Phenotyping of Rare CFTR Mutations Reveals Distinct Trafficking and Functional Defects


 , ,
, ,
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Lentiviral Vector (LV) Production
2.2. Human Embryonic Kidney 293T (HEK293T) Stable Cell Lines
2.2.1. Stable Cell Line Generation
2.2.2. RT-PCR for CFTR mRNA Expression Levels
2.2.3. Western Blotting
2.2.4. Immunocytochemistry
2.2.5. Flow Cytometry
2.2.6. Halide Sensitive (HS) YFP Assay
2.3. Intestinal Organoids
2.3.1. Viral Vector Transduction
2.3.2. Forskolin Induced Swelling (FIS) Assay to Quantify CFTR Activity
2.4. Statistics
3. Results
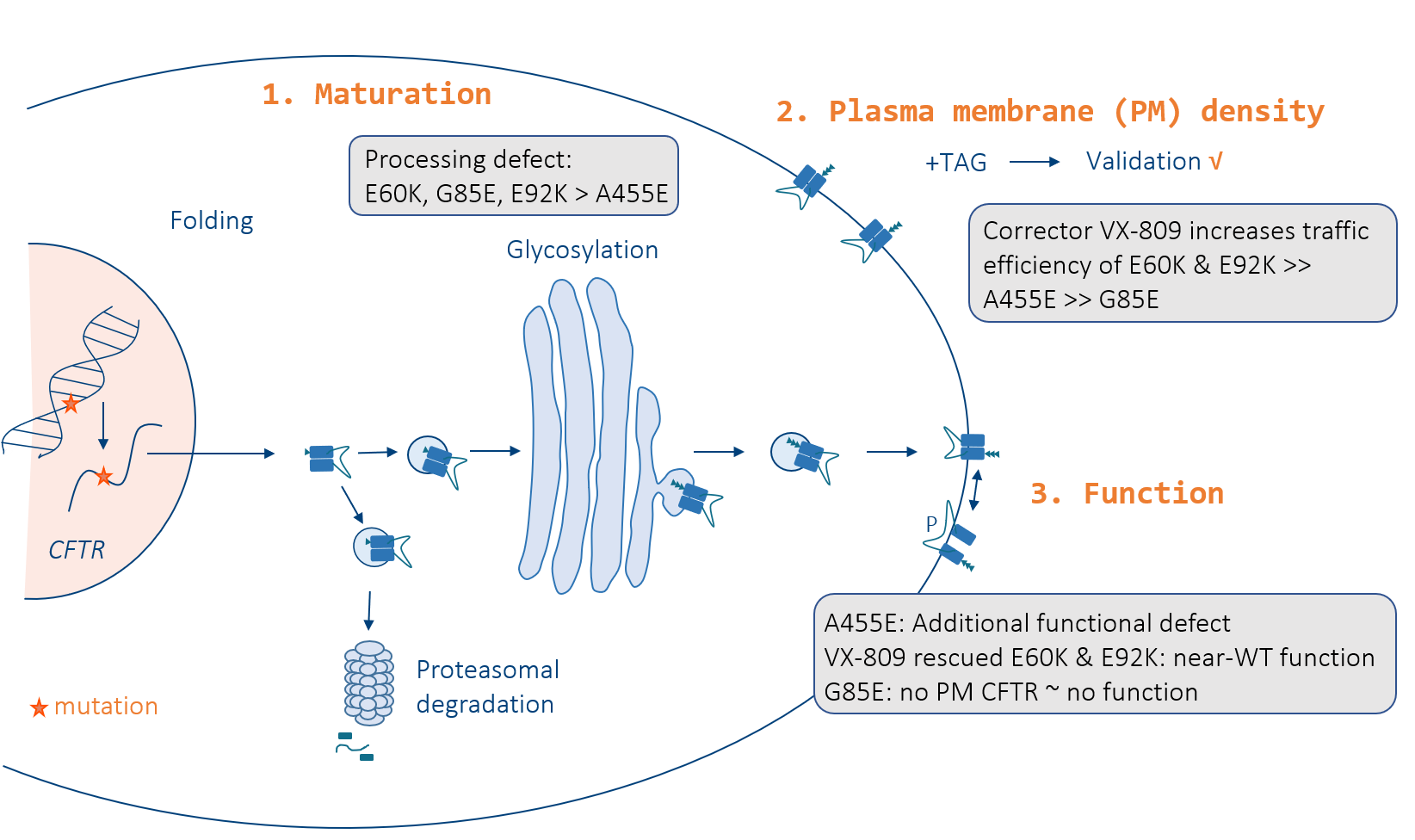
3.1. E60K, G85E, E92K and A455E Lead to Impaired Processing and Endoplasmic Reticulum Retention of CFTR
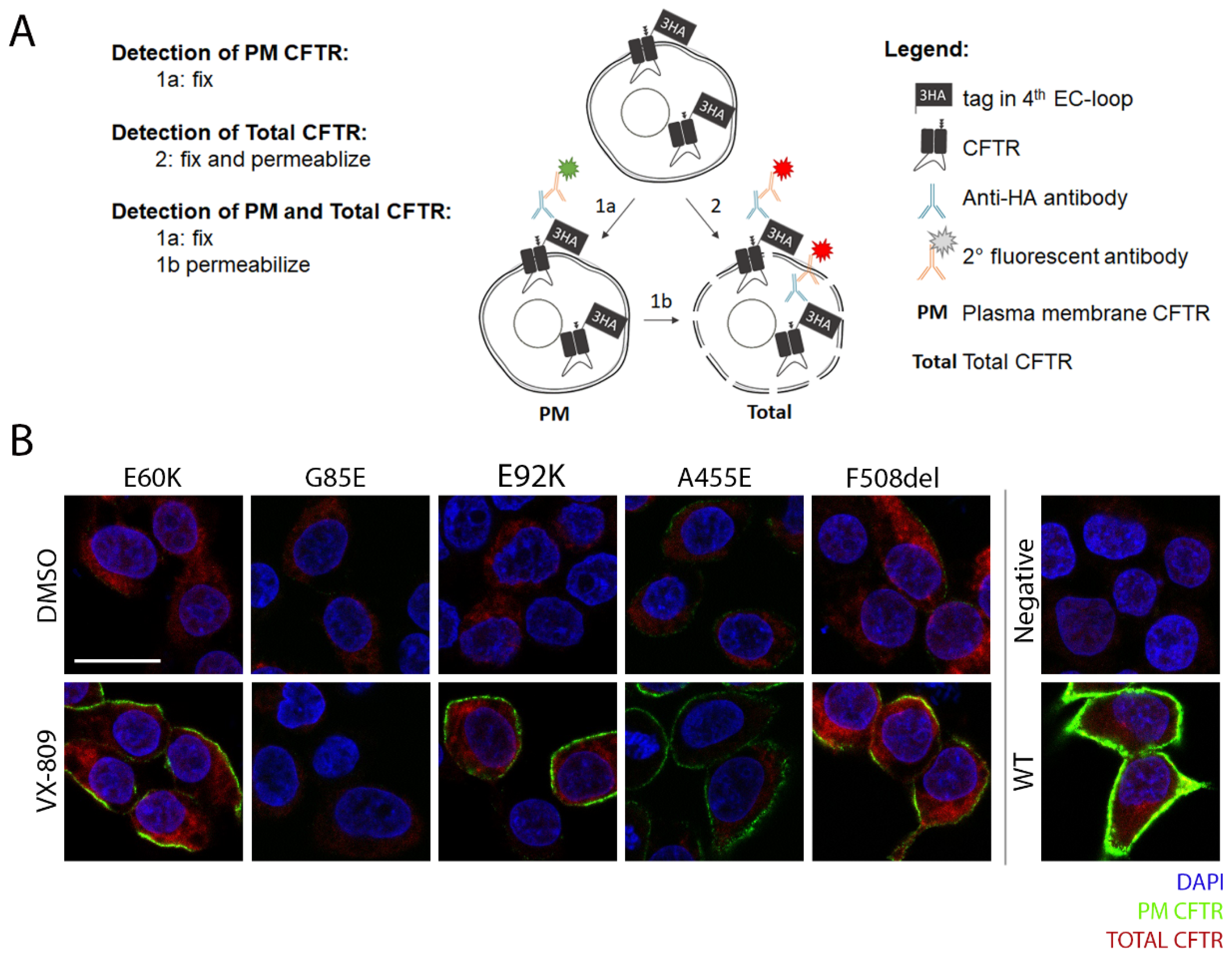
3.2. Tagging CFTR in the 4th Extracellular Loop Minimally Affects CFTR Maturation and Function
3.3. Corrector VX-809 Restores E60K and E92K Function
3.4. A Medium-Throughput Flow Cytometry Assay for Quantifying CFTR Plasma Membrane Density and Traffic Efficiency
