Targeted Phototherapy for Malignant Pleural Mesothelioma: Near-Infrared Photoimmunotherapy Targeting Podoplanin



 ,
,  ,
,  and
and 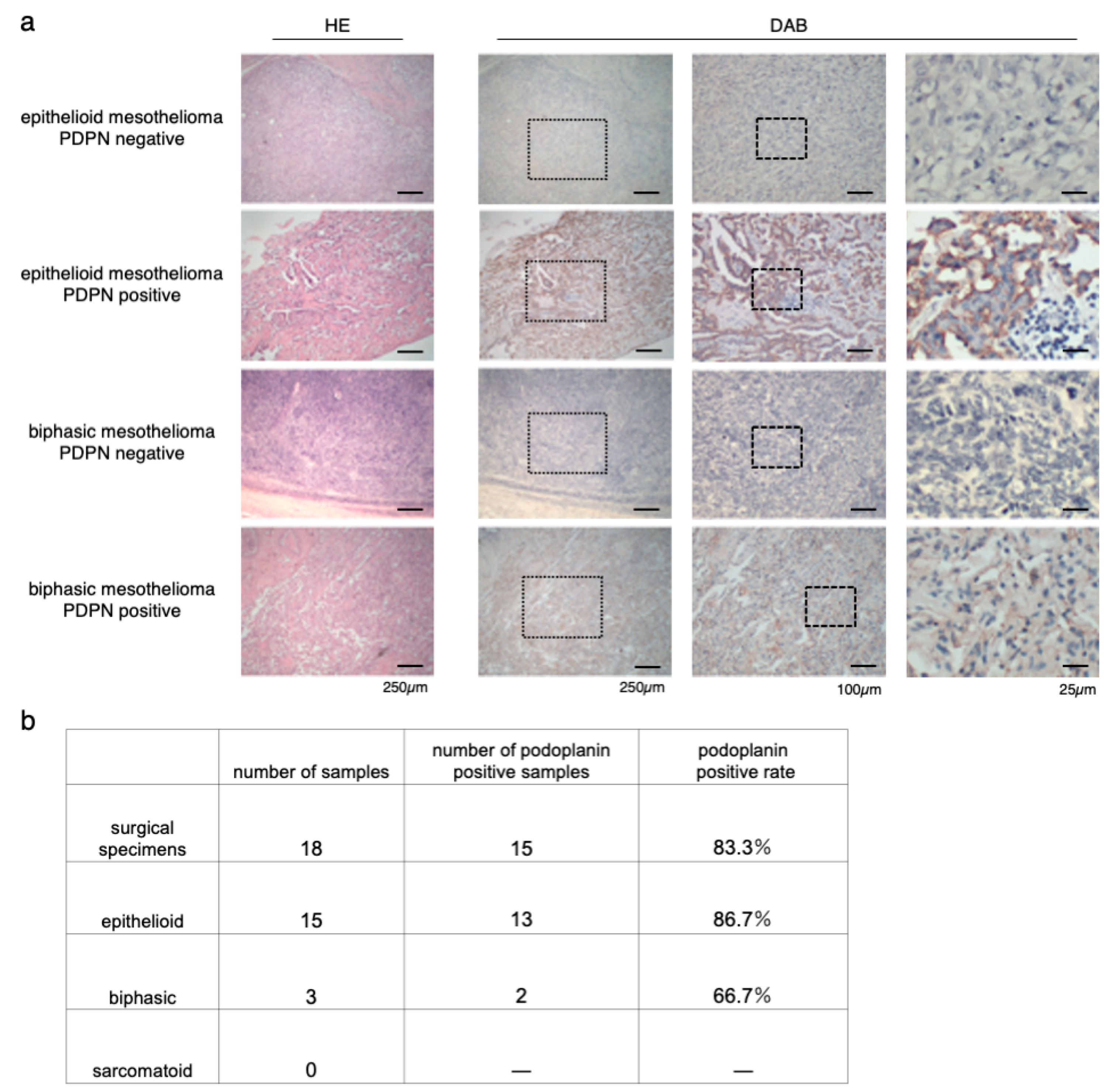{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Immunostaining of Surgical Human Resected MPM Samples
2.3. Reagents
2.4. Synthesis of IR700-Conjugated NZ-1
2.5. Cell Lines
2.6. Cell Culture
2.7. Flow Cytometry
2.8. Fluorescence Microscopy
2.9. In Vitro NIR-PIT
2.10. Cytotoxicity or Phototoxicity Assay
2.11. Animal and Tumor Models
2.12. Characterization of Mouse Model
2.13. In Vivo Local PDPN-Targeted NIR-PIT
2.14. Statistical Analysis
3. Results
3.1. Immunostaining of Surgical Specimens of Human-Resected MPM
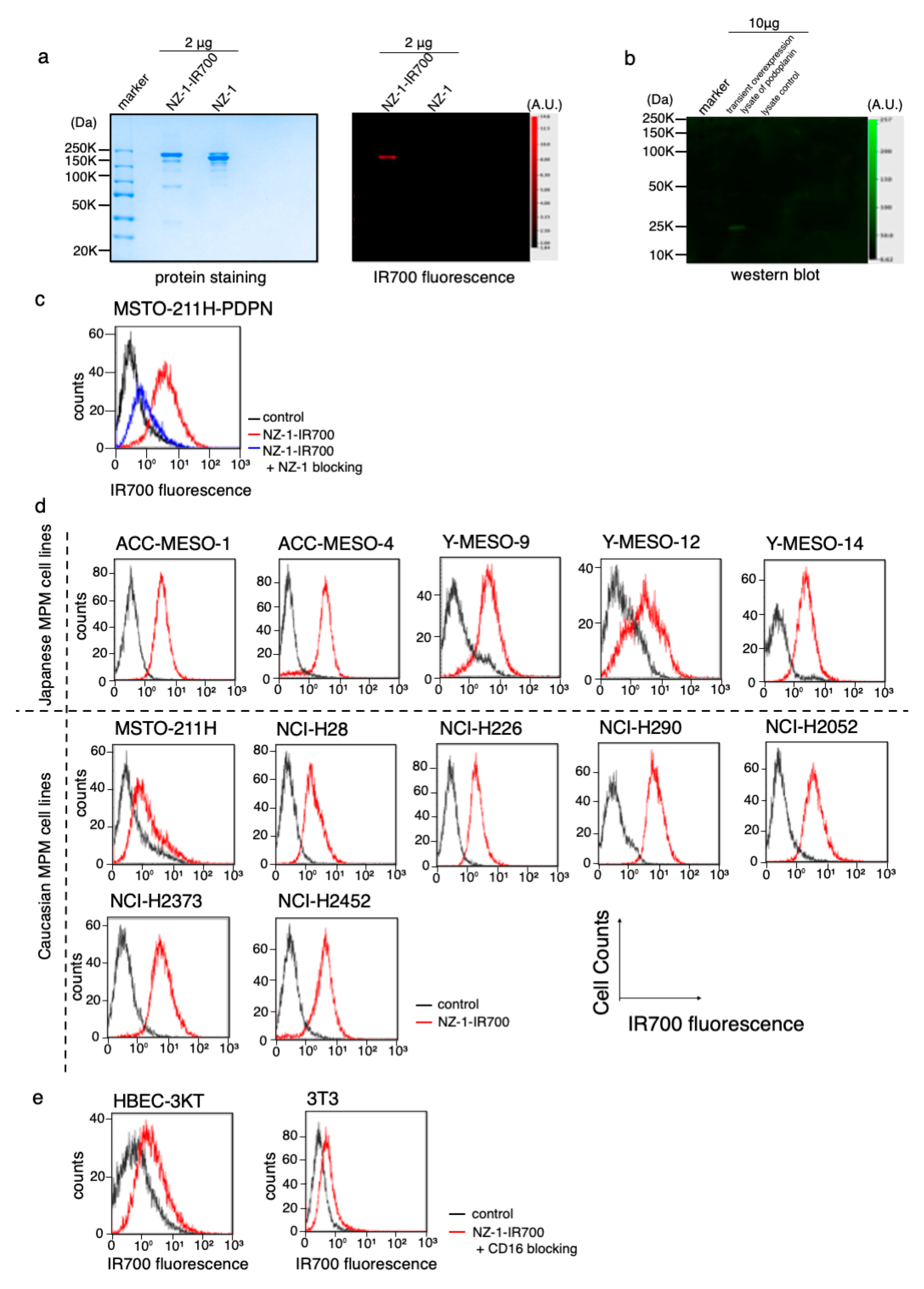
3.2. Conjugation of NZ-1 with IR700DX
3.3. PDPN Expression in the MPM Cell Lines
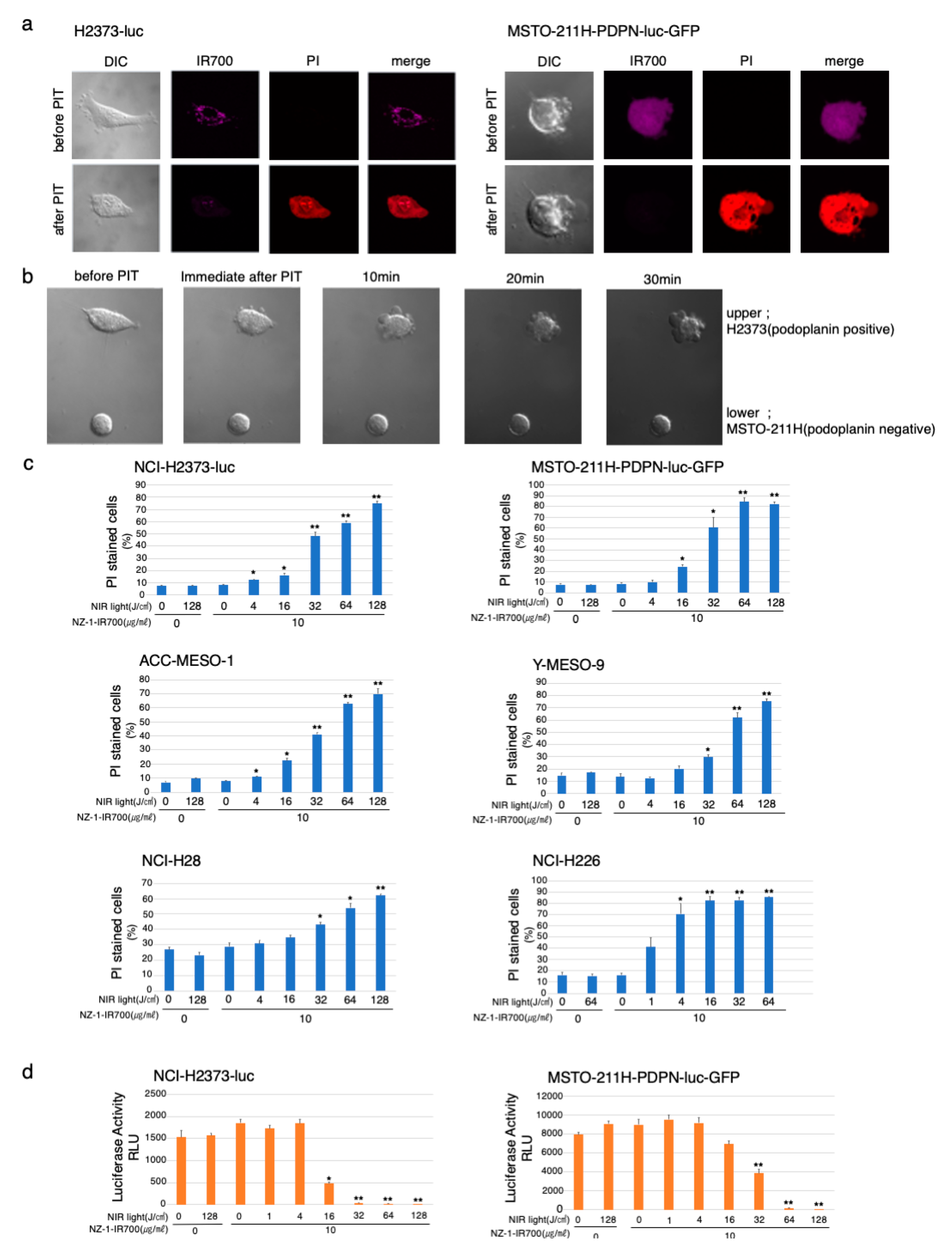
3.4. In Vitro Microscopic Observation of NIR-PIT with NZ-1-IR700 on MPM Cell Lines
3.5. In Vitro NIR-PIT Effects with NZ-1-IR700 in MPM Cell Lines
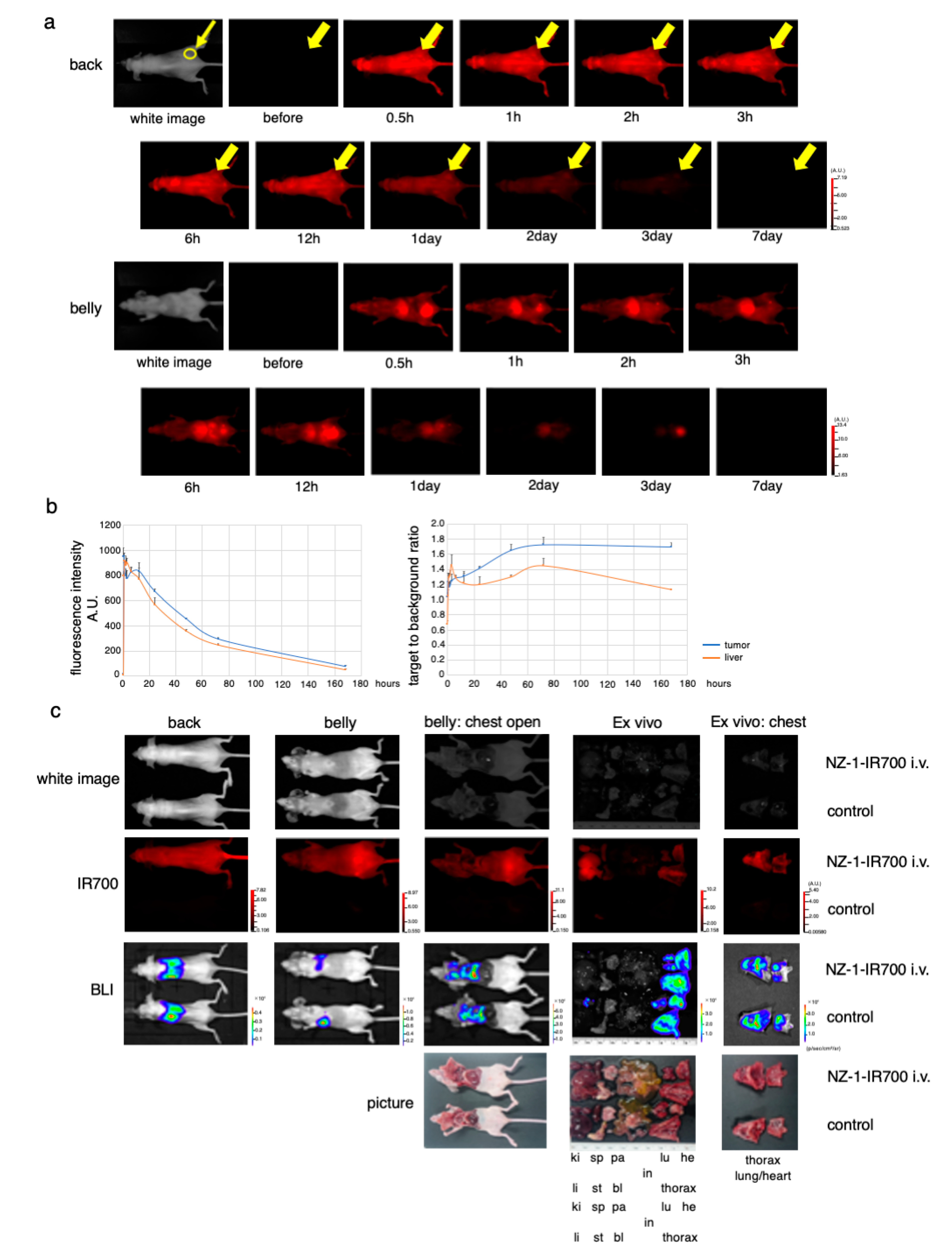
3.6. In Vivo Biodistribution of NZ-1-IR700
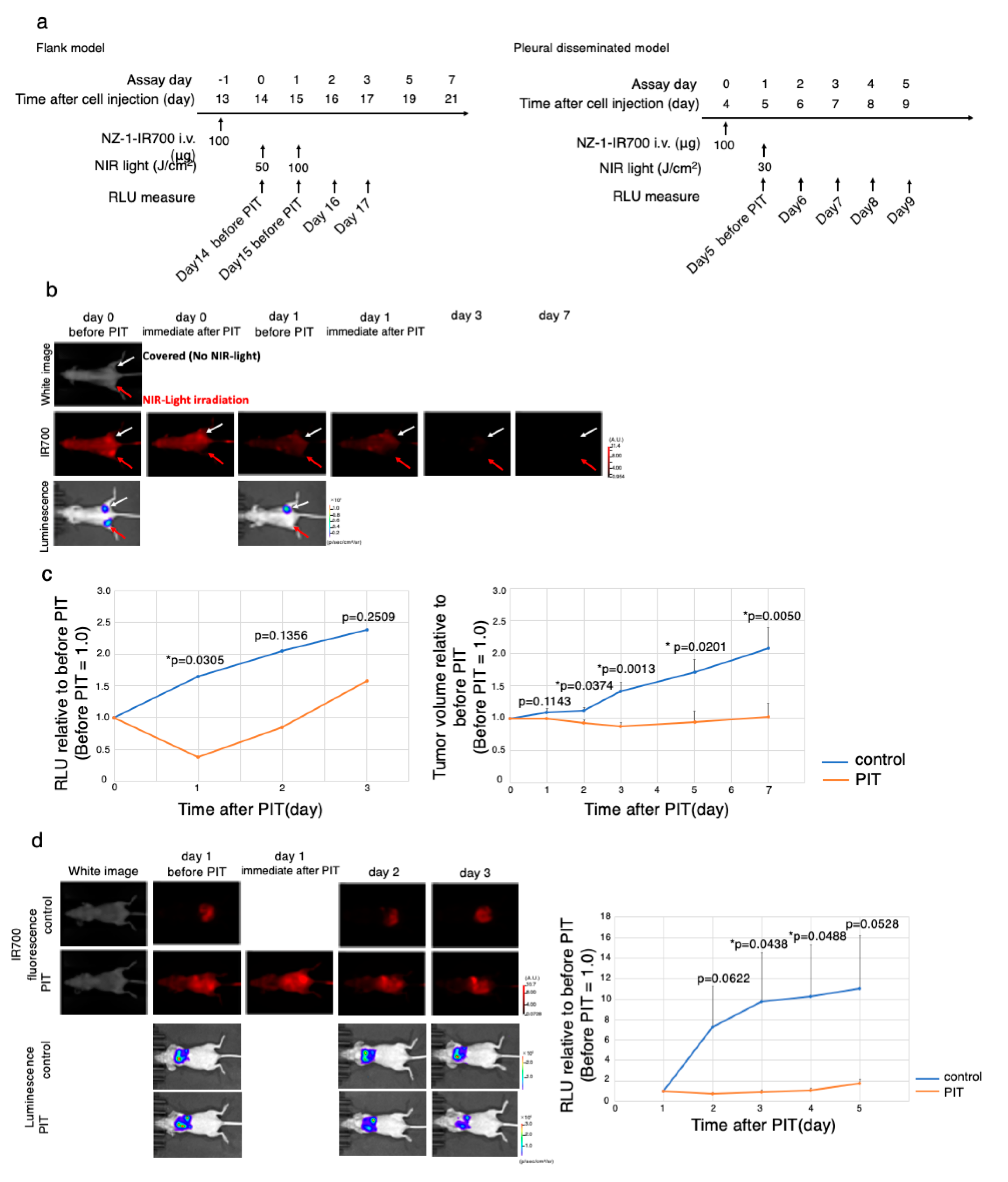
3.7. In Vivo Antitumor Effect of PDPN-Targeted NIR-PIT
3.8. Characterization of the Pleural Disseminated MPM Model
3.9. In Vivo PDPN-Targeted NIR-PIT Effect on MPM Pleural Disseminated Orthotopic Model
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rudd, R.M. Malignant mesothelioma. Br. Med. Bull. 2010, 93, 105–123. [Google Scholar] [CrossRef] [PubMed]
- Bibby, A.C.; Tsim, S.; Kanellakis, N.; Ball, H.; Talbot, D.C.; Blyth, K.G.; Maskell, N.A.; Psallidas, I. Malignant pleural mesothelioma: An update on investigation, diagnosis and treatment. Eur. Respir. Rev. 2016, 25, 472–486. [Google Scholar] [CrossRef] [PubMed]
