The Immune Response Against Human Cytomegalovirus Links Cellular to Systemic Senescence


{kind=link}
{kind=link}
Abstract
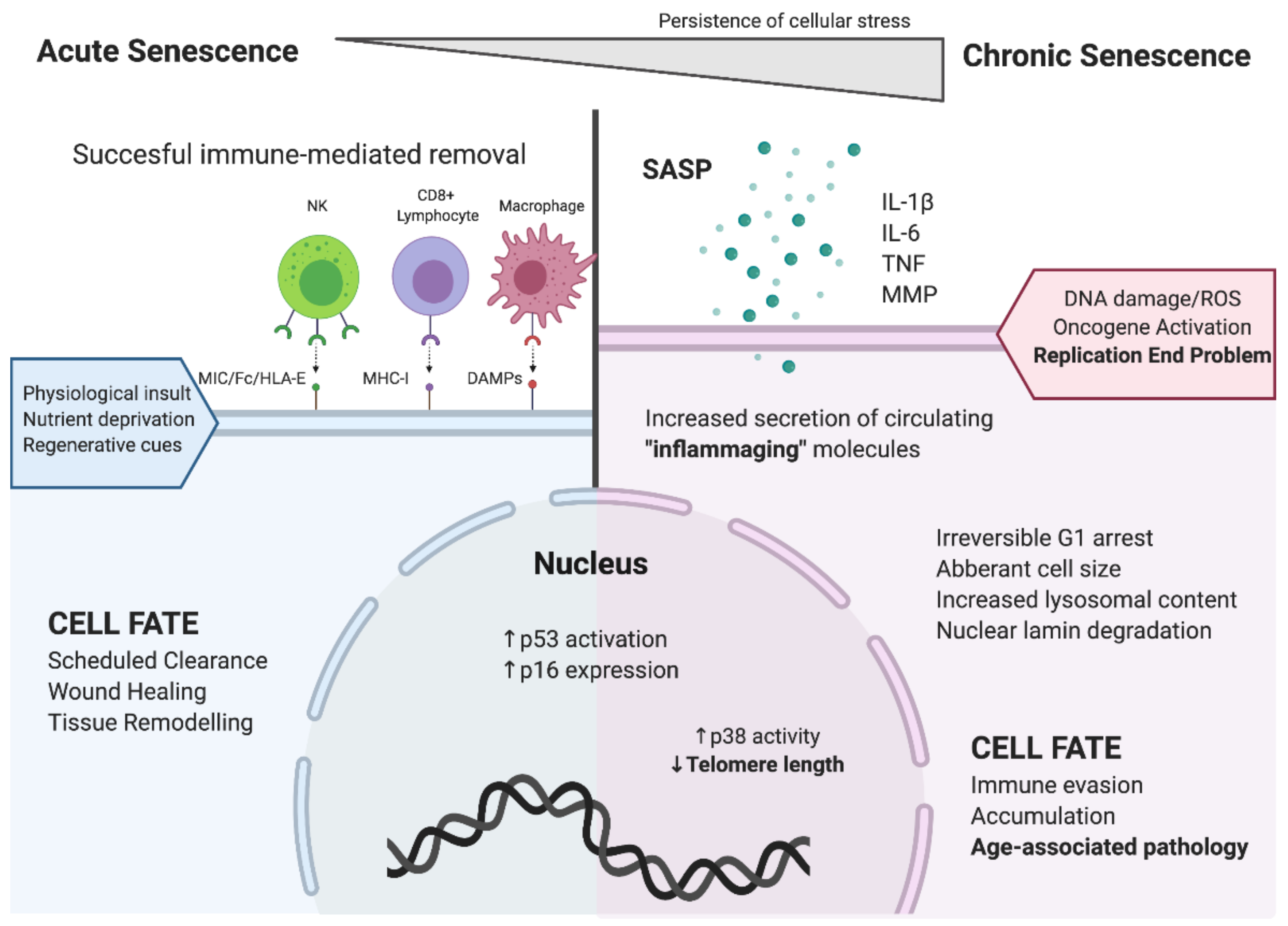
1. Cellular Senescence
2. Inflammaging
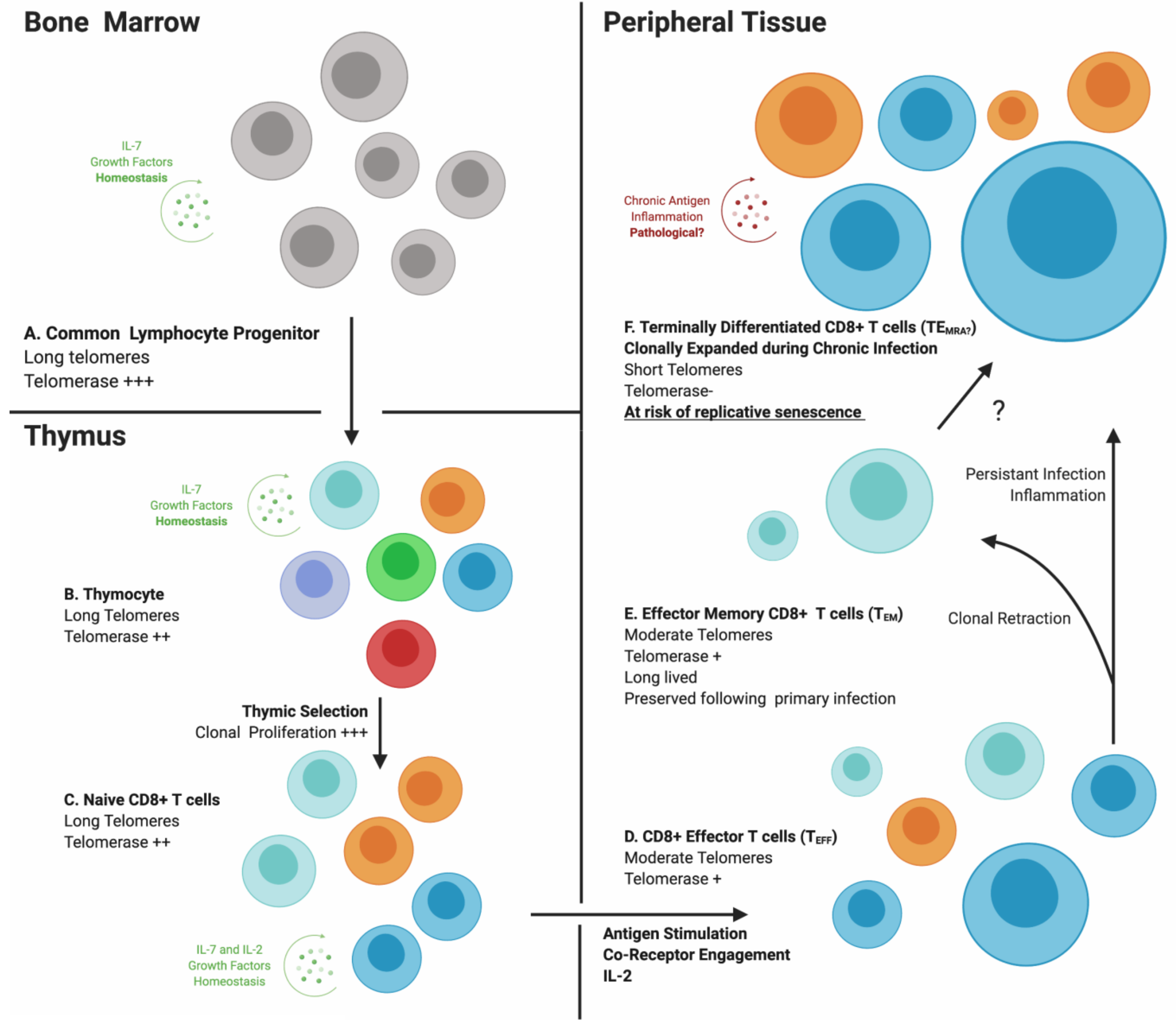
3. Immunosenescence
4. Cytomegalovirus Infection Generates a Unique CD8+ T-Cell Subset
5. CMV-Specific CD8+ T Cells: The Link Between CMV Infection and Age-Related Morbidity
6. HIV-Associated “Premature” Aging and CMV Co-Infection
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Hayflick, L. The limited in vitro lifetime of human diploid cell strains. Exp. Cell Res. 1965, 37, 614–636. [Google Scholar] [CrossRef]
- Coppé, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef]
- Howcroft, T.K.; Campisi, J.; Louis, G.B.; Smith, M.T.; Wise, B.; Wyss-Coray, T.; Augustine, A.D.; McElhaney, J.E.; Kohanski, R.; Sierra, F. The role of inflammation in age-related disease. Aging 2013, 5, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Eming, S.A.; Martin, P.; Tomic-Canic, M. Wound repair and regeneration: Mechanisms, signaling, and translation. Sci. Transl. Med. 2014, 6, 265sr6. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Espín, D.; Cañamero, M.; Maraver, A.; Gómez-López, G.; Contreras, J.; Murillo-Cuesta, S.; Rodríguez-Baeza, A.; Varela-Nieto, I.; Ruberte, J.; Collado, M.; et al. Programmed cell senescence during mammalian embryonic development. Cell 2013, 155, 1104–1118. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, S.; Long, E.O. Cellular senescence induced by CD158d reprograms natural killer cells to promote vascular remodeling. Proc. Natl. Acad. Sci. USA 2012, 109, 20596–20601. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, S. HLA-G-mediated NK cell senescence promotes vascular remodeling: Implications for reproduction. Cell. Mol. Immunol. 2014, 11, 460–466. [Google Scholar] [CrossRef]
- Harley, C.B.; Futcher, A.B.; Greider, C.W. Telomeres shorten during ageing of human fibroblasts. Nature 1990, 345, 458–460. [Google Scholar] [CrossRef]
- Hewitt, G.; Jurk, D.; Marques, F.D.; Correia-Melo, C.; Hardy, T.; Gackowska, A.; Anderson, R.; Taschuk, M.; Mann, J.; Passos, J.F. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat. Commun. 2012, 3, 708. [Google Scholar] [CrossRef]
- Friedrich, U.; Griese, E.-U.; Schwab, M.; Fritz, P.; Thon, K.-P.; Klotz, U. Telomere length in different tissues of elderly patients. Mech. Ageing Dev. 2000, 119, 89–99. [Google Scholar] [CrossRef]
- Okuda, K.; Bardeguez, A.; Gardner, J.P.; Rodriguez, P.; Ganesh, V.; Kimura, M.; Skurnick, J.; Awad, G.; Aviv, A. Telomere Length in the Newborn. Pediatr. Res. 2002, 52, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.Z.; Allsopp, R.C.; Futcher, A.B.; Greider, C.W.; Harley, C.B. Telomere end-replication problem and cell aging. J. Mol. Biol. 1992, 225, 951–960. [Google Scholar] [CrossRef]
- Wright, W.E.; Piatyszek, M.A.; Rainey, W.E.; Byrd, W.; Shay, J.W. Telomerase activity in human germline and embryonic tissues and cells. Dev. Genet. 1996, 18, 173–179. [Google Scholar] [CrossRef]
- Yasumoto, S.; Kunimura, C.; Kikuchi, K.; Tahara, H.; Ohji, H.; Yamamoto, H.; Ide, T.; Utakoji, T. Telomerase activity in normal human epithelial cells. Oncogene 1996, 13, 433–439. [Google Scholar]
- Hiyama, K.; Hirai, Y.; Kyoizumi, S.; Akiyama, M.; Hiyama, E.; Piatyszek, M.A.; Shay, J.W.; Ishioka, S.; Yamakido, M. Activation of telomerase in human lymphocytes and hematopoietic progenitor cells. J. Immunol. 1995, 155, 3711–3715. [Google Scholar]