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wanyama, S.S.; Thomas, M. Annual Report Belgian Cystic Fibrosis Registry 2016. 2018. Available online: https://www.muco.be/wp-content/uploads/2019/07/Report-Belgian-CF-registry-2016-EN_final.pdf (accessed on 27 February 2020).
- Kerem, B.; Rommens, J.M.; Buchanan, J.A.; Markiewicz, D.; Cox, T.K.; Chakravarti, A.; Buchwald, M.; Tsui, L.C. Identification of the cystic fibrosis gene: Genetic analysis. Science 1989, 245, 1073–1080. [Google Scholar] [CrossRef] [PubMed]
- Riordan, J.R.; Rommens, J.M.; Kerem, B.; Alon, N.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.; Plavsic, N.; Chou, J.L.; et al. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science 1989, 245, 1066–1073. [Google Scholar] [CrossRef] [PubMed]
- Rommens, J.M.; Iannuzzi, M.C.; Kerem, B.; Drumm, M.L.; Melmer, G.; Dean, M.; Rozmahel, R.; Cole, J.L.; Kennedy, D.; Hidaka, N.; et al. Identification of the cystic fibrosis gene: Chromosome walking and jumping. Science 1989, 245, 1059–1065. [Google Scholar] [CrossRef] [PubMed]
- De Boeck, K.; Amaral, M.D. Progress in therapies for cystic fibrosis. Lancet Respir. Med. 2016, 4, 662–674. [Google Scholar] [CrossRef]
