Cardiotoxicity and Heart Failure: Lessons from Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes and Anticancer Drugs


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Anticancer Drug-Induced Cardiotoxicity
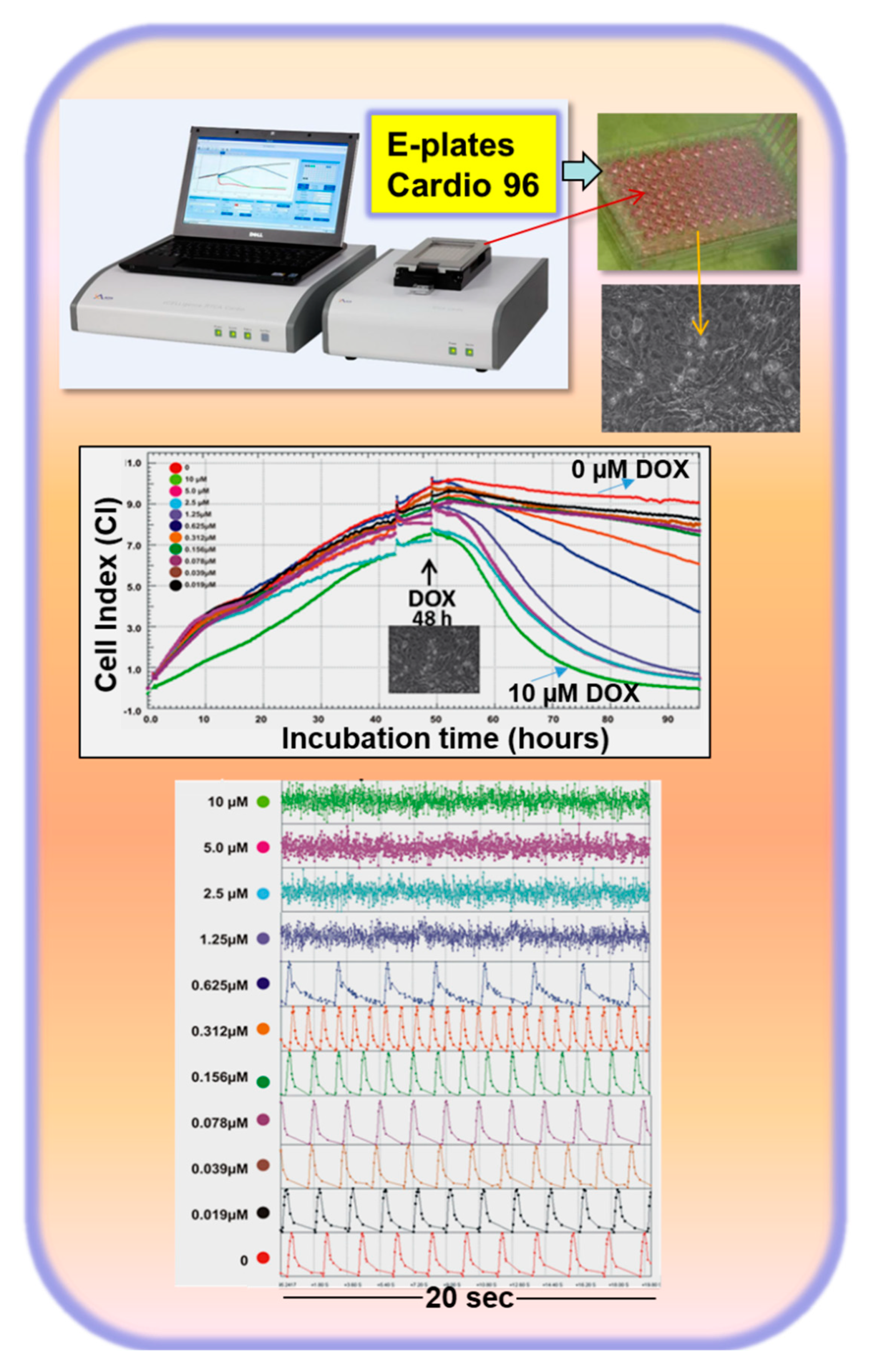