- Hu, B.T.; Lee, S.C.; Marin, E.; Ryan, D.H.; Insel, R.A. Telomerase is up-regulated in human germinal center B cells in vivo and can be re-expressed in memory B cells activated in vitro. J. Immunol. 1997, 159, 1068–1071. [Google Scholar]
- Rodier, F.; Coppé, J.-P.; Patil, C.K.; Hoeijmakers, W.A.; Muñoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 2009, 11, 973–979. [Google Scholar] [CrossRef]
- Chung, H.Y.; Cesari, M.; Anton, S.; Marzetti, E.; Giovannini, S.; Seo, A.Y.; Carter, C.; Yu, B.P.; Leeuwenburgh, C. Molecular inflammation: Underpinnings of aging and age-related diseases. Ageing Res. Rev. 2009, 8, 18–30. [Google Scholar] [CrossRef]
- Freund, A.; Orjalo, A.V.; Desprez, P.Y.; Campisi, J. Inflammatory networks during cellular senescence: Causes and consequences. Trends Mol. Med. 2010, 16, 238–246. [Google Scholar] [CrossRef]
- Zhang, J.; Rane, G.; Dai, X.; Shanmugam, M.K.; Arfuso, F.; Samy, R.P.; Lai, M.K.; Kappei, D.; Kumar, A.P.; Sethi, G. Ageing and the telomere connection: An intimate relationship with inflammation. Ageing Res. Rev. 2016, 25, 55–69. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, J.; Ramsey, M.R.; Ligon, K.L.; Torrice, C.; Koh, A.; Bonner-Weir, S.; Sharpless, N.E. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature 2006, 443, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Le, O.N.; Rodier, F.; Fontaine, F.; Coppe, J.P.; Campisi, J.; DeGregori, J.; Laverdière, C.; Kokta, V.; Haddad, E.; Beauséjour, C.M. Ionizing radiation-induced long-term expression of senescence markers in mice is independent of p53 and immune status. Aging Cell 2010, 9, 398–409. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.P.; Rodier, F.; Patil, C.K.; Freund, A.; Desprez, P.Y.; Campisi, J. Tumor suppressor and aging biomarker p16(INK4a) induces cellular senescence without the associated inflammatory secretory phenotype. J. Biol. Chem. 2011, 286, 36396–36403. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; Van de Sluis, B.; Kirkland, J.L.; Van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef]
- Franceschi, C.; Campisi, J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. A. Biol. Sci. Med. Sci. 2014, 69, S4–S9. [Google Scholar] [CrossRef]
- Libby, P.; Okamoto, Y.; Rocha, V.Z.; Folco, E. Inflammation in atherosclerosis: Transition from theory to practice. Circ. J. 2010, 74, 213–220. [Google Scholar] [CrossRef]
- Parrinello, S.; Coppe, J.-P.; Krtolica, A.; Campisi, J. Stromal-epithelial interactions in aging and cancer: Senescent fibroblasts alter epithelial cell differentiation. J. Cell Sci. 2005, 118, 485–496. [Google Scholar] [CrossRef]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Invest. 2003, 112, 1821–1830. [Google Scholar] [CrossRef]
- Tracy, R.P. Emerging relationships of inflammation, cardiovascular disease and chronic diseases of aging. Int. J. Obes. Relat. Metab. Disord. 2003, 27, S29–S34. [Google Scholar] [CrossRef]
- Galkina, E.; Ley, K. Immune and inflammatory mechanisms of atherosclerosis (*). Annu. Rev. Immunol. 2009, 27, 165–197. [Google Scholar] [CrossRef] [PubMed]
- Abu-Remaileh, M.; Bender, S.; Raddatz, G.; Ansari, I.; Cohen, D.; Gutekunst, J.; Musch, T.; Linhart, H.; Breiling, A.; Pikarsky, E.; et al. Chronic inflammation induces a novel epigenetic program that is conserved in intestinal adenomas and in colorectal cancer. Cancer Res. 2015, 75, 2120–2130. [Google Scholar] [CrossRef]