- Carioli, G.; Bonifazi, M.; Rossi, M.; Zambelli, A.; Franchi, M.; Zocchetti, C.; Gasparini, S.; Corrao, G.; La Vecchia, C.; Negri, E. Management and survival of pleural mesothelioma: A record linkage study. Respiration 2018, 95, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Sayan, M.; Eren, M.F.; Gupta, A.; Ohri, N.; Kotek, A.; Babalioglu, I.; Kaplan, S.O.; Duran, O.; Or, O.D.; Cukurcayir, F.; et al. Current treatment strategies in malignant pleural mesothelioma with a treatment algorithm. Adv. Respir. Med. 2019, 87, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Olsen, N.J.; Franklin, P.J.; Reid, A.; de Klerk, N.H.; Threlfall, T.J.; Shilkin, K.; Musk, B. Increasing incidence of malignant mesothelioma after exposure to asbestos during home maintenance and renovation. Med. J. Aust. 2011, 195, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Frank, A.L.; Joshi, T.K. The global spread of asbestos. Ann. Glob. Health 2014, 80, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Hashim, D.; Boffetta, P. Occupational and environmental exposures and cancers in developing countries. Ann. Glob. Health 2014, 80, 393–411. [Google Scholar] [CrossRef]
- Kindler, H.L.; Ismaila, N.; Armato, S.G.; Bueno, R.; Hesdorffer, M.; Jahan, T.; Jones, C.M.; Miettinen, M.; Pass, H.; Rimner, A.; et al. Treatment of malignant pleural mesothelioma: American society of clinical oncology clinical practice guideline. J. Clin. Oncol. 2018, 36, 1343–1373. [Google Scholar] [CrossRef]
- Quintanilla, M.; Montero, L.M.; Renart, J.; Villar, E.M. Podoplanin in inflammation and cancer. Int. J. Mol. Sci. 2019, 20, 707. [Google Scholar] [CrossRef]
- Kato, Y.; Kaneko, M.K.; Kuno, A.; Uchiyama, N.; Amano, K.; Chiba, Y.; Hasegawa, Y.; Hirabayashi, J.; Narimatsu, H.; Mishima, K.; et al. Inhibition of tumor cell-induced platelet aggregation using a novel anti-podoplanin antibody reacting with its platelet-aggregation-stimulating domain. Biochem. Biophys. Res. Commun. 2006, 349, 1301–1307. [Google Scholar] [CrossRef]
