The Leader Position of Mesenchymal Cells Expressing N-Cadherin in the Collective Migration of Epithelial Cancer



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Tumor-Spheres Generation and Migration Analysis
2.3. Immunohistochemistry and Immunofluorescence
2.4. Time-Lapse Microscopy
2.5. Statistical Analysis
3. Results
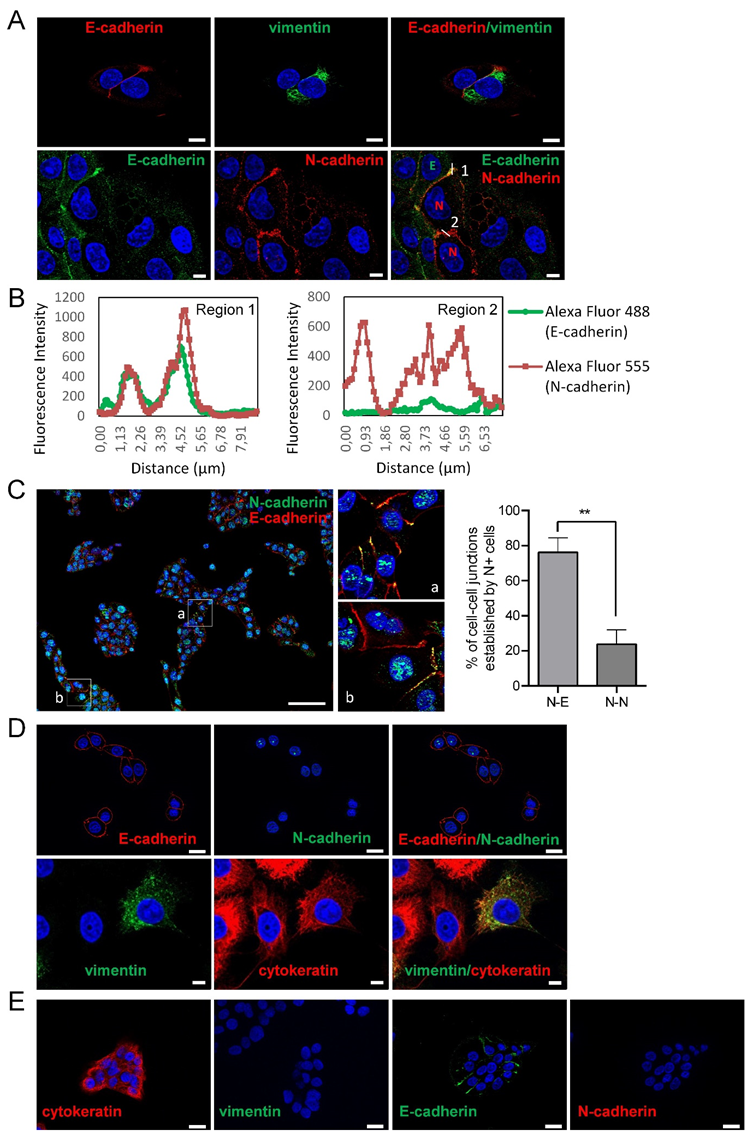
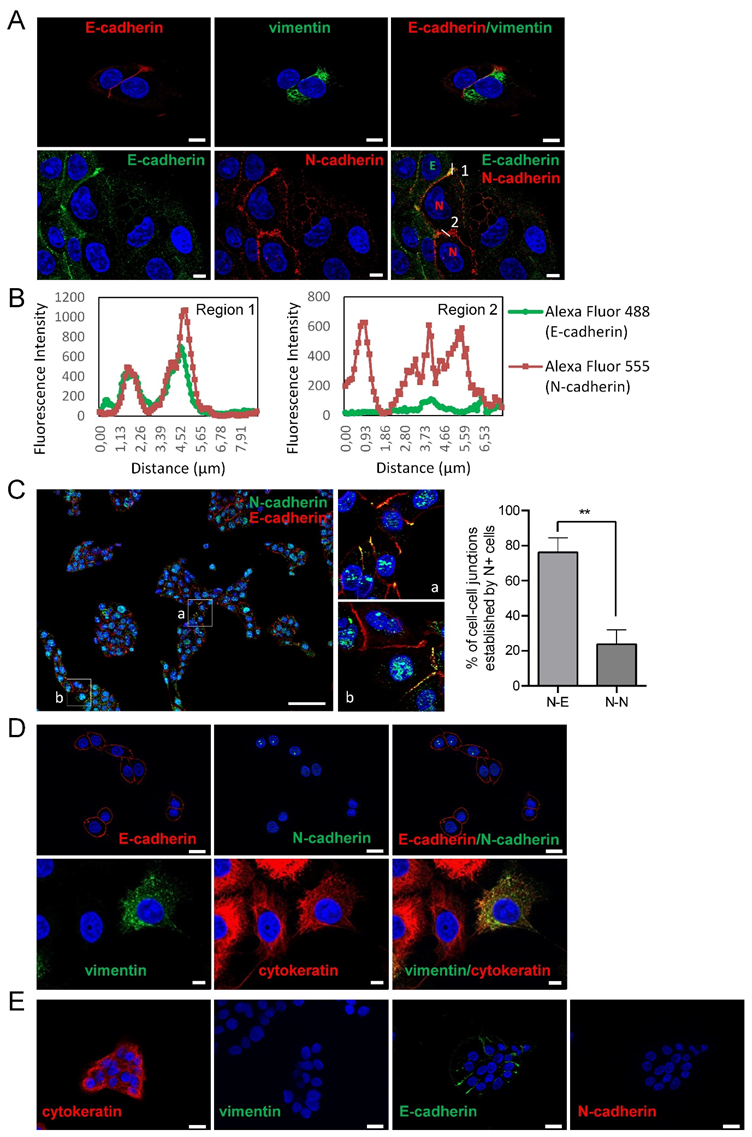
3.1. Phenotypic Heterogeneity and