- Franceschi, C.; Capri, M.; Monti, D.; Giunta, S.; Olivieri, F.; Sevini, F.; Panourgia, M.P.; Invidia, L.; Celani, L.; Scurti, M.; et al. Inflammaging and anti-inflammaging: A systemic perspective on aging and longevity emerged from studies in humans. Mech. Ageing Dev. 2007, 128, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, D.B.; Firth, C.M.; Phillips, A.C.; Moss, P.; Baylis, D.; Syddall, H.; Sayer, A.A.; Cooper, C.; Lord, J.M. The age-related increase in low-grade systemic inflammation (Inflammaging) is not driven by cytomegalovirus infection. Aging Cell 2012, 11, 912–915. [Google Scholar] [CrossRef] [PubMed]
- Ovadya, Y.; Landsberger, T.; Leins, H.; Vadai, E.; Gal, H.; Biran, A.; Yosef, R.; Sagiv, A.; Agrawal, A.; Shapira, A.; et al. Impaired immune surveillance accelerates accumulation of senescent cells and aging. Nat. Commun. 2018, 9, 5435. [Google Scholar] [CrossRef] [PubMed]
- Makinodan, T.; Kay, M.M.B. Age influence on the immune system. Adv. Immunol. 1980, 29, 287–330. [Google Scholar]
- Pawelec, G.; Adibzadeh, M.; Pohla, H.; Schaudt, K. Immunosenescence: Ageing of the immune system. Immunol. Today 1995, 16, 420–422. [Google Scholar] [CrossRef]
- Franceschi, C.; Valensin, S.; Fagnoni, F.; Barbi, C.; Bonafè, M. Biomarkers of immunosenescence within an evolutionary perspective: The challenge of heterogeneity and the role of antigenic load. Exp. Gerontol. 1999, 34, 911–921. [Google Scholar] [CrossRef]
- Gray, D.; Abramson, J.; Benoist, C.; Mathis, D. Proliferative arrest and rapid turnover of thymic epithelial cells expressing Aire. J. Exp. Med. 2007, 204, 2521–2528. [Google Scholar] [CrossRef]
- Nasi, M.; Troiano, L.; Lugli, E.; Pinti, M.; Ferraresi, R.; Monterastelli, E.; Mussi, C.; Salvioli, G.; Franceschi, C.; Cossarizza, A. Thymic output and functionality of the IL-7/IL-7 receptor system in centenarians: Implications for the neolymphogenesis at the limit of human life. Aging Cell 2006, 5, 167–175. [Google Scholar] [CrossRef]
- Yang, Y.; An, J.; Weng, N.-P. Telomerase is involved in IL-7-mediated differential survival of naive and memory CD4+ T cells. J. Immunol. 2008, 180, 3775–3781. [Google Scholar] [CrossRef] [PubMed]
- Yamada, O.; Motoji, T.; Mizoguchi, H. Up-regulation of telomerase activity in human lymphocytes. Biochim. Biophys. Acta 1996, 1314, 260–266. [Google Scholar] [CrossRef][Green Version]
- Parish, S.T.; Kim, S.; Sekhon, R.K.; Wu, J.E.; Kawakatsu, Y.; Effros, R.B. Adenosine deaminase modulation of telomerase activity and replicative senescence in human CD8 T lymphocytes. J. Immunol. 2010, 184, 2847–2854. [Google Scholar] [CrossRef] [PubMed]
- Janeway, C.A., Jr. How the immune system protects the host from infection. Microbes Infect. 2001, 3, 1167–1171. [Google Scholar] [CrossRef]
- Prlic, M.; Bevan, M.J. Exploring regulatory mechanisms of CD8+ T cell contraction. PNAS 2008, 105, 16689–16694. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.; Lin, J.X.; Leonard, W.J. Interleukin-2 at the crossroads of effector responses, tolerance, and immunotherapy. Immunity 2013, 38, 13–25. [Google Scholar] [CrossRef]
- Effros, R.B. Telomerase induction in T cells: A cure for aging and disease? Exp. Gerontol. 2007, 42, 416–420. [Google Scholar] [CrossRef]
- Plunkett, F.J.; Franzese, O.; Finney, H.M.; Fletcher, J.M.; Belaramani, L.L.; Salmon, M.; Dokal, I.; Webster, D.; Lawson, A.D.; Akbar, A.N. The Loss of Telomerase Activity in Highly Differentiated CD8+CD28−CD27− T Cells Is Associated with Decreased Akt (Ser473) Phosphorylation. J. Immunol. 2007, 178, 7710–7719. [Google Scholar] [CrossRef]