- Kato, Y.; Kaneko, M.K.; Kunita, A.; Ito, H.; Kameyama, A.; Ogasawara, S.; Matsuura, N.; Hasegawa, Y.; Suzuki-inoue, K.; Inoue, O.; et al. Molecular analysis of the pathophysiological binding of the platelet aggregation-inducing factor podoplanin to the C-type lectin-like receptor CLEC-2. Cancer Sci. 2008, 99, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Abe, S.; Morita, Y.; Kaneko, M.K.; Hanibuchi, M.; Tsujimoto, Y.; Goto, H.; Kakiuchi, S.; Aono, Y.; Huang, J.; Sato, S.; et al. A novel targeting therapy of malignant mesothelioma using anti-podoplanin antibody. J. Immunol. 2013, 190, 6239–6249. [Google Scholar] [CrossRef] [PubMed]
- Mitsunaga, M.; Ogawa, M.; Kosaka, N.; Rosenblum, L.T.; Choyke, P.L.; Kobayashi, H. Cancer cell-selective in vivo near infrared photoimmunotherapy targeting specific membrane molecules. Nat. Med. 2011, 17, 1685–1691. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Nagaya, T.; Choyke, P.L.; Kobayashi, H. Near infrared photoimmunotherapy in the treatment of pleural disseminated NSCLC: Preclinical experience. Theranostics 2015, 5, 698–709. [Google Scholar] [CrossRef]
- Sato, K.; Hanaoka, H.; Watanabe, R.; Nakajima, T.; Choyke, P.L.; Kobayashi, H. Near infrared photoimmunotherapy in the treatment of disseminated peritoneal ovarian cancer. Mol. Cancer Ther. 2014, 14, 141–150. [Google Scholar] [CrossRef]
- Sato, K.; Watanabe, R.; Hanaoka, H.; Harada, T.; Nakajima, T.; Kim, I.; Paik, C.H.; Choyke, P.L.; Kobayashi, H. Photoimmunotherapy: Comparative effectiveness of two monoclonal antibodies targeting the epidermal growth factor receptor. Mol. Oncol. 2014, 8, 620–632. [Google Scholar] [CrossRef]
- Nagaya, T.; Nakamura, Y.; Sato, K.; Zhang, Y.F.; Ni, M.; Choyke, P.L.; Ho, M.; Kobayashi, H. Near infrared photoimmunotherapy with an anti-mesothelin antibody. Oncotarget 2016, 7, 23361–23369. [Google Scholar] [CrossRef]
