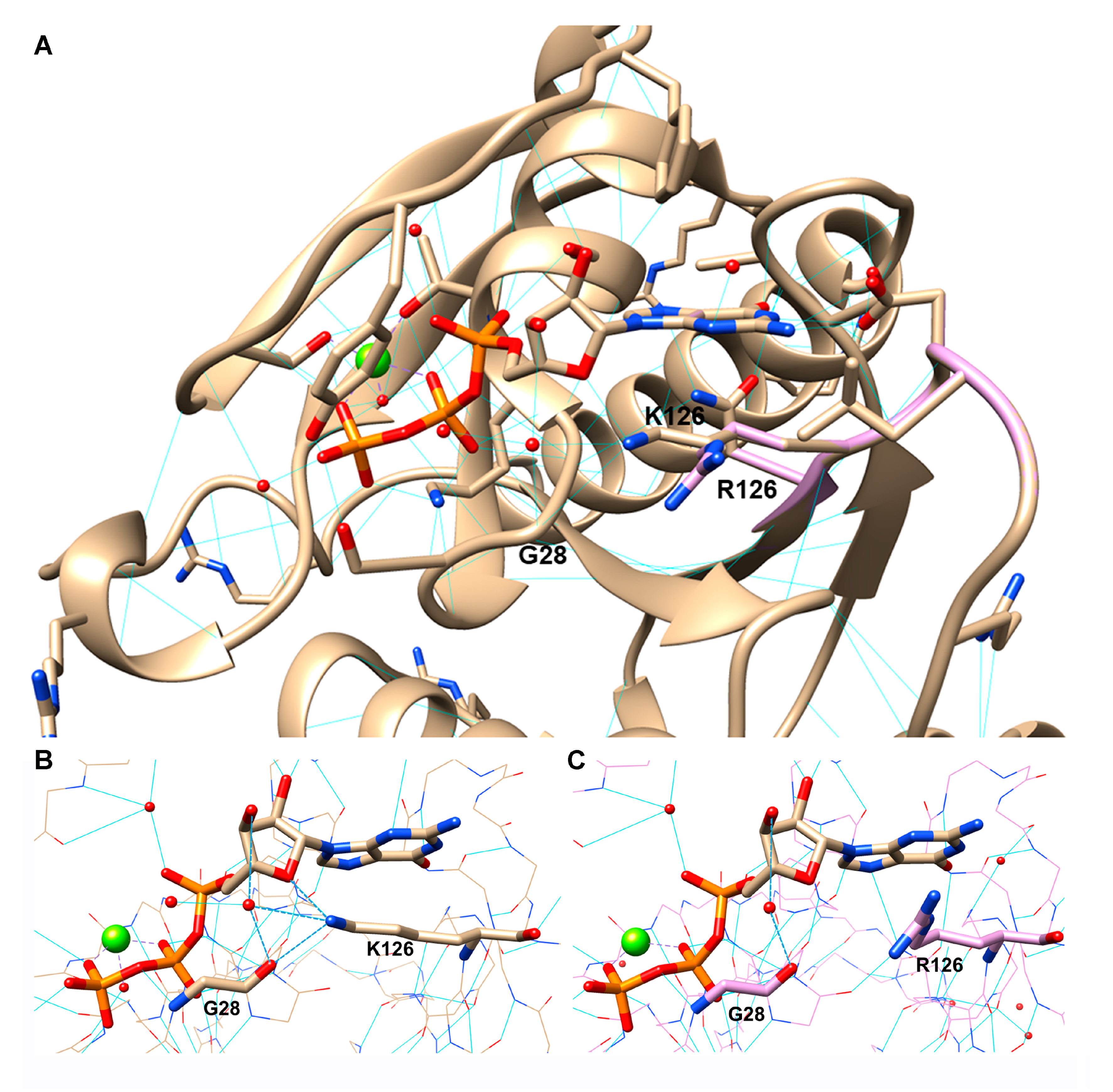
3.9. RAB7K126R Computational Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
List of Abbreviations
References
- Verhoeven, K.; De Jonghe, P.; Coen, K.; Verpoorten, N.; Auer-Grumbach, M.; Kwon, J.M.; FitzPatrick, D.; Schmedding, E.; De Vriendt, E.; Jacobs, A.; et al. Mutations in the small gtp-ase late endosomal protein rab7 cause charcot-marie-tooth type 2b neuropathy. Am. J. Hum. Genet. 2003, 72, 722–727. [Google Scholar] [CrossRef] [PubMed]
- Houlden, H.; King, R.H.; Muddle, J.R.; Warner, T.T.; Reilly, M.M.; Orrell, R.W.; Ginsberg, L. A novel rab7 mutation associated with ulcero-mutilating neuropathy. Ann. Neurol. 2004, 56, 586–590. [Google Scholar] [CrossRef] [PubMed]
- Meggouh, F.; Bienfait, H.M.; Weterman, M.A.; de Visser, M.; Baas, F. Charcot-marie-tooth disease due to a de novo mutation of the rab7 gene. Neurology 2006, 67, 1476–1478. [Google Scholar] [CrossRef] [PubMed]
- Manganelli, F.; Pisciotta, C.; Provitera, V.; Taioli, F.; Iodice, R.; Topa, A.; Fabrizi, G.M.; Nolano, M.; Santoro, L. Autonomic nervous system involvement in a new cmt2b family. J. Peripher. Nerv. Syst. 2012, 17, 361–364. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Han, C.; Liu, W.; Wang, P.; Zhang, X. A novel rab7 mutation in a chinese family with charcot-marie-tooth type 2b disease. Gene 2014, 534, 431–434. [Google Scholar] [CrossRef]
- Pareyson, D.; Saveri, P.; Piscosquito, G. Charcot-marie-tooth disease and related hereditary neuropathies: From gene function to associated phenotypes. Curr. Mol. Med. 2014, 14, 1009–1103. [Google Scholar] [CrossRef]
- Kwon, J.M.; Elliott, J.L.; Yee, W.C.; Ivanovich, J.; Scavarda, N.J.; Moolsintong, P.J.; Goodfellow, P.J. Assignment of a second charcot-marie-tooth type ii locus to chromosome 3q. Am. J. Hum. Genet. 1995, 57, 853–858. [Google Scholar]
- Auer-Grumbach, M.; De Jonghe, P.; Wagner, K.; Verhoeven, K.; Hartung, H.P.; Timmerman, V. Phenotype-genotype correlations in a cmt2b family with refined 3q13-q22 locus. Neurology 2000, 55, 1552–1557. [Google Scholar] [CrossRef]
- De Jonghe, P.V.T.; FitzPatrick, D.; Spoelders, P.; Martin, J.J.; Van Broeckhoven, C. Mutilating neuropathic ulcerations in a chromosome 3q13-q22 linked charcot-marie-tooth disease type 2b family. J. Neurol. Neurosurg. Psychiatry 1997, 62, 570–573. [Google Scholar] [CrossRef]
- Auer-Grumbach, M.; Wagner, K.; Timmerman, V.; De Jonghe, P.; Hartung, H.P. Ulcero-mutilating neuropathy in an austrian kinship without linkage to hereditary motor and sensory neuropathy iib and hereditary sensory neuropathy I loci. Neurology 2000, 54, 45–52. [Google Scholar] [CrossRef]