3. Introduction of an In Vitro Cardiotoxic Model to Recapitulate the Mechanisms Involved in the Development of HF
3.1. Identification of Biological Processes and Signal Transduction Pathways by Anticancer Drugs in hiPSC-Derived CMs Applying Transcriptomics
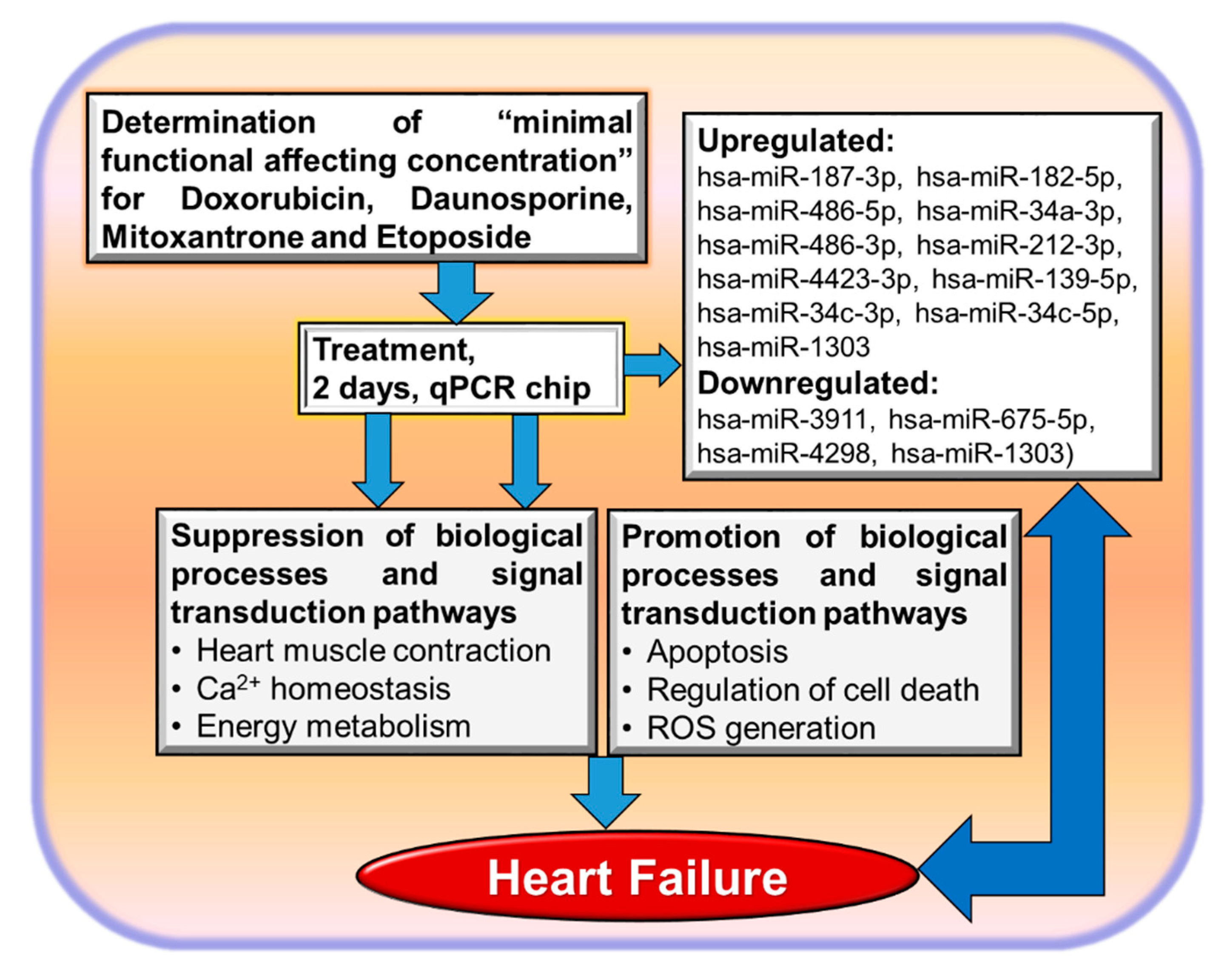
3.2. Non-Coding RNAs and Cardiotoxicity