- Saltzman, R.L.; Peterson, P.K. Immunodeficiency of the elderly. Rev. Infect. Dis. 1987, 9, 1127–1139. [Google Scholar] [CrossRef]
- Cawthon, R.M.; Smith, K.R.; O’Brien, E.; Sivatchenko, A.; Kerber, R.A. Association between telomere length in blood and mortality in people aged 60 years or older. Lancet 2003, 361, 393–395. [Google Scholar] [CrossRef]
- Van de Berg, P.J.; Van Stijn, A.; Ten Berge, I.J.; Van Lier, R.A. A fingerprint left by cytomegalovirus infection in the human T cell compartment. J. Clin. Virol. 2008, 41, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Pourgheysari, B.; Khan, N.; Best, D.; Bruton, R.; Nayak, L.; Moss, P.A. The cytomegalovirus-specific CD4+ T-cell response expands with age and markedly alters the CD4+ T-cell repertoire. J. Virol. 2007, 81, 7759–7765. [Google Scholar] [CrossRef] [PubMed]
- Effros, R.B.; Boucher, N.; Porter, V.; Zhu, X.; Spaulding, C.; Walford, R.L.; Kronenberg, M.; Cohen, D.; Schächter, F. Decline in CD28+ T cells in centenarians and in long-term T cell cultures: A possible cause for both in vivo and in vitro immunosenescence. Exp. Gerontol. 1994, 29, 601–609. [Google Scholar] [CrossRef]
- Hoji, A.; Connolly, N.C.; Buchanan, W.G.; Rinaldo, C.R., Jr. CD27 and CD57 expression reveals atypical differentiation of human immunodeficiency virus type 1-specific memory CD8+ T cells. Clin. Vaccine Immunol. 2007, 14, 74–80. [Google Scholar] [CrossRef]
- Van Leeuwen, E.M.; Gamadia, L.E.; Baars, P.A.; Remmerswaal, E.B.; ten Berge, I.J.; van Lier, R.A. Proliferation Requirements of Cytomegalovirus-Specific, Effector-Type Human CD8+ T Cells. J. Immunol. 2002, 169, 5838–5843. [Google Scholar] [CrossRef]
- Topp, M.S.; Riddell, S.R.; Akatsuka, Y.; Jensen, M.C.; Blattman, J.N.; Greenberg, P.D. Restoration of CD28 Expression in CD28− CD8+ Memory Effector T Cells Reconstitutes Antigen-induced IL-2 Production. J. Exp. Med. 2003, 198, 947–955. [Google Scholar] [CrossRef]
- Mojumdar, K.; Vajpayee, M.; Chauhan, N.K.; Singh, A.; Singh, R.; Kurapati, S. Loss of CD127 & increased immunosenescence of T cell subsets in HIV infected individuals. Indian. J. Med. Res. 2011, 134, 972–981. [Google Scholar]
- Cicin-Sain, L.; Brien, J.D.; Uhrlaub, J.L.; Drabig, A.; Marandu, T.F.; Nikolich-Zugich, J. Cytomegalovirus infection impairs immune responses and accentuates T-cell pool changes observed in mice with aging. PLoS Pathog. 2012, 8, e1002849. [Google Scholar] [CrossRef]
- Khan, N.; Shariff, N.; Cobbold, M.; Bruton, R.; Ainsworth, J.A.; Sinclair, A.J.; Nayak, L.; Moss, P.A. Cytomegalovirus Seropositivity Drives the CD8 T Cell Repertoire Toward Greater Clonality in Healthy Elderly Individuals. J. Immunol. 2002, 169, 1984–1992. [Google Scholar] [CrossRef]
- Lopez-Vergès, S.; Milush, J.M.; Pandey, S.; York, V.A.; Arakawa-Hoyt, J.; Pircher, H.; Norris, P.J.; Nixon, D.F.; Lanier, L.L. CD57 defines a functionally distinct population of mature NK cells in the human CD56dimCD16+ NK-cell subset. Blood 2010, 116, 3865–3874. [Google Scholar] [CrossRef]
- Waller, E.C.; McKinney, N.; Hicks, R.; Carmichael, A.J.; Sissons, J.G.; Wills, M.R. Differential costimulation through CD137 (4-1BB) restores proliferation of human virus-specific “effector memory” (CD28(-) CD45RA(HI)) CD8(+) T cells. Blood 2007, 110, 4360–4366. [Google Scholar] [CrossRef] [PubMed]