- Houlden, H.; King, R.; Blake, J.; Groves, M.; Love, S.; Woodward, C.; Hammans, S.; Nicoll, J.; Lennox, G.; O’Donovan, D.G.; et al. Clinical, pathological and genetic characterization of hereditary sensory and autonomic neuropathy type 1 (hsan i). Brain 2006, 129, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Auer-Grumbach, M.; De Jonghe, P.; Verhoeven, K.; Timmerman, V.; Wagner, K.; Hartung, H.P.; Nicholson, G.A. Autosomal dominant inherited europathies with prominent sensory loss and mutilations: A review. Arch. Neurol. 2003, 60, 329–334. [Google Scholar] [CrossRef]
- Kugathasan, U.; Evans, M.R.B.; Morrow, J.M.; Sinclair, C.D.J.; Thornton, J.S.; Yousry, T.A.; Hornemann, T.; Suriyanarayanan, S.; Owusu-Ansah, K.; Lauria, G.; et al. Development of mrc centre mri calf muscle fat fraction protocol as a sensitive outcome measure in hereditary sensory neuropathy type 1. J. Neurol. Neurosurg. Psychiatry 2019, 90, 895–906. [Google Scholar] [CrossRef]
- Bucci, C.; Thomsen, P.; Nicoziani, P.; McCarthy, J.; van Deurs, B. Rab7: A key to lysosome biogenesis. Mol. Biol. Cell 2000, 11, 467–480. [Google Scholar] [CrossRef]
- Mateus, D.; Marini, E.S.; Progida, C.; Bakke, O. Rab7a modulates er stress and er morphology. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 781–793. [Google Scholar] [CrossRef]
- Wong, Y.C.; Ysselstein, D.; Krainc, D. Mitochondria-lysosome contacts regulate mitochondrial fission via rab7 gtp hydrolysis. Nature 2018, 554, 382–386. [Google Scholar] [CrossRef]
- Deinhardt, K.; Salinas, S.; Verastegui, C.; Watson, R.; Worth, D.; Hanrahan, S.; Bucci, C.; Schiavo, G. Rab5 and rab7 control endocytic sorting along the axonal retrograde transport pathway. Neuron 2006, 52, 293–305. [Google Scholar] [CrossRef]
- Saxena, S.; Bucci, C.; Weis, J.; Kruttgen, A. The small gtpase rab7 controls the endosomal trafficking and neuritogenic signaling of the nerve growth factor receptor trka. J. Neurosci. 2005, 25, 10930–10940. [Google Scholar] [CrossRef]
- Margiotta, A.; Progida, C.; Bakke, O.; Bucci, C. Rab7a regulates cell migration through rac1 and vimentin. Biochim. Biophys. Acta 2017, 1864, 367–381. [Google Scholar] [CrossRef]
- Kawauchi, T.; Sekine, K.; Shikanai, M.; Chihama, K.; Tomita, K.; Kubo, K.; Nakajima, K.; Nabeshima, Y.; Hoshino, M. Rab gtpases-dependent endocytic pathways regulate neuronal migration and maturation through n-cadherin trafficking. Neuron 2010, 67, 588–602. [Google Scholar] [CrossRef]
- Cogli, L.; Progida, C.; Thomas, C.L.; Spencer-Dene, B.; Donno, C.; Schiavo, G.; Bucci, C. Charcot-marie-tooth type 2b disease-causing rab7a mutant proteins show altered interaction with the neuronal intermediate filament peripherin. Acta Neuropathol. 2013, 25, 257–272. [Google Scholar] [CrossRef] [PubMed]
- Spinosa, M.R.; Progida, C.; De Luca, A.; Colucci, A.M.R.; Alifano, P.; Bucci, C. Functional characterization of rab7 mutant proteins associated with charcot-marie-tooth type 2b disease. J. Neurosci. 2008, 28, 1640–1648. [Google Scholar] [CrossRef] [PubMed]