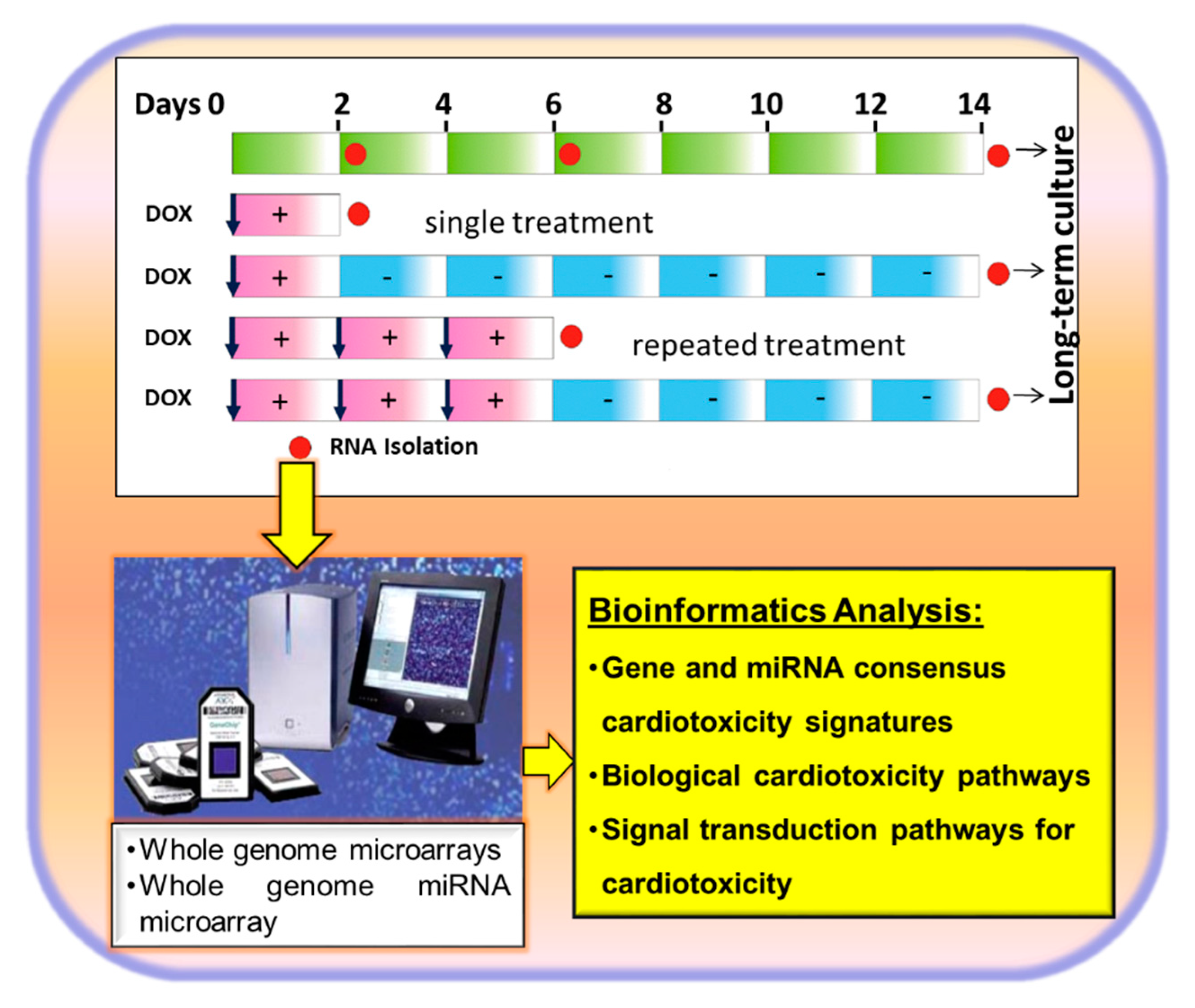
3.3. Transcriptome-Based Quantification of the Cardiotoxicity Capacity of Different Compounds
4. Is It Possible to Identify Common Factors Playing a Role in Heart Failure and Cardiotoxicity Using In Vitro Cardiotoxicity Methods?