- Sato, K.; Nakajima, T.; Choyke, P.L.; Kobayashi, H. Selective cell elimination in vitro and in vivo from tissues and tumors using antibodies conjugated with a near infrared phthalocyanine. RSC Adv. 2015, 5, 25105–25114. [Google Scholar] [CrossRef]
- Sato, K.; Ando, K.; Okuyama, S.; Moriguchi, S.; Ogura, T.; Totoki, S.; Hanaoka, H.; Nagaya, T.; Kokawa, R.; Takakura, H.; et al. Photoinduced ligand release from a silicon phthalocyanine dye conjugated with monoclonal antibodies: A mechanism of cancer cell cytotoxicity after near-infrared photoimmunotherapy. ACS Cent. Sci. 2018, 4, 1559–1569. [Google Scholar] [CrossRef]
- Kobayashi, H.; Choyke, P.L. Near-infrared photoimmunotherapy of cancer. Acc. Chem. Res. 2019, 52, 2332–2339. [Google Scholar] [CrossRef]
- Isobe, Y.; Sato, K.; Nishinaga, Y.; Takahashi, K.; Taki, S.; Yasui, H.; Shimizu, M.; Endo, R.; Koike, C.; Kuramoto, N.; et al. Near infrared photoimmunotherapy targeting DLL3 for small cell lung cancer. EBioMedicine 2020, 52, 102632. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Nagaya, T.; Mitsunaga, M.; Choyke, P.L.; Kobayashi, H. Near infrared photoimmunotherapy for lung metastases. Cancer Lett. 2015, 365, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Nagaya, T.; Nakamura, Y.; Harada, T.; Choyke, P.L.; Kobayashi, H. Near infrared photoimmunotherapy prevents lung cancer metastases in a murine model. Oncotarget 2015, 6, 19747–19758. [Google Scholar] [CrossRef]
- Sato, K.; Choyke, P.L.; Hisataka, K. Selective cell elimination from mixed 3D culture using a near infrared photoimmunotherapy technique. J. Vis. Exp. 2016, 2016, 8–11. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Gorka, A.P.; Nagaya, T.; Michie, M.S.; Nakamura, Y.; Nani, R.R.; Coble, V.L.; Vasalatiy, O.V.; Swenson, R.E.; Choyke, P.L.; et al. Effect of charge localization on the: In vivo optical imaging properties of near-infrared cyanine dye/monoclonal antibody conjugates. Mol. Biosyst. 2016, 12, 3046–3056. [Google Scholar] [CrossRef]