- De Luca, A.; Progida, C.; Spinosa, M.R.; Alifano, P.; Bucci, C. Characterization of the rab7k157n mutant protein associated with charcot-marie-tooth type 2b. Biochem. Biophys. Res. Commun. 2008, 372, 283–287. [Google Scholar] [CrossRef] [PubMed]
- McCray, B.A.; Skordalakes, E.; Taylor, J.P. Disease mutations in rab7 result in unregulated nucleotide exchange and inappropriate activation. Hum. Mol. Genet. 2010, 19, 1033–1047. [Google Scholar] [CrossRef] [PubMed]
- Cogli, L.; Progida, C.; Lecci, R.; Bramato, R.; Krüttgen, A.; Bucci, C. Cmt2b-associated rab7 mutants inhibit neurite outgrowth. Acta Neuropathol. 2010, 120, 491–501. [Google Scholar] [CrossRef]
- Yamauchi, J.; Torii, T.; Kusakawa, S.; Sanbe, A.; Nakamura, K.; Takashima, S.; Hamasaki, H.; Kawaguchi, S.; Miyamoto, Y.; Tanoue, A. The mood stabilizer valproic acid improves defective neurite formation caused by charcot-marie-tooth disease-associated mutant rab7 through the jnk signaling pathway. J. Neurosci. Res. 2010, 88, 3189–3197. [Google Scholar] [CrossRef]
- Progida, C.; Malerød, L.; Stuffers, S.; Brech, A.; Bucci, C.; Stenmark, H. Rilp is required for proper morphology and function of late endosomes. J. Cell Sci. 2007, 120, 3729–3737. [Google Scholar] [CrossRef]
- Ceresa, B.P.; Bahr, S.J. Rab7 activity affects epidermal growth factor: Epidermal growth factor receptor degradation by regulating endocytic trafficking from the late endosome. J. Biol. Chem. 2006, 281, 1099–1106. [Google Scholar] [CrossRef]
- Cantalupo, G.; Alifano, P.; Roberti, V.; Bruni, C.B.; Bucci, C. Rab-interacting lysosomal protein (rilp): The rab7 effector required for transport to lysosomes. EMBO J. 2001, 20, 683–693. [Google Scholar] [CrossRef]
- Xian, C.J.; Zhou, X.F. Egf family of growth factors: Essential roles and functional redundancy in the nerve system. Front. Biosci. 2004, 9, 85–92. [Google Scholar] [CrossRef][Green Version]
- Yamada, M.; Ikeuchi, T.; Hatanaka, H. The neurotrophic action and signalling of epidermal growth factor. Prog. Neurobiol. 1997, 51, 19–37. [Google Scholar] [CrossRef]
- Basuray, S.; Mukherjee, S.; Seaman, M.N.; Wandinger-Ness, A. Rab7 mutants associated with charcot-marie-tooth disease cause delayed growth factor receptor transport and altered endosomal and nuclear signaling. J. Biol. Chem. 2013, 288, 1135–1149. [Google Scholar] [CrossRef] [PubMed]
- Fabrizi, G.M.; Simonati, A.; Morbin, M.; Cavallaro, T.; Taioli, F.; Benedetti, M.D.; Edomi, P.; Rizzuto, N. Clinical and pathological correlations in charcot-marie-tooth neuropathy type 1a with the 17p11.2p12 duplication: A cross-sectional morphometric and immunohistochemical study in twenty cases. Muscle Nerve 1998, 21, 869–877. [Google Scholar] [CrossRef]
- Lauria, G.; Hsieh, S.T.; Johansson, O.; Kennedy, W.R.; Leger, J.M.; Mellgren, S.I.; Nolano, M.; Merkies, I.S.; Polydefkis, M.; Smith, A.G.; et al. European federation of neurological societies/peripheral nerve society guideline on the use of skin biopsy in the diagnosis of small fiber neuropathy. Report of a joint task force of the european federation of neurological societies and the peripheral nerve society. Eur. J. Neurol. 2010, 17, e903–e949. [Google Scholar]