5. Conclusions and Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Sachinidis, A.; Albrecht, W.; Nell, P.; Cherianidou, A.; Hewitt, N.J.; Edlund, K.; Hengstler, J.G. Road Map for Development of Stem Cell-Based Alternative Test Methods. Trends Mol. Med. 2019, 25, 470–481. [Google Scholar] [CrossRef]
- Doss, M.X.; Sachinidis, A. Current Challenges of iPSC-Based Disease Modeling and Therapeutic Implications. Cells 2019, 8, 403. [Google Scholar] [CrossRef]
- Lazzarini, V.; Mentz, R.J.; Fiuzat, M.; Metra, M.; O‘Connor, C.M. Heart failure in elderly patients: Distinctive features and unresolved issues. Eur. J. Heart Fail. 2013, 15, 717–723. [Google Scholar] [CrossRef]
- Kostin, S. Types of cardiomyocyte death and clinical outcomes in patients with heart failure. J. Am. Coll. Cardiol. 2011, 57, 1532–1534. [Google Scholar] [CrossRef]
- McMurray, J.J.; Adamopoulos, S.; Anker, S.D.; Auricchio, A.; Bohm, M.; Dickstein, K.; Falk, V.; Filippatos, G.; Fonseca, C.; Gomez-Sanchez, M.A.; et al. ESC guidelines for the diagnosis and treatment of acute and chronic heart failure 2012: The Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2012 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association (HFA) of the ESC. Eur. J. Heart Fail. 2012, 14, 803–869. [Google Scholar] [CrossRef] [PubMed]
- Upadhya, B.; Kitzman, D.W. Heart failure with preserved ejection fraction: New approaches to diagnosis and management. Clin. Cardiol. 2020, 43, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Ciutac, A.M.; Dawson, D. The role of inflammation in stress cardiomyopathy. Trends Cardiovasc. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Cojan-Minzat, B.O.; Zlibut, A.; Agoston-Coldea, L. Non-ischemic dilated cardiomyopathy and cardiac fibrosis. Heart Fail. Rev. 2020. [Google Scholar] [CrossRef] [PubMed]
- Harvey, P.A.; Leinwand, L.A. The cell biology of disease: Cellular mechanisms of cardiomyopathy. J. Cell Biol. 2011, 194, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Chiong, M.; Wang, Z.V.; Pedrozo, Z.; Cao, D.J.; Troncoso, R.; Ibacache, M.; Criollo, A.; Nemchenko, A.; Hill, J.A.; Lavandero, S. Cardiomyocyte death: Mechanisms and translational implications. Cell Death Dis. 2011, 2, e244. [Google Scholar] [CrossRef]
- Zeitz, M.J.; Smyth, J.W. Translating Translation to Mechanisms of Cardiac Hypertrophy. J Cardiovasc Dev Dis. 2020, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Menna, P.; Paz, O.G.; Chello, M.; Covino, E.; Salvatorelli, E.; Minotti, G. Anthracycline cardiotoxicity. Expert Opin. Drug Saf. 2012, 11 (Suppl. 1), S21–S36. [Google Scholar] [CrossRef] [PubMed]
- Agunbiade, T.A.; Zaghlol, R.Y.; Barac, A. Heart Failure in Relation to Anthracyclines and Other Chemotherapies. Methodist Debakey Cardiovasc. J. 2019, 15, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Cardinale, D.; Iacopo, F.; Cipolla, C.M. Cardiotoxicity of Anthracyclines. Front. Cardiovasc. Med. 2020, 7, 26. [Google Scholar] [CrossRef]
- Singal, P.K.; Iliskovic, N. Doxorubicin-induced cardiomyopathy. N. Engl. J. Med. 1998, 339, 900–905. [Google Scholar] [CrossRef]
- Stewart, D.J.; Grewaal, D.; Green, R.M.; Mikhael, N.; Goel, R.; Montpetit, V.A.; Redmond, M.D. Concentrations of doxorubicin and its metabolites in human autopsy heart and other tissues. Anticancer Res. 1993, 13, 1945–1952. [Google Scholar]
- Smith, L.A.; Cornelius, V.R.; Plummer, C.J.; Levitt, G.; Verrill, M.; Canney, P.; Jones, A. Cardiotoxicity of anthracycline agents for the treatment of cancer: Systematic review and meta-analysis of randomised controlled trials. BMC Cancer 2010, 10, 337. [Google Scholar] [CrossRef]