- Hadrup, S.R.; Strindhall, J.; Kollgaard, T.; Seremet, T.; Johansson, B.; Pawelec, G.; thor Straten, P.; Wikby, A. Longitudinal studies of clonally expanded CD8 T cells reveal a repertoire shrinkage predicting mortality and an increased number of dysfunctional cytomegalovirus-specific T cells in the very elderly. J. Immunol. 2006, 176, 2645–2653. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, Q.; Wagner, W.M.; Voehringer, D.; Wikby, A.; Klatt, T.; Walter, S.; Muller, C.A.; Pircher, H.; Pawelec, G. Age-associated accumulation of CMV-specific CD8+ T cells expressing the inhibitory killer cell lectin-like receptor G1 (KLRG1). Exp. Gerontol. 2003, 38, 911–920. [Google Scholar] [CrossRef]
- Scherrenburg, J.; Pietersma, F.; Jacobi, R.; Schuurman, R.; Meijer, E.; Van Baarle, D. Cytomegalovirus (CMV)-Reactivation Influences T-Cell Differentiation and CMV-Specific T-Cell Reconstitution after Stem Cell Transplantation. J. Clin. Cell. Immunol. 2013, 1–9. [Google Scholar] [CrossRef]
- Jackson, S.E.; Sedikides, G.X.; Okecha, G.; Poole, E.L.; Sinclair, J.H.; Wills, M.R. Latent Cytomegalovirus (CMV) Infection Does Not Detrimentally Alter T Cell Responses in the Healthy Old, But Increased Latent CMV Carriage Is Related to Expanded CMV-Specific T Cells. Front. Immunol. 2017, 8, 733. [Google Scholar] [CrossRef]
- Chiu, Y.L.; Lin, C.H.; Sung, B.Y.; Chuang, Y.F.; Schneck, J.P.; Kern, F.; Pawelec, G.; Wang, G.C. Cytotoxic polyfunctionality maturation of cytomegalovirus-pp65-specific CD4 + and CD8 + T-cell responses in older adults positively correlates with response size. Sci. Rep. 2016, 6, 19227. [Google Scholar] [CrossRef]
- Sandberg, J.K.; Fast, N.M.; Nixon, D.F. Functional heterogeneity of cytokines and cytolytic effector molecules in human CD8+ T lymphocytes. J. Immunol. 2001, 167, 181–187. [Google Scholar] [CrossRef]
- Makedonas, G.; Banerjee, P.P.; Pandey, R.; Hersperger, A.R.; Sanborn, K.B.; Hardy, G.A.; Orange, J.S.; Betts, M.R. Rapid up-regulation and granule-independent transport of perforin to the immunological synapse define a novel mechanism of antigen-specific CD8+ T cell cytotoxic activity. J. Immunol. 2009, 182, 5560–5569. [Google Scholar] [CrossRef]
- Appay, V.; Nixon, D.F.; Donahoe, S.M.; Gillespie, G.M.; Dong, T.; King, A.; Ogg, G.S.; Spiegel, H.M.; Conlon, C.; Spina, C.A.; et al. HIV-specific CD8(+) T cells produce antiviral cytokines but are impaired in cytolytic function. J. Exp. Med. 2000, 192, 63–75. [Google Scholar] [CrossRef]
- Heath, J.J.; Fudge, N.J.; Gallant, M.E.; Grant, M.D. Proximity of Cytomegalovirus-Specific CD8(+) T Cells to Replicative Senescence in Human Immunodeficiency Virus-Infected Individuals. Front. Immunol. 2018, 9, 201. [Google Scholar] [CrossRef]
- Staras, S.A.; Dollard, S.C.; Radford, K.W.; Flanders, W.D.; Pass, R.F.; Cannon, M.J. Seroprevalence of cytomegalovirus infection in the United States, 1988–1994. Clin. Infect. Dis. 2006, 43, 1143–1151. [Google Scholar] [CrossRef] [PubMed]
- Simanek, A.M.; Dowd, J.B.; Pawelec, G.; Melzer, D.; Dutta, A.; Aiello, A.E. Seropositivity to cytomegalovirus, inflammation, all-cause and cardiovascular disease-related mortality in the United States. PLoS ONE 2011, 6, e16103. [Google Scholar] [CrossRef] [PubMed]
- Palmer, D.B. The effect of age on thymic function. Front. Immunol. 2013, 4, 316. [Google Scholar] [CrossRef] [PubMed]
- Sauce, D.; Larsen, M.; Fastenackels, S.; Duperrier, A.; Keller, M.; Grubeck-Loebenstein, B.; Ferrand, C.; Debre, P.; Sidi, D.; Appay, V. Evidence of premature immune aging in patients thymectomized during early childhood. J. Clin. Invest. 2009, 119, 3070–3078. [Google Scholar] [CrossRef]