- Edgar, R.C. Muscle: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, A.D.; Riederer, M.A.; Pfeffer, S.R. Biochemical analysis of rab9, a ras-like gtpase involved in protein transport from late endosomes to the trans golgi metwork. J. Biol. Chem. 1993, 268, 6925–6931. [Google Scholar]
- Stenmark, H.; Parton, R.G.; Steele-Mortimer, O.; Lutcke, A.; Gruenberg, J.; Zerial, M. Inhibition of rab5 gtpase activity stimulates membrane fusion in endocytosis. EMBO J. 1994, 13, 1287–1296. [Google Scholar] [CrossRef]
- Kjeldgaard, M.; Nyborg, J.; Clark, B.F. The gtp binding motif: Variations on a theme. FASEB J. 1996, 10, 1347–1368. [Google Scholar] [CrossRef]
- Kerr, B.; Delrue, M.A.; Sigaudy, S.; Perveen, R.; Marche, M.; Burgelin, I.; Stef, M.; Tang, B.; Eden, O.B.; O’Sullivan, J.; et al. Genotype-phenotype correlation in costello syndrome: Hras mutation analysis in 43 cases. J. Med. Genet. 2006, 43, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Mahar, M.; Cavalli, V. Intrinsic mechanisms of neuronal axon regeneration. Nat. Rev. Neurosci. 2018, 19, 323–337. [Google Scholar] [CrossRef]
- Kirkcaldie, M.T.K.; Dwyer, S.T. The third wave: Intermediate filaments in the maturing nervous system. Mol. Cell Neurosci. 2017, 84, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Parlakian, A.; Paulin, D.; Izmiryan, A.; Xue, Z.; Li, Z. Intermediate filaments in peripheral nervous system: Their expression, dysfunction and diseases. Rev. Neurol. 2016, 172, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Progida, C.; Spinosa, M.; De Luca, A.; Bucci, C. Rilp interacts with the vps22 component of the escrt-ii complex. Biochem. Biophys. Res. Commun. 2006, 347, 1074–1079. [Google Scholar] [CrossRef] [PubMed]
- Maklad, A.; Nicolai, J.R.; Bichsel, K.J.; Evenson, J.E.; Lee, T.C.; Threadgill, D.W.; Hansen, L.A. The egfr is required for proper innervation to the skin. J. Invest. Dermatol. 2009, 129, 690–698. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Neufeld, A.H. Activation of epidermal growth factor receptors in astrocytes: From development to neural injury. J. Neurosci. Res. 2007, 85, 3523–3529. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.M.; Strunz, C. Growth factor and cytokine regulation of chondroitin sulfate proteoglycans by astrocytes. Glia 2005, 52, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Erschbamer, M.; Pernold, K.; Olson, L. Inhibiting epidermal growth factor receptor improves structural, locomotor, sensory, and bladder recovery from experimental spinal cord injury. J. Neurosci. 2007, 27, 6428–6435. [Google Scholar] [CrossRef]