- Chatterjee, K.; Zhang, J.; Honbo, N.; Karliner, J.S. Doxorubicin Cardiomyopathy. Cardiology 2010, 115, 155–162. [Google Scholar] [CrossRef]
- Qiu, Z.; Wei, Y.; Song, Q.; Du, B.; Wang, H.; Chu, Y.; Hu, Y. The Role of Myocardial Mitochondrial Quality Control in Heart Failure. Front. Pharmacol. 2019, 10, 1404. [Google Scholar] [CrossRef]
- O’Brien, P.J. Cardiac troponin is the most effective translational safety biomarker for myocardial injury in cardiotoxicity. Toxicology 2008, 245, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, H.; Maddock, H. Molecular basis of cancer-therapy-induced cardiotoxicity: Introducing microRNA biomarkers for early assessment of subclinical myocardial injury. Clin. Sci. 2014, 126, 377–400. [Google Scholar] [CrossRef] [PubMed]
- Prathumsap, N.; Shinlapawittayatorn, K.; Chattipakorn, S.C.; Chattipakorn, N. Effects of doxorubicin on the heart: From molecular mechanisms to intervention strategies. Eur. J. Pharmacol. 2020, 866, 172818. [Google Scholar] [CrossRef] [PubMed]
- Wenningmann, N.; Knapp, M.; Ande, A.; Vaidya, T.R.; Ait-Oudhia, S. Insights into Doxorubicin-induced Cardiotoxicity: Molecular Mechanisms, Preventive Strategies, and Early Monitoring. Mol. Pharmacol. 2019, 96, 219–232. [Google Scholar] [CrossRef]
- Anson, B.D.; Kolaja, K.L.; Kamp, T.J. Opportunities for Use of Human iPS Cells in Predictive Toxicology. Clin. Pharmacol. Ther. 2011, 89, 754–758. [Google Scholar] [CrossRef]
- Meganathan, K.; Jagtap, S.; Wagh, V.; Winkler, J.; Gaspar, J.A.; Hildebrand, D.; Trusch, M.; Lehmann, K.; Hescheler, J.; Schluter, H.; et al. Identification of thalidomide-specific transcriptomics and proteomics signatures during differentiation of human embryonic stem cells. PLoS ONE 2012, 7, e44228. [Google Scholar] [CrossRef]
- Shinde, V.; Hoelting, L.; Srinivasan, S.P.; Meisig, J.; Meganathan, K.; Jagtap, S.; Grinberg, M.; Liebing, J.; Bluethgen, N.; Rahnenfuhrer, J.; et al. Definition of transcriptome-based indices for quantitative characterization of chemically disturbed stem cell development: Introduction of the STOP-Toxukn and STOP-Toxukk tests. Arch. Toxicol. 2017, 91, 839–864. [Google Scholar] [CrossRef]
- Shinde, V.; Perumal Srinivasan, S.; Henry, M.; Rotshteyn, T.; Hescheler, J.; Rahnenfuhrer, J.; Grinberg, M.; Meisig, J.; Bluthgen, N.; Waldmann, T.; et al. Comparison of a teratogenic transcriptome-based predictive test based on human embryonic versus inducible pluripotent stem cells. Stem Cell Res. Ther. 2016, 7, 190. [Google Scholar] [CrossRef]
- Meganathan, K.; Jagtap, S.; Srinivasan, S.P.; Wagh, V.; Hescheler, J.; Hengstler, J.; Leist, M.; Sachinidis, A. Neuronal developmental gene and miRNA signatures induced by histone deacetylase inhibitors in human embryonic stem cells. Cell Death Dis. 2015, 6, e1756. [Google Scholar] [CrossRef]
- Tafuri, G.; Trotta, F.; Leufkens, H.G.M.; Pani, L. Disclosure of grounds of European withdrawn and refused applications: A step forward on regulatory transparency. Br. J. Clin. Pharmacol. 2013, 75, 1149–1151. [Google Scholar] [CrossRef]
- Shinde, V.; Sureshkumar, P.; Sotiriadou, I.; Hescheler, J.; Sachinidis, A. Human Embryonic and Induced Pluripotent Stem Cell Based Toxicity Testing Models: Future Applications in New Drug Discovery. Curr. Med. Chem. 2016, 23, 3495–3509. [Google Scholar] [CrossRef] [PubMed]
- del Álamo, J.C.; Lemons, D.; Serrano, R.; Savchenko, A.; Cerignoli, F.; Bodmer, R.; Mercola, M. High throughput physiological screening of iPSC-derived cardiomyocytes for drug development. Biochim. Biophys. Acta Mol. Cell Res. 2016, 1863, 1717–1727. [Google Scholar] [CrossRef] [PubMed]