- Sato, K.; Gorka, A.P.; Nagaya, T.; Michie, M.S.; Nani, R.R.; Nakamura, Y.; Coble, V.L.; Vasalatiy, O.V.; Swenson, R.E.; Choyke, P.L.; et al. Role of fluorophore charge on the in vivo optical imaging properties of near-infrared cyanine dye/monoclonal antibody conjugates. Bioconjug. Chem. 2016, 27, 404–413. [Google Scholar] [CrossRef]
- Sato, K.; Nagaya, T.; Nakamura, Y.; Harada, T.; Nani, R.R.; Shaum, J.B.; Gorka, A.P.; Kim, I.; Paik, C.H.; Choyke, P.L.; et al. Impact of C4’-O-Alkyl linker on in vivo pharmacokinetics of near-infrared cyanine/monoclonal antibody conjugates. Mol. Pharm. 2015, 12, 3303–3311. [Google Scholar] [CrossRef]
- Sato, K.; Choyke, P.L.; Kobayashi, H. Photoimmunotherapy of gastric cancer peritoneal carcinomatosis in a mouse model. PLoS ONE 2014, 9, e113276. [Google Scholar] [CrossRef]
- Sato, K.; Watanabe, T.; Wang, S.; Kakeno, M.; Matsuzawa, K.; Matsui, T.; Yokoi, K.; Murase, K.; Sugiyama, I.; Ozawa, M.; et al. Numb controls E-cadherin endocytosis through p120 catenin with aPKC. Mol. Biol. Cell 2011, 22, 3103–3119. [Google Scholar] [CrossRef]
- Usami, N.; Fukui, T.; Kondo, M.; Taniguchi, T.; Yokoyama, T.; Mori, S.; Yokoi, K.; Horio, Y.; Shimokata, K.; Sekido, Y.; et al. Establishment and characterization of four malignant pleural mesothelioma cell lines from Japanese patients. Cancer Sci. 2006, 97, 387–394. [Google Scholar] [CrossRef]
- Kawaguchi, K.; Murakami, H.; Taniguchi, T.; Fujii, M.; Kawata, S.; Fukui, T.; Kondo, Y.; Osada, H.; Usami, N.; Yokoi, K.; et al. Combined inhibition of MET and EGFR suppresses proliferation of malignant mesothelioma cells. Carcinogenesis 2009, 30, 1097–1105. [Google Scholar] [CrossRef]