- Torfadottir, H.; Freysdottir, J.; Skaftadottir, I.; Haraldsson, A.; Sigfusson, G.; Ogmundsdottir, H.M. Evidence for extrathymic T cell maturation after thymectomy in infancy. Clin. Exp. Immunol. 2006, 145, 407–412. [Google Scholar] [CrossRef]
- Lindau, P.; Mukherjee, R.; Gutschow, M.V.; Vignali, M.; Warren, E.H.; Riddell, S.R.; Makar, K.W.; Turtle, C.J.; Robins, H.S. Cytomegalovirus Exposure in the Elderly Does Not Reduce CD8 T Cell Repertoire Diversity. J. Immunol. 2019, 202, 476–483. [Google Scholar] [CrossRef]
- Van de Berg, P.J.; Heutinck, K.M.; Raabe, R.; Minnee, R.C.; Young, S.L.; van Donselaar-van der Pant, K.A.; Bemelman, F.J.; Van Lier, R.A.; Ten Berge, I.J. Human cytomegalovirus induces systemic immune activation characterized by a type 1 cytokine signature. J. Infect. Dis. 2010, 202, 690–699. [Google Scholar] [CrossRef]
- Lagrand, W.K.; Visser, C.A.; Hermens, W.T.; Niessen, H.W.; Verheugt, F.W.; Wolbink, G.J.; Hack, C.E. C-reactive protein as a cardiovascular risk factor: More than an epiphenomenon? Circulation 1999, 100, 96–102. [Google Scholar] [CrossRef]
- Vaes, B.; Pasquet, A.; Wallemacq, P.; Rezzoug, N.; Mekouar, H.; Olivier, P.A.; Legrand, D.; Mathei, C.; Van Pottelbergh, G.; Degryse, J. The BELFRAIL (BFC80+) study: A population-based prospective cohort study of the very elderly in Belgium. BMC Geriatr. 2010, 10, 39. [Google Scholar] [CrossRef]
- Roberts, E.T.; Haan, M.N.; Dowd, J.B.; Aiello, A.E. Cytomegalovirus antibody levels, inflammation, and mortality among elderly Latinos over 9 years of follow-up. Am. J. Epidemiol. 2010, 172, 363–371. [Google Scholar] [CrossRef]
- Spyridopoulos, I.; Martin-Ruiz, C.; Hilkens, C.; Yadegarfar, M.E.; Isaacs, J.; Jagger, C.; Kirkwood, T.; von Zglinicki, T. CMV seropositivity and T-cell senescence predict increased cardiovascular mortality in octogenarians: Results from the Newcastle 85+ study. Aging Cell 2016, 15, 389–392. [Google Scholar] [CrossRef] [PubMed]
- Savva, G.M.; Pachnio, A.; Kaul, B.; Morgan, K.; Huppert, F.A.; Brayne, C.; Moss, P.A.H.; The Medical Research Council Cognitive Function and Ageing Study. Cytomegalovirus infection is associated with increased mortality in the older population. Aging Cell 2013, 12, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Dedinska, I.; Laca, L.; Miklusica, J.; Kantarova, D.; Galajda, P.; Mokan, M. Correlation between CMV Infection and Post-transplantation New-onset Diabetes Mellitus. Int. J. Organ Transplant. Med. 2016, 7, 173–182. [Google Scholar] [PubMed]
- Shimamura, M. The contribution of cytomegalovirus to atherosclerotic events after kidney transplantation. J. Infect. Dis. 2013, 207, 1487–1490. [Google Scholar] [CrossRef]
- Blum, A.; Peleg, A.; Weinberg, M. Anti-cytomegalovirus (CMV) IgG antibody titer in patients with risk factors to atherosclerosis. Clin. Exp.l Med. 2003, 3, 157–160. [Google Scholar] [CrossRef]
- Hansen, S.G.; Ford, J.C.; Lewis, M.S.; Ventura, A.B.; Hughes, C.M.; Coyne-Johnson, L.; Whizin, N.; Oswald, K.; Shoemaker, R.; Swanson, T.; et al. Profound early control of highly pathogenic SIV by an effector memory T-cell vaccine. Nature 2011, 473, 523–527. [Google Scholar] [CrossRef]
- Hansen, S.G.; Sacha, J.B.; Hughes, C.M.; Ford, J.C.; Burwitz, B.J.; Scholz, I.; Gilbride, R.M.; Lewis, M.S.; Gilliam, A.N.; Ventura, A.B.; et al. Cytomegalovirus vectors violate CD8+ T cell epitope recognition paradigms. Science 2013, 340, 1237874. [Google Scholar] [CrossRef]
- Barton, E.S.; White, D.W.; Cathelyn, J.S.; Brett-McClellan, K.A.; Engle, M.; Diamond, M.S.; Miller, V.L.; Virgin, H.W.t. Herpesvirus latency confers symbiotic protection from bacterial infection. Nature 2007, 447, 326–329. [Google Scholar] [CrossRef]