- Burridge, P.W.; Matsa, E.; Shukla, P.; Lin, Z.C.; Churko, J.M.; Ebert, A.D.; Lan, F.; Diecke, S.; Huber, B.; Mordwinkin, N.M.; et al. Chemically defined generation of human cardiomyocytes. Nat. Methods 2014, 11, 855–860. [Google Scholar] [CrossRef] [PubMed]
- Lian, X.; Hsiao, C.; Wilson, G.; Zhu, K.; Hazeltine, L.B.; Azarin, S.M.; Raval, K.K.; Zhang, J.; Kamp, T.J.; Palecek, S.P. Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc. Natl. Acad. Sci. USA 2012, 109, E1848–E1857. [Google Scholar] [CrossRef]
- Hirt, M.N.; Hansen, A.; Eschenhagen, T. Cardiac Tissue Engineering State of the Art. Circ. Res. 2014, 114, 354–367. [Google Scholar] [CrossRef]
- Matsa, E.; Burridge, P.W.; Wu, J.C. Human Stem Cells for Modeling Heart Disease and for Drug Discovery. Sci. Trans. Med. 2014, 6, 239ps6. [Google Scholar] [CrossRef]
- Chaudhari, U.; Nemade, H.; Gaspar, J.A.; Hescheler, J.; Hengstler, J.G.; Sachinidis, A. MicroRNAs as early toxicity signatures of doxorubicin in human-induced pluripotent stem cell-derived cardiomyocytes. Arch. Toxicol. 2016, 90, 3087–3098. [Google Scholar] [CrossRef]
- Chaudhari, U.; Nemade, H.; Sureshkumar, P.; Vinken, M.; Ates, G.; Rogiers, V.; Hescheler, J.; Hengstler, J.G.; Sachinidis, A. Functional cardiotoxicity assessment of cosmetic compounds using human-induced pluripotent stem cell-derived cardiomyocytes. Arch. Toxicol. 2018, 92, 371–381. [Google Scholar] [CrossRef]
- Chaudhari, U.; Nemade, H.; Wagh, V.; Gaspar, J.A.; Ellis, J.K.; Srinivasan, S.P.; Spitkovski, D.; Nguemo, F.; Louisse, J.; Bremer, S.; et al. Identification of genomic biomarkers for anthracycline-induced cardiotoxicity in human iPSC-derived cardiomyocytes: An in vitro repeated exposure toxicity approach for safety assessment. Arch. Toxicol. 2016, 90, 2763–2777. [Google Scholar] [CrossRef]
- Clements, M.; Thomas, N. High-Throughput Multi-Parameter Profiling of Electrophysiological Drug Effects in Human Embryonic Stem Cell Derived Cardiomyocytes Using Multi-Electrode Arrays. Toxicol. Sci. 2014, 140, 445–461. [Google Scholar] [CrossRef]
- Nemade, H.; Chaudhari, U.; Acharya, A.; Hescheler, J.; Hengstler, J.G.; Papadopoulos, S.; Sachinidis, A. Cell death mechanisms of the anti-cancer drug etoposide on human cardiomyocytes isolated from pluripotent stem cells. Arch. Toxicol. 2018, 92, 1507–1524. [Google Scholar] [CrossRef] [PubMed]
- Matsa, E.; Rajamohan, D.; Dick, E.; Young, L.; Mellor, I.; Staniforth, A.; Denning, C. Drug evaluation in cardiomyocytes derived from human induced pluripotent stem cells carrying a long QT syndrome type 2 mutation. Eur. Heart J. 2011, 32, 952–962. [Google Scholar] [CrossRef] [PubMed]
- Liang, P.; Lan, F.; Lee, A.S.; Gong, T.; Sanchez-Freire, V.; Wang, Y.; Diecke , S.; Sallam, K.; Knowles, J.W.; Wang, P.J.; et al. Drug screening using a library of human induced pluripotent stem cell-derived cardiomyocytes reveals disease-specific patterns of cardiotoxicity. Circulation 2013, 127, 1677–1691. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Srivastava, D. A developmental view of microRNA function. Trends Biochem. Sci. 2007, 32, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Barraclough, J.Y.; Joan, M.; Joglekar, M.V.; Hardikar, A.A.; Patel, S. MicroRNAs as Prognostic Markers in Acute Coronary Syndrome Patients-A Systematic Review. Cells 2019, 8, 1572. [Google Scholar] [CrossRef]
- Mirna, M.; Paar, V.; Rezar, R.; Topf, A.; Eber, M.; Hoppe, U.C.; Lichtenauer, M.; Jung, C. MicroRNAs in Inflammatory Heart Diseases and Sepsis-Induced Cardiac Dysfunction: A Potential Scope for the Future? Cells 2019, 8, 1352. [Google Scholar] [CrossRef]