- Sibilia, M.; Wagner, E.F. Strain-dependent epithelial defects in mice lacking the egf receptor. Science 1995, 269, 234–238. [Google Scholar] [CrossRef]
- Sibilia, M.; Steinbach, J.P.; Stingl, L.; Aguzzi, A.; Wagner, E.F. A strain-independent postnatal neurodegeneration in mice lacking the egf receptor. EMBO J. 1998, 17, 719–731. [Google Scholar] [CrossRef]
- Hatanpaa, K.J.; Burma, S.; Zhao, D.; Habib, A.A. Epidermal growth factor receptor in glioma: Signal transduction, neuropathology, imaging, and radioresistance. Neoplasia 2010, 12, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, S.; Fang, M.; Ni, B.; Lu, D.; Martin, B.; Maudsley, S. Central role of the egf receptor in neurometabolic aging. Int. J. Endocrinol. 2012, 2012, 739428. [Google Scholar] [CrossRef] [PubMed]
- Hendry, J.M.; Alvarez-Veronesi, M.C.; Placheta, E.; Zhang, J.J.; Gordon, T.; Borschel, G.H. Erbb2 blockade with herceptin (trastuzumab) enhances peripheral nerve regeneration after repair of acute or chronic peripheral nerve injury. Ann. Neurol. 2016, 80, 112–126. [Google Scholar] [CrossRef] [PubMed]
- Placheta, E.; Hendry, J.M.; Wood, M.D.; Lafontaine, C.W.; Liu, E.H.; Alvarez Veronesi, M.C.; Frey, M.; Gordon, T.; Borschel, G.H. The erbb2 inhibitor herceptin (trastuzumab) promotes axonal outgrowth four weeks after acute nerve transection and repair. Neurosci. Lett. 2014, 582, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.P.; Wu, J.; Johansson, G.; Rizvi, T.A.; Miller, S.C.; Geiger, H.; Malik, P.; Li, W.; Mukouyama, Y.S.; Cancelas, J.A.; et al. Nf1 mutation expands an egfr-dependent peripheral nerve progenitor that confers neurofibroma tumorigenic potential. Cell Stem Cell 2008, 3, 658–669. [Google Scholar] [CrossRef][Green Version]
- Gu, Y.; Chen, C.; Yi, S.; Wang, S.; Gong, L.; Liu, J.; Gu, X.; Zhao, Q.; Li, S. Mir-sc8 inhibits schwann cell proliferation and migration by targeting egfr. PLoS ONE 2015, 10, e0145185. [Google Scholar] [CrossRef]
- Dahlhoff, M.; Emrich, D.; Wolf, E.; Schneider, M.R. Increased activation of the epidermal growth factor receptor in transgenic mice overexpressing epigen causes peripheral neuropathy. Biochim. Biophys. Acta 2013, 1832, 2068–2076. [Google Scholar] [CrossRef][Green Version]
- Kanehisa, M.; Goto, S. Kegg: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Furumichi, M.; Morishima, K.; Tanabe, M. New approach for understanding genome variations in kegg. Nucleic Acids Res. 2019, 47, D590–D595. [Google Scholar] [CrossRef]
- Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 2019, 28, 1947–1951. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation | Clinical Phenotype | Families/Sporadic Cases | Age of Onset | Reference |
|---|---|---|---|---|
| L129F | CMT2B ulcero-mutilating features | Three related Austrian families | Adolescence—adulthood | [1,8] |
| K157N | CMT2B ulcerations, osteomyelitis, amputation | A patient (de novo mutation) | Adolescence | [3] |