- Elmaagacli, A.H.; Koldehoff, M. Cytomegalovirus replication reduces the relapse incidence in patients with acute myeloid leukemia. Blood 2016, 128, 456–459. [Google Scholar] [CrossRef]
- Elmaagacli, A.H.; Steckel, N.K.; Koldehoff, M.; Hegerfeldt, Y.; Trenschel, R.; Ditschkowski, M.; Christoph, S.; Gromke, T.; Kordelas, L.; Ottinger, H.D.; et al. Early human cytomegalovirus replication after transplantation is associated with a decreased relapse risk: Evidence for a putative virus-versus-leukemia effect in acute myeloid leukemia patients. Blood 2011, 118, 1402–1412. [Google Scholar] [CrossRef]
- Koldehoff, M.; Ross, S.R.; Duhrsen, U.; Beelen, D.W.; Elmaagacli, A.H. Early CMV-replication after allogeneic stem cell transplantation is associated with a reduced relapse risk in lymphoma. Leukemia Lymphoma 2017, 58, 822–833. [Google Scholar] [CrossRef] [PubMed]
- Della Chiesa, M.; Falco, M.; Muccio, L.; Bertaina, A.; Locatelli, F.; Moretta, A. Impact of HCMV Infection on NK Cell Development and Function after HSCT. Front. Immunol. 2013, 4, 458. [Google Scholar] [CrossRef] [PubMed]
- Koldehoff, M.; Lindemann, M.; Ross, S.R.; Elmaagacli, A.H. Cytomegalovirus induces HLA-class-II-restricted alloreactivity in an acute myeloid leukemia cell line. PLoS ONE 2018, 13, e0191482. [Google Scholar] [CrossRef] [PubMed]
- Papagno, L.; Spina, C.A.; Marchant, A.; Salio, M.; Rufer, N.; Little, S.; Dong, T.; Chesney, G.; Waters, A.; Easterbrook, P.; et al. Immune Activation and CD8+ T-Cell Differentiation towards Senescence in HIV-1 Infection. PLoS Biol. 2004, 2, e20. [Google Scholar] [CrossRef] [PubMed]
- Jabs, D.A.; Enger, C.; Bartlett, J.G. Cytomegalovirus Retinitis and Acquired Immunodeficiency Syndrome. Arch. Ophthalmol. 1989, 107, 75–80. [Google Scholar] [CrossRef]
- Ballegaard, V.; Braendstrup, P.; Pedersen, K.K.; Kirkby, N.; Stryhn, A.; Ryder, L.P.; Gerstoft, J.; Nielsen, S.D. Cytomegalovirus-specific T-cells are associated with immune senescence, but not with systemic inflammation, in people living with HIV. Sci. Rep. 2018, 8, 3778. [Google Scholar] [CrossRef] [PubMed]
- Wittkop, L.; Bitard, J.; Lazaro, E.; Neau, D.; Bonnet, F.; Mercie, P.; Dupon, M.; Hessamfar, M.; Ventura, M.; Malvy, D.; et al. Effect of cytomegalovirus-induced immune response, self antigen-induced immune response, and microbial translocation on chronic immune activation in successfully treated HIV type 1-infected patients: The ANRS CO3 Aquitaine Cohort. J. Infect. Dis. 2013, 207, 622–627. [Google Scholar] [CrossRef] [PubMed]
- Lurain, N.S.; Hanson, B.A.; Hotton, A.L.; Weber, K.M.; Cohen, M.H.; Landay, A.L. The Association of Human Cytomegalovirus with Biomarkers of Inflammation and Immune Activation in HIV-1-Infected Women. AIDS Res. Hum. Retrovir. 2016, 32, 134–143. [Google Scholar] [CrossRef]
- Freeman, M.L.; Mudd, J.C.; Shive, C.L.; Younes, S.A.; Panigrahi, S.; Sieg, S.F.; Lee, S.A.; Hunt, P.W.; Calabrese, L.H.; Gianella, S.; et al. CD8 T-Cell Expansion and Inflammation Linked to CMV Coinfection in ART-treated HIV Infection. Clin. Infect. Dis. 2016, 62, 392–396. [Google Scholar] [CrossRef]
- Lichtner, M.; Cicconi, P.; Vita, S.; Cozzi-Lepri, A.; Galli, M.; Lo Caputo, S.; Saracino, A.; De Luca, A.; Moioli, M.; Maggiolo, F.; et al. Cytomegalovirus coinfection is associated with an increased risk of severe non-AIDS-defining events in a large cohort of HIV-infected patients. J. Infect. Dis. 2015, 211, 178–186. [Google Scholar] [CrossRef]