- Pang, J.K.S.; Phua, Q.H.; Soh, B.S. Applications of miRNAs in cardiac development, disease progression and regeneration. Stem Cell Res. Ther. 2019, 10, 336. [Google Scholar] [CrossRef]
- Chan, M.M.; Santhanakrishnan, R.; Chong, J.P.; Chen, Z.; Tai, B.C.; Liew, O.W.; Ng, T.P.; Ling, L.H.; Sim, D.; Leong, K.T.; et al. Growth differentiation factor 15 in heart failure with preserved vs. reduced ejection fraction. Eur. J. Heart Fail. 2016, 18, 81–88. [Google Scholar] [CrossRef]
- Goletti, S.; Gruson, D. Personalized risk assessment of heart failure patients: More perspectives from transforming growth factor super-family members. Clin. Chim. Acta 2015, 443, 94–99. [Google Scholar] [CrossRef]
- Wollert, K.C.; Kempf, T. Growth differentiation factor 15 in heart failure: An update. Curr. Heart Fail. Rep. 2012, 9, 337–345. [Google Scholar] [CrossRef]
- Wiklund, F.E.; Bennet, A.M.; Magnusson, P.K.; Eriksson, U.K.; Lindmark, F.; Wu, L.; Yaghoutyfam, N.; Marquis, C.P.; Stattin, P.; Pedersen, N.L.; et al. Macrophage inhibitory cytokine-1 (MIC-1/GDF15): A new marker of all-cause mortality. Aging Cell 2010, 9, 1057–1064. [Google Scholar] [CrossRef] [PubMed]
- Bettencourt, P.; Ferreira-Coimbra, J.; Rodrigues, P.; Marques, P.; Moreira, H.; Pinto, M.J.; Guimaraes, J.T.; Lourenco, P. Towards a multi-marker prognostic strategy in acute heart failure: A role for GDF-15. ESC Heart Fail. 2018, 5, 1017–1022. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, E.C.; Koniaris, L.G.; Zimmers-Koniaris, T.; Sebald, S.M.; Huynh, T.V.; Lee, S.J. Characterization of growth-differentiation factor 15, a transforming growth factor beta superfamily member induced following liver injury. Mol. Cell. Biol. 2000, 20, 3742–3751. [Google Scholar] [CrossRef] [PubMed]
- Zimmers, T.A.; Jim, X.L.; Hsiao, E.C.; McGrath, S.A.; Esquela, A.F.; Koniaris, L.G. Growth differentiation factor-15/macrophage inhibitory cytokine-1 induction after kidney and lung injury. Shock 2005, 23, 543–548. [Google Scholar] [PubMed]
- Yatsuga, S.; Fujita, Y.; Ishii, A.; Fukumoto, Y.; Arahata, H.; Kakuma, T.; Kojima, T.; Ito, M.; Tanaka, M.; Saiki, R.; et al. Growth differentiation factor 15 as a useful biomarker for mitochondrial disorders. Ann. Neurol. 2015, 78, 814–823. [Google Scholar] [CrossRef]
- Fujita, Y.; Taniguchi, Y.; Shinkai, S.; Tanaka, M.; Ito, M. Secreted growth differentiation factor 15 as a potential biomarker for mitochondrial dysfunctions in aging and age-related disorders. Geriatr Gerontol. Int. 2016, 16 (Suppl. 1), 17–29. [Google Scholar] [CrossRef]
- Liu, X.; Chua, C.C.; Gao, J.; Chen, Z.; Landy, C.L.; Hamdy, R.; Chua, B.H. Pifithrin-alpha protects against doxorubicin-induced apoptosis and acute cardiotoxicity in mice. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H933–H939. [Google Scholar] [CrossRef]
- Li, J.; Ghiani, C.A.; Kim, J.Y.; Liu, A.; Sandoval, J.; DeVellis, J.; Casaccia-Bonnefil, P. Inhibition of p53 transcriptional activity: A potential target for future development of therapeutic strategies for primary demyelination. J. Neurosci. 2008, 28, 6118–6127. [Google Scholar] [CrossRef]
- Li, Y.-L.; Chang, J.T.; Lee, L.-Y.; Fan, K.-H.; Lu, Y.-C.; Li, Y.-C.; Chiang, C.-H.; You, G.-R.; Chen, H.-Y.; Cheng, A.-J. GDF15 contributes to radioresistance and cancer stemness of head and neck cancer by regulating cellular reactive oxygen species via a SMAD-associated signaling pathway. Oncotarget 2017, 8, 1508–1528. [Google Scholar] [CrossRef]