- Veit, G.; Avramescu, R.G.; Chiang, A.N.; Houck, S.A.; Cai, Z.; Peters, K.W.; Hong, J.S.; Pollard, H.B.; Guggino, W.B.; Balch, W.E.; et al. From CFTR biology toward combinatorial pharmacotherapy: Expanded classification of cystic fibrosis mutations. Mol. Biol. Cell 2016, 27, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Heijerman, H.G.M.; McKone, E.F.; Downey, D.G.; Van Braeckel, E.; Rowe, S.M.; Tullis, E.; Mall, M.A.; Welter, J.J.; Ramsey, B.W.; McKee, C.M.; et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: A double-blind, randomised, phase 3 trial. Lancet 2019, 394, 1940–1948. [Google Scholar] [CrossRef]
- Middleton, P.G.; Mall, M.A.; Drevinek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor-tezacaftor-ivacaftor for cystic fibrosis with a single Phe508del allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef]
- HIGHLIGHTS OF PRESCRIBING INFORMATION—KALYDECO® (Ivacaftor) Tablets, for Oral Use. 2017. Available online: https://pi.vrtx.com/files/uspi_ivacaftor.pdf (accessed on 27 February 2020).
- HIGHLIGHTS OF PRESCRIBING INFORMATION—SYMDEKO™ (Tezacaftor/Ivacaftor) Tablets; (Ivacaftor) Tablets, for Oral Use. 2018. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/210491Orig1s000LBL.pdf (accessed on 27 February 2020).