- Blackman, M.A.; Woodland, D.L. The narrowing of the CD8 T cell repertoire in old age. Curr. Opin. Immunol. 2011, 23, 537–542. [Google Scholar] [CrossRef]
- Barrett, L.; Stapleton, S.N.; Fudge, N.J.; Grant, M.D. Immune resilience in HIV-infected individuals seronegative for cytomegalovirus. Aids 2014, 28, 2045–2049. [Google Scholar] [CrossRef] [PubMed]
- Baum, P.D.; Young, J.J.; Schmidt, D.; Zhang, Q.; Hoh, R.; Busch, M.; Martin, J.; Deeks, S.; McCune, J.M. Blood T-cell receptor diversity decreases during the course of HIV infection, but the potential for a diverse repertoire persists. Blood 2012, 119, 3469–3477. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Heath, J.; Newhook, N.; Comeau, E.; Gallant, M.; Fudge, N.; Grant, M. NKG2C(+)CD57(+) Natural Killer Cell Expansion Parallels Cytomegalovirus-Specific CD8(+) T Cell Evolution towards Senescence. J. Immunol. Res. 2016, 2016, 7470124. [Google Scholar] [CrossRef]
- Serrano-Villar, S.; Perez-Elias, M.J.; Dronda, F.; Casado, J.L.; Moreno, A.; Royuela, A.; Perez-Molina, J.A.; Sainz, T.; Navas, E.; Hermida, J.M.; et al. Increased risk of serious non-AIDS-related events in HIV-infected subjects on antiretroviral therapy associated with a low CD4/CD8 ratio. PLoS ONE 2014, 9, e85798. [Google Scholar] [CrossRef] [PubMed]
- Wolthers, K.C.; Bea, G.; Wisman, A.; Otto, S.A.; De Roda Husman, A.M.; Schaft, N.; De Wolf, F.; Goudsmit, J.; Coutinho, R.A.; Van der Zee, A.G.; et al. T cell telomere length in HIV-1 infection: No evidence for increased CD4+ T cell turnover. Science 1996, 274, 1543–1547. [Google Scholar] [CrossRef] [PubMed]
- Barrett, L.; Fudge, N.J.; Heath, J.J.; Grant, M.D. Cytomegalovirus Immunity and Exhaustive CD8+ T Cell Proliferation in Treated Human Immunodeficiency Virus Infection. Clin. Infect. Dis. 2016, 62, 1467–1468. [Google Scholar] [CrossRef]
- Riddell, N.E.; Griffiths, S.J.; Rivino, L.; King, D.C.; Teo, G.H.; Henson, S.M.; Cantisan, S.; Solana, R.; Kemeny, D.M.; MacAry, P.A.; et al. Multifunctional cytomegalovirus (CMV)-specific CD8(+) T cells are not restricted by telomere-related senescence in young or old adults. Immunology 2015, 144, 549–560. [Google Scholar] [CrossRef]
- Dock, J.N.; Effros, R.B. Role of CD8 T Cell Replicative Senescence in Human Aging and in HIV-mediated Immunosenescence. Aging Dis. 2011, 2, 382–397. [Google Scholar]
- Zanet, D.L.; Thorne, A.; Singer, J.; Maan, E.J.; Sattha, B.; Le Campion, A.; Soudeyns, H.; Pick, N.; Murray, M.; Money, D.M.; et al. Association between short leukocyte telomere length and HIV infection in a cohort study: No evidence of a relationship with antiretroviral therapy. Clin. Infect. Dis. 2014, 58, 1322–1332. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
J. Heath, J.; D. Grant, M. The Immune Response Against Human Cytomegalovirus Links Cellular to Systemic Senescence. Cells 2020, 9, 766. https://doi.org/10.3390/cells9030766
J. Heath J, D. Grant M. The Immune Response Against Human Cytomegalovirus Links Cellular to Systemic Senescence. Cells. 2020; 9(3):766. https://doi.org/10.3390/cells9030766
Chicago/Turabian StyleJ. Heath, John, and Michael D. Grant. 2020. "The Immune Response Against Human Cytomegalovirus Links Cellular to Systemic Senescence" Cells 9, no. 3: 766. https://doi.org/10.3390/cells9030766
APA StyleJ. Heath, J., & D. Grant, M. (2020). The Immune Response Against Human Cytomegalovirus Links Cellular to Systemic Senescence. Cells, 9(3), 766. https://doi.org/10.3390/cells9030766