- Li, J.; Yang, L.; Qin, W.; Zhang, G.; Yuan, J.; Wang, F. Adaptive Induction of Growth Differentiation Factor 15 Attenuates Endothelial Cell Apoptosis in Response to High Glucose Stimulus. PLoS ONE 2013, 8, e65549. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Madungwe, N.B.; Imam Aliagan, A.D.; Tombo, N.; Bopassa, J.C. Liproxstatin-1 protects the mouse myocardium against ischemia/reperfusion injury by decreasing VDAC1 levels and restoring GPX4 levels. Biochem. Biophys. Res. Commun. 2019, 520, 606–611. [Google Scholar] [CrossRef]
- Zilka, O.; Shah, R.; Li, B.; Friedmann Angeli, J.P.; Griesser, M.; Conrad, M.; Pratt, D.A. On the Mechanism of Cytoprotection by Ferrostatin-1 and Liproxstatin-1 and the Role of Lipid Peroxidation in Ferroptotic Cell Death. ACS Cent. Sci. 2017, 3, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Abulizi, P.; Loganathan, N.; Zhao, D.; Mele, T.; Zhang, Y.; Zwiep, T.; Liu, K.; Zheng, X. Growth Differentiation Factor-15 Deficiency Augments Inflammatory Response and Exacerbates Septic Heart and Renal Injury Induced by Lipopolysaccharide. Sci. Rep. 2017, 7, 1037. [Google Scholar] [CrossRef] [PubMed]
- Vegter, E.L.; van der Meer, P.; de Windt, L.J.; Pinto, Y.M.; Voors, A.A. MicroRNAs in heart failure: From biomarker to target for therapy. Eur. J. Heart Fail. 2016, 18, 457–468. [Google Scholar] [CrossRef]
- Elzenaar, I.; Pinto, Y.M.; van Oort, R.J. MicroRNAs in heart failure: New targets in disease management. Clin. Pharmacol. Ther. 2013, 94, 480–489. [Google Scholar] [CrossRef]
- Bernardo, B.C.; Gao, X.M.; Winbanks, C.E.; Boey, E.J.; Tham, Y.K.; Kiriazis, H.; Gregorevic, P.; Obad, S.; Kauppinen, S.; Du, X.J.; et al. Therapeutic inhibition of the miR-34 family attenuates pathological cardiac remodeling and improves heart function. Proc. Natl. Acad. Sci. USA 2012, 109, 17615–17620. [Google Scholar] [CrossRef]
- Greco, S.; Fasanaro, P.; Castelvecchio, S.; D’Alessandra, Y.; Arcelli, D.; Di Donato, M.; Malavazos, A.; Capogrossi, M.C.; Menicanti, L.; Martelli, F. MicroRNA dysregulation in diabetic ischemic heart failure patients. Diabetes 2012, 61, 1633–1641. [Google Scholar] [CrossRef]
- Matsumoto, S.; Sakata, Y.; Suna, S.; Nakatani, D.; Usami, M.; Hara, M.; Kitamura, T.; Hamasaki, T.; Nanto, S.; Kawahara, Y.; et al. Circulating p53-responsive microRNAs are predictive indicators of heart failure after acute myocardial infarction. Circ. Res. 2013, 113, 322–326. [Google Scholar] [CrossRef]
- Ruggeri, C.; Gioffre, S.; Achilli, F.; Colombo, G.I.; D’Alessandra, Y. Role of microRNAs in doxorubicin-induced cardiotoxicity: An overview of preclinical models anPratd cancer patients. Heart Fail. Rev. 2018, 23, 109–122. [Google Scholar] [CrossRef]



© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sachinidis, A. Cardiotoxicity and Heart Failure: Lessons from Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes and Anticancer Drugs. Cells 2020, 9, 1001. https://doi.org/10.3390/cells9041001
Sachinidis A. Cardiotoxicity and Heart Failure: Lessons from Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes and Anticancer Drugs. Cells. 2020; 9(4):1001. https://doi.org/10.3390/cells9041001
Chicago/Turabian StyleSachinidis, Agapios. 2020. "Cardiotoxicity and Heart Failure: Lessons from Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes and Anticancer Drugs" Cells 9, no. 4: 1001. https://doi.org/10.3390/cells9041001
APA StyleSachinidis, A. (2020). Cardiotoxicity and Heart Failure: Lessons from Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes and Anticancer Drugs. Cells, 9(4), 1001. https://doi.org/10.3390/cells9041001