- Ordóñez, N.G. D24-0 and podoplanin are highly specific and sensitive immunohistochemical markers of epithelioid malignant mesothelioma. Hum. Pathol. 2005, 36, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Padgett, D.M.; Cathro, H.P.; Wick, M.R.; Mills, S.E. Podoplanin is a better immunohistochemical marker for sarcomatoid mesothelioma than calretinin. Am. J. Surg. Pathol. 2008, 32, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Yang, Q.; McMahon, L.A.; Wang, H.L.; Xu, H. Value of D24-0 in the differential diagnosis of pleural neoplasms with emphasis on its positivity in solitary fibrous tumor. Appl. Immunohistochem. Mol. Morphol. 2010, 18, 411–413. [Google Scholar] [PubMed]
- Zalcman, G.; Mazieres, J.; Margery, J.; Greillier, L.; Audigier-Valette, C.; Moro-Sibilot, D.; Molinier, O.; Corre, R.; Monnet, I.; Gounant, V.; et al. Bevacizumab for newly diagnosed pleural mesothelioma in the Mesothelioma Avastin Cisplatin Pemetrexed Study (MAPS): A randomised, controlled, open-label, phase 3 trial. Lancet 2016, 387, 1405–1414. [Google Scholar] [CrossRef]
- Lievense, L.A.; Sterman, D.H.; Cornelissen, R.; Aerts, J.G. Checkpoint blockade in lung cancer and mesothelioma. Am. J. Respir. Crit. Care Med. 2017, 196, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Balar, A.V.; Weber, J.S. PD-1 and PD-L1 antibodies in cancer: Current status and future directions. Cancer Immunol. Immunother. 2017, 66, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Nicolini, F.; Bocchini, M.; Bronte, G.; Delmonte, A.; Guidoboni, M.; Crinò, L.; Mazza, M. Malignant pleural mesothelioma: State-of-the-art on current therapies and promises for the future. Front. Oncol. 2020, 9. [Google Scholar] [CrossRef]
- Kato, Y.; Kaneko, M.K. A Cancer-specific monoclonal antibody recognizes the aberrantly glycosylated podoplanin. Sci. Rep. 2014, 4, 5924. [Google Scholar] [CrossRef]
- Pula, B.; Witkiewicz, W.; Dziegiel, P.; Podhorska-Okolow, M. Significance of podoplanin expression in cancer-associated fibroblasts: A comprehensive review. Int. J. Oncol. 2013, 42, 1849–1857. [Google Scholar] [CrossRef] [PubMed]