| N161I | CMT2B pain, no muscle atrophy, and (except for the proband) ulcero-mutilating features | A Chinese family | Teens or later | [5] |
| N161T | CMT2B pain, ulcers, gangrene, and amputations | An English family | Adolescence | [2] |
| V162M | CMT2B ulcero-mutilating features (except for pt I-2 of the Scottish family who had drop feet but probably no ulcers) | Five unrelated families (from USA, Scotland, Austria, Belgium, Italy) | Adolescence—adulthood | [1,4,7,9,10] |
| K126R | CMT2 Predominantly motor phenotype with little sensory involvement | One Italian family | Adolescence | Present paper |
| Patients | Proband | Father |
| Age at examination, yrs. | 39 | 55 |
| Onset age/symptoms | 14/Cramps, gait difficulties | 11/Gait difficulties |
| Motor symptoms legs | Complete footdrop, AFOs | Complete footdrop, AFOs |
| Sensory symptoms legs | Slight sensory loss feet | N |
| Proximal/Distal weakness UL | −/+- (4+; 4+) | −/+ (2;1) |
| Proximal/Distal weakness LL | −/+ (0;3) | −/+ (0;0) |
| Ankle deep tendon reflexes | Absent | Absent |
| Pinprick UL/LL | Normal/Ankles | Normal/Normal |
| Joint position sense UL/LL | Normal/Normal | Normal/Toes |
| Vibration sense UL/LL | Normal/Toes | Normal/Ankles |
| Pes cavus | N | Y |
| Additional features | Mild toenail dystrophy | Hand tremor |
| Ulnar nerve SAP amplitude | 5.9 µV (ulnar nerve) | 0.8 µV (median nerve) |
| CMTES | 9/28 | 11/28 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saveri, P.; De Luca, M.; Nisi, V.; Pisciotta, C.; Romano, R.; Piscosquito, G.; Reilly, M.M.; Polke, J.M.; Cavallaro, T.; Fabrizi, G.M.; et al. Charcot-Marie-Tooth Type 2B: A New Phenotype Associated with a Novel RAB7A Mutation and Inhibited EGFR Degradation. Cells 2020, 9, 1028. https://doi.org/10.3390/cells9041028
Saveri P, De Luca M, Nisi V, Pisciotta C, Romano R, Piscosquito G, Reilly MM, Polke JM, Cavallaro T, Fabrizi GM, et al. Charcot-Marie-Tooth Type 2B: A New Phenotype Associated with a Novel RAB7A Mutation and Inhibited EGFR Degradation. Cells. 2020; 9(4):1028. https://doi.org/10.3390/cells9041028
Chicago/Turabian StyleSaveri, Paola, Maria De Luca, Veronica Nisi, Chiara Pisciotta, Roberta Romano, Giuseppe Piscosquito, Mary M. Reilly, James M. Polke, Tiziana Cavallaro, Gian Maria Fabrizi, and et al. 2020. "Charcot-Marie-Tooth Type 2B: A New Phenotype Associated with a Novel RAB7A Mutation and Inhibited EGFR Degradation" Cells 9, no. 4: 1028. https://doi.org/10.3390/cells9041028
APA StyleSaveri, P., De Luca, M., Nisi, V., Pisciotta, C., Romano, R., Piscosquito, G., Reilly, M. M., Polke, J. M., Cavallaro, T., Fabrizi, G. M., Fossa, P., Cichero, E., Lombardi, R., Lauria, G., Magri, S., Taroni, F., Pareyson, D., & Bucci, C. (2020). Charcot-Marie-Tooth Type 2B: A New Phenotype Associated with a Novel RAB7A Mutation and Inhibited EGFR Degradation. Cells, 9(4), 1028. https://doi.org/10.3390/cells9041028