- Randell, S.H.; Fulcher, M.L.; O’Neal, W.; Olsen, J.C. Primary epithelial cell models for cystic fibrosis research. Methods Mol. Biol. 2011, 742, 285–310. [Google Scholar]
- Dekkers, J.F.; Wiegerinck, C.L.; de Jonge, H.R.; Bronsveld, I.; Janssens, H.M.; de Winter-de Groot, K.M.; Brandsma, A.M.; de Jong, N.W.; Bijvelds, M.J.; Scholte, B.J.; et al. A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nat. Med. 2013, 19, 939–945. [Google Scholar] [CrossRef]
- Pranke, I.M.; Hatton, A.; Simonin, J.; Jais, J.P.; Le Pimpec-Barthes, F.; Carsin, A.; Bonnette, P.; Fayon, M.; Stremler-Le Bel, N.; Grenet, D.; et al. Correction of CFTR function in nasal epithelial cells from cystic fibrosis patients predicts improvement of respiratory function by CFTR modulators. Sci. Rep. 2017, 7, 7375. [Google Scholar] [CrossRef]
- Van Goor, F.; Yu, H.; Burton, B.; Hoffman, B.J. Effect of ivacaftor on CFTR forms with missense mutations associated with defects in protein processing or function. J. Cyst. Fibros. 2014, 13, 29–36. [Google Scholar] [CrossRef]
- Clancy, J.P.; Cotton, C.U.; Donaldson, S.H.; Solomon, G.M.; Van Devanter, D.R.; Boyle, M.P.; Gentzsch, M.; Nick, J.A.; Illek, B.; Wallenburg, J.C.; et al. CFTR modulator theratyping: Current status, gaps and future directions. J. Cyst. Fibros. 2019, 18, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Vidovic, D.; Carlon, M.S.; da Cunha, M.F.; Dekkers, J.F.; Hollenhorst, M.I.; Bijvelds, M.J.; Ramalho, A.S.; Van den Haute, C.; Ferrante, M.; Baekelandt, V.; et al. rAAV-CFTRDeltaR Rescues the cystic fibrosis phenotype in human intestinal organoids and cystic fibrosis mice. Am. J. Respir. Crit. Care Med. 2016, 193, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Pampinella, F.; Nemes, C.; Benharouga, M.; So, J.; Du, K.; Bache, K.G.; Papsin, B.; Zerangue, N.; Stenmark, H.; et al. Misfolding diverts CFTR from recycling to degradation: Quality control at early endosomes. J. Cell Biol. 2004, 164, 923–933. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Aleksandrov, L.; Chang, X.B.; Hou, Y.X.; He, L.; Hegedus, T.; Gentzsch, M.; Aleksandrov, A.; Balch, W.E.; Riordan, J.R. Domain interdependence in the biosynthetic assembly of CFTR. J. Mol. Biol. 2007, 365, 981–994. [Google Scholar] [CrossRef] [PubMed]
- Gees, M.; Musch, S.; Van der Plas, S.; Wesse, A.S.; Vandevelde, A.; Verdonck, K.; Mammoliti, O.; Hwang, T.C.; Sonck, K.; Stouten, P.; et al. Identification and characterization of novel CFTR potentiators. Front. Pharmacol. 2018, 9, 1221. [Google Scholar] [CrossRef]
- Galietta, L.J.; Haggie, P.M.; Verkman, A.S. Green fluorescent protein-based halide indicators with improved chloride and iodide affinities. FEBS Lett. 2001, 499, 220–224. [Google Scholar] [CrossRef]
- Kim, S.J.; Skach, W.R. Mechanisms of CFTR folding at the endoplasmic reticulum. Front. Pharmacol. 2012, 3, 201. [Google Scholar] [CrossRef]