- Ugorski, M.; Dziegiel, P.; Suchanski, J. Podoplanin—A small glycoprotein with many faces. Am. J. Cancer Res. 2016, 6, 370–386. [Google Scholar] [PubMed]
- Kitano, H.; Kageyama, S.I.; Hewitt, S.M.; Hayashi, R.; Doki, Y.; Ozaki, Y.; Fujino, S.; Takikita, M.; Kubo, H.; Fukuoka, J. Podoplanin expression in cancerous stroma induces lymphangiogenesis and predicts lymphatic spread and patient survival. Arch. Pathol. Lab. Med. 2010, 134, 1520–1527. [Google Scholar] [PubMed]
- Danielli, R.; Patuzzo, R.; Ruffini, P.A.; Maurichi, A.; Giovannoni, L.; Elia, G.; Neri, D.; Santinami, M. Armed antibodies for cancer treatment: A promising tool in a changing era. Cancer Immunol. Immunother. 2015, 64, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Sudo, H.; Tsuji, A.B.; Sugyo, A.; Saga, T.; Kaneko, M.K.; Kato, Y.; Higashi, T. Therapeutic efficacy evaluation of radioimmunotherapy with 90Y-labeled anti-podoplanin antibody NZ-12 for mesothelioma. Cancer Sci. 2019, 110, 1653–1664. [Google Scholar] [CrossRef] [PubMed]
- Wayne, A.S.; Fitzgerald, D.J.; Kreitman, R.J.; Pastan, I. Antibody derivatives as new therapeutics for hematologic. Immunotoxins for leukemia. Blood 2015, 123, 2470–2478. [Google Scholar] [CrossRef]
- Pacheco, J.M.; Camidge, D.R. Antibody drug conjugates in thoracic malignancies. Lung Cancer 2018, 124, 260–269. [Google Scholar] [CrossRef]
- Ito, K.; Mitsunaga, M.; Nishimura, T.; Saruta, M.; Iwamoto, T.; Kobayashi, H.; Tajiri, H. Near-Infrared photochemoimmunotherapy by photoactivatable bifunctional antibody-drug conjugates targeting human epidermal growth factor receptor 2 positive cancer. Bioconjug. Chem. 2017, 28, 1458–1469. [Google Scholar] [CrossRef]