- Ren, H.Y.; Grove, D.E.; De La Rosa, O.; Houck, S.A.; Sopha, P.; Van Goor, F.; Hoffman, B.J.; Cyr, D.M. VX-809 corrects folding defects in cystic fibrosis transmembrane conductance regulator protein through action on membrane-spanning domain 1. Mol. Biol. Cell 2013, 24, 3016–3024. [Google Scholar]
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.; Burton, B.; Stack, J.H.; Straley, K.S.; Decker, C.J.; Miller, M.; McCartney, J.; Olson, E.R.; et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc. Natl. Acad. Sci. USA 2011, 108, 18843–18848. [Google Scholar] [CrossRef]
- Okiyoneda, T.; Veit, G.; Dekkers, J.F.; Bagdany, M.; Soya, N.; Xu, H.; Roldan, A.; Verkman, A.S.; Kurth, M.; Simon, A.; et al. Mechanism-based corrector combination restores DeltaF508-CFTR folding and function. Nat. Chem. Biol. 2013, 9, 444–454. [Google Scholar] [CrossRef] [PubMed]
- Botelho, H.M.; Uliyakina, I.; Awatade, N.T.; Proenca, M.C.; Tischer, C.; Sirianant, L.; Kunzelmann, K.; Pepperkok, R.; Amaral, M.D. Protein traffic disorders: An effective high-throughput fluorescence microscopy pipeline for drug discovery. Sci. Rep. 2015, 5, 9038. [Google Scholar] [CrossRef] [PubMed]
- Perkins, L.A.; Fisher, G.W.; Naganbabu, M.; Schmidt, B.F.; Mun, F.; Bruchez, M.P. High-content surface and total expression siRNA kinase library screen with VX-809 treatment reveals kinase targets that enhance F508del-CFTR rescue. Mol. Pharm. 2018, 15, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Carlile, G.W.; Keyzers, R.A.; Teske, K.A.; Robert, R.; Williams, D.E.; Linington, R.G.; Gray, C.A.; Centko, L.; Yan, R.M.; Anjos, S.M.; et al. Correction of F508del-CFTR trafficking by the sponge alkaloid latonduine is modulated by interaction with PARP. Chem. Biol. 2012, 19, 1288–1299. [Google Scholar] [PubMed]
- Wang, C.; Balch, W.E. Bridging genomics to phenomics at atomic resolution through variation spatial profiling. Cell Rep. 2018, 24, 2013–2028. [Google Scholar] [CrossRef] [PubMed]
- Avramescu, R.G.; Kai, Y.; Xu, H.; Bidaud-Meynard, A.; Schnur, A.; Frenkiel, S.; Matouk, E.; Veit, G.; Lukacs, G.L. Mutation-specific downregulation of CFTR2 variants by gating potentiator. Hum. Mol. Genet. 2017, 26, 4873–4885. [Google Scholar] [CrossRef]
- Ramalho, A.S.; Fürstová, E.; Vermeulen, F.; Vonk, A.M.; Ferrante, M.; Dupont, L.; Proesmans, M.; Boon, M.; Sarouk, I.; Cordero, C.V.; et al. Correction of CFTR function in intestinal organoids to guide precision treatment of Cystic Fibrosis. Eur. Respir. J. 2020. under review. [Google Scholar]
- Angles, F.; Hutt, D.M.; Balch, W.E. HDAC inhibitors rescue multiple disease-causing CFTR variants. Hum. Mol. Genet. 2019, 28, 1982–2000. [Google Scholar] [CrossRef]
- Lopes-Pacheco, M.; Boinot, C.; Sabirzhanova, I.; Rapino, D.; Cebotaru, L. Combination of correctors rescues CFTR transmembrane-domain mutants by mitigating their interactions with proteostasis. Cell. Physiol. Biochem. 2017, 41, 2194–2210. [Google Scholar] [CrossRef]
- Muraglia, K.A.; Chorghade, R.S.; Kim, B.R.; Tang, X.X.; Shah, V.S.; Grillo, A.S.; Daniels, P.N.; Cioffi, A.G.; Karp, P.H.; Zhu, L.; et al. Small-molecule ion channels increase host defences in cystic fibrosis airway epithelia. Nature 2019, 567, 405–408. [Google Scholar] [CrossRef]
- Alton, E.W.; Beekman, J.M.; Boyd, A.C.; Brand, J.; Carlon, M.S.; Connolly, M.M.; Chan, M.; Conlon, S.; Davidson, H.E.; Davies, J.C.; et al. Preparation for a first-in-man lentivirus trial in patients with cystic fibrosis. Thorax 2017, 72, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Haque, A.; Dewerth, A.; Antony, J.S.; Riethmuller, J.; Schweizer, G.R.; Weinmann, P.; Latifi, N.; Yasar, H.; Pedemonte, N.; Sondo, E.; et al. Chemically modified hCFTR mRNAs recuperate lung function in a mouse model of cystic fibrosis. Sci. Rep. 2018, 8, 16776. [Google Scholar] [CrossRef] [PubMed]
- Schonberger, M.; Althaus, M.; Fronius, M.; Clauss, W.; Trauner, D. Controlling epithelial sodium channels with light using photoswitchable amilorides. Nat. Chem. 2014, 6, 712–719. [Google Scholar] [CrossRef] [PubMed]
- Cebotaru, L.; Rapino, D.; Cebotaru, V.; Guggino, W.B. Correcting the cystic fibrosis disease mutant, A455E CFTR. PLoS ONE 2014, 9, e85183. [Google Scholar] [CrossRef]
- Lopes-Pacheco, M.; Sabirzhanova, I.; Rapino, D.; Morales, M.M.; Guggino, W.B.; Cebotaru, L. Correctors rescue CFTR mutations in nucleotide-binding domain 1 (NBD1) by modulating proteostasis. Chembiochem 2016, 17, 493–505. [Google Scholar] [CrossRef]
- Garg, V.; Shen, J.; Li, C.; Agarwal, S.; Gebre, A.; Robertson, S.; Huang, J.; Han, L.; Jiang, L.; Stephan, K.; et al. Pharmacokinetic and drug-drug interaction profiles of the combination of tezacaftor/ivacaftor. Clin. Transl. Sci. 2019, 12, 267–275. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ensinck, M.; De Keersmaecker, L.; Heylen, L.; Ramalho, A.S.; Gijsbers, R.; Farré, R.; De Boeck, K.; Christ, F.; Debyser, Z.; Carlon, M.S. Phenotyping of Rare CFTR Mutations Reveals Distinct Trafficking and Functional Defects. Cells 2020, 9, 754. https://doi.org/10.3390/cells9030754
Ensinck M, De Keersmaecker L, Heylen L, Ramalho AS, Gijsbers R, Farré R, De Boeck K, Christ F, Debyser Z, Carlon MS. Phenotyping of Rare CFTR Mutations Reveals Distinct Trafficking and Functional Defects. Cells. 2020; 9(3):754. https://doi.org/10.3390/cells9030754
Chicago/Turabian StyleEnsinck, Marjolein, Liesbeth De Keersmaecker, Lise Heylen, Anabela S. Ramalho, Rik Gijsbers, Ricard Farré, Kris De Boeck, Frauke Christ, Zeger Debyser, and Marianne S. Carlon. 2020. "Phenotyping of Rare CFTR Mutations Reveals Distinct Trafficking and Functional Defects" Cells 9, no. 3: 754. https://doi.org/10.3390/cells9030754
APA StyleEnsinck, M., De Keersmaecker, L., Heylen, L., Ramalho, A. S., Gijsbers, R., Farré, R., De Boeck, K., Christ, F., Debyser, Z., & Carlon, M. S. (2020). Phenotyping of Rare CFTR Mutations Reveals Distinct Trafficking and Functional Defects. Cells, 9(3), 754. https://doi.org/10.3390/cells9030754