- Okada, M.; Kijima, T.; Aoe, K.; Kato, T.; Fujimoto, N.; Nakagawa, K.; Takeda, Y.; Hida, T.; Kanai, K.; Imamura, F.; et al. Clinical efficacy and safety of nivolumab: Results of a multicenter, open-label, single-arm, Japanese phase II study in malignant pleural mesothelioma (MERIT). Clin. Cancer Res. 2019, 25, 5485–5492. [Google Scholar] [CrossRef]
- Sato, K.; Sato, N.; Xu, B.; Nakamura, Y.; Nagaya, T.; Choyke, P.L.; Hasegawa, Y.; Kobayashi, H. Spatially selective depletion of tumor-associated regulatory T cells with near-infrared photoimmunotherapy. Sci. Transl. Med. 2016, 8, 352ra110. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nishinaga, Y.; Sato, K.; Yasui, H.; Taki, S.; Takahashi, K.; Shimizu, M.; Endo, R.; Koike, C.; Kuramoto, N.; Nakamura, S.; et al. Targeted Phototherapy for Malignant Pleural Mesothelioma: Near-Infrared Photoimmunotherapy Targeting Podoplanin. Cells 2020, 9, 1019. https://doi.org/10.3390/cells9041019
Nishinaga Y, Sato K, Yasui H, Taki S, Takahashi K, Shimizu M, Endo R, Koike C, Kuramoto N, Nakamura S, et al. Targeted Phototherapy for Malignant Pleural Mesothelioma: Near-Infrared Photoimmunotherapy Targeting Podoplanin. Cells. 2020; 9(4):1019. https://doi.org/10.3390/cells9041019
Chicago/Turabian StyleNishinaga, Yuko, Kazuhide Sato, Hirotoshi Yasui, Shunichi Taki, Kazuomi Takahashi, Misae Shimizu, Rena Endo, Chiaki Koike, Noriko Kuramoto, Shota Nakamura, and et al. 2020. "Targeted Phototherapy for Malignant Pleural Mesothelioma: Near-Infrared Photoimmunotherapy Targeting Podoplanin" Cells 9, no. 4: 1019. https://doi.org/10.3390/cells9041019
APA StyleNishinaga, Y., Sato, K., Yasui, H., Taki, S., Takahashi, K., Shimizu, M., Endo, R., Koike, C., Kuramoto, N., Nakamura, S., Fukui, T., Yukawa, H., Baba, Y., K. Kaneko, M., Chen-Yoshikawa, T. F., Kobayashi, H., Kato, Y., & Hasegawa, Y. (2020). Targeted Phototherapy for Malignant Pleural Mesothelioma: Near-Infrared Photoimmunotherapy Targeting Podoplanin. Cells, 9(4), 1019. https://doi.org/10.3390/cells9041019