Cell-To-Cell Interactions of Human SCC-Derived Cells
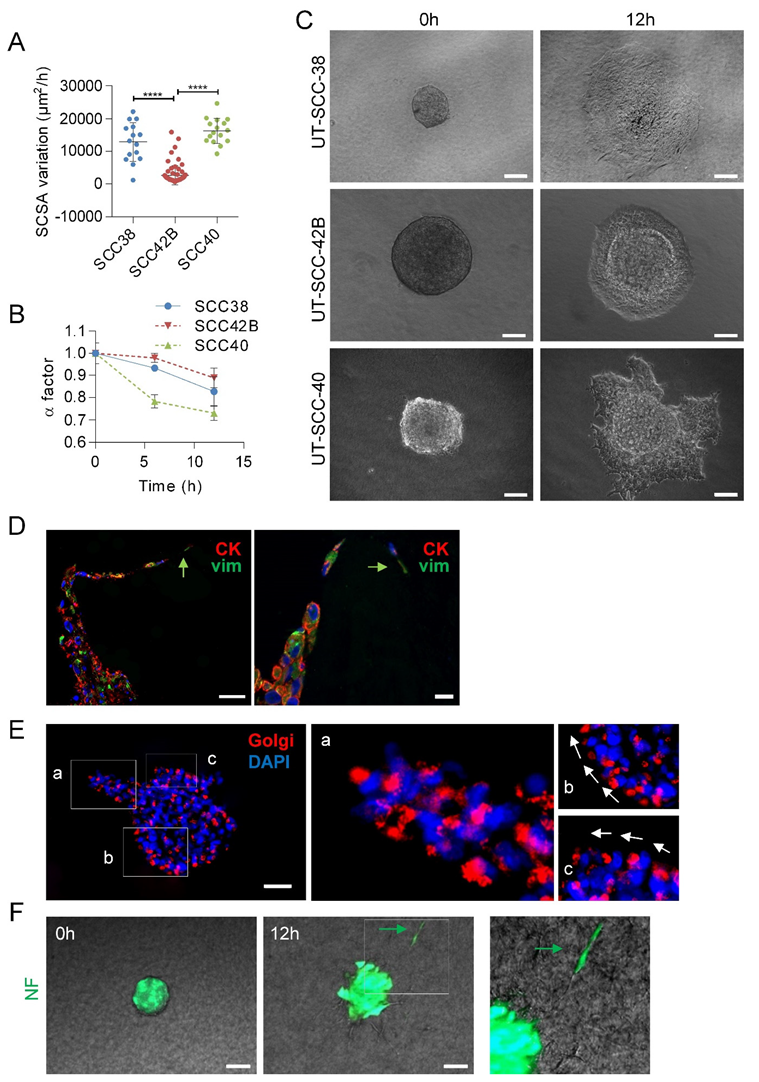
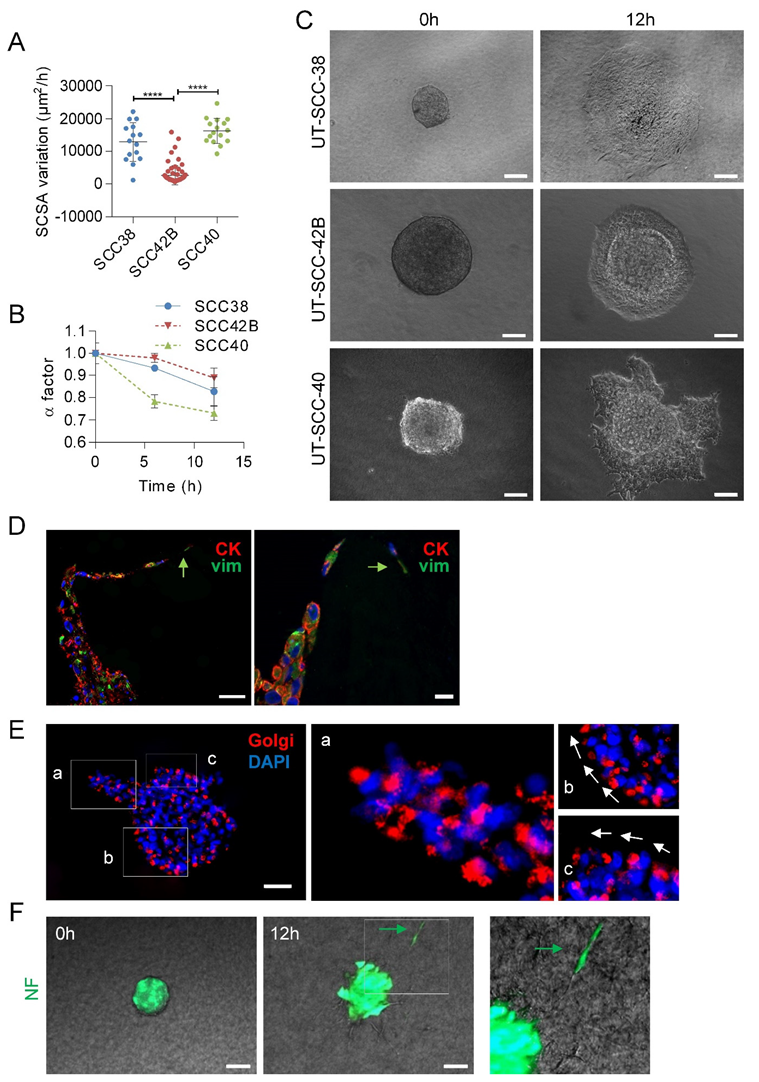
3.2. Human SCC-Derived Cells, but not Fibroblasts, Display a Collective Mode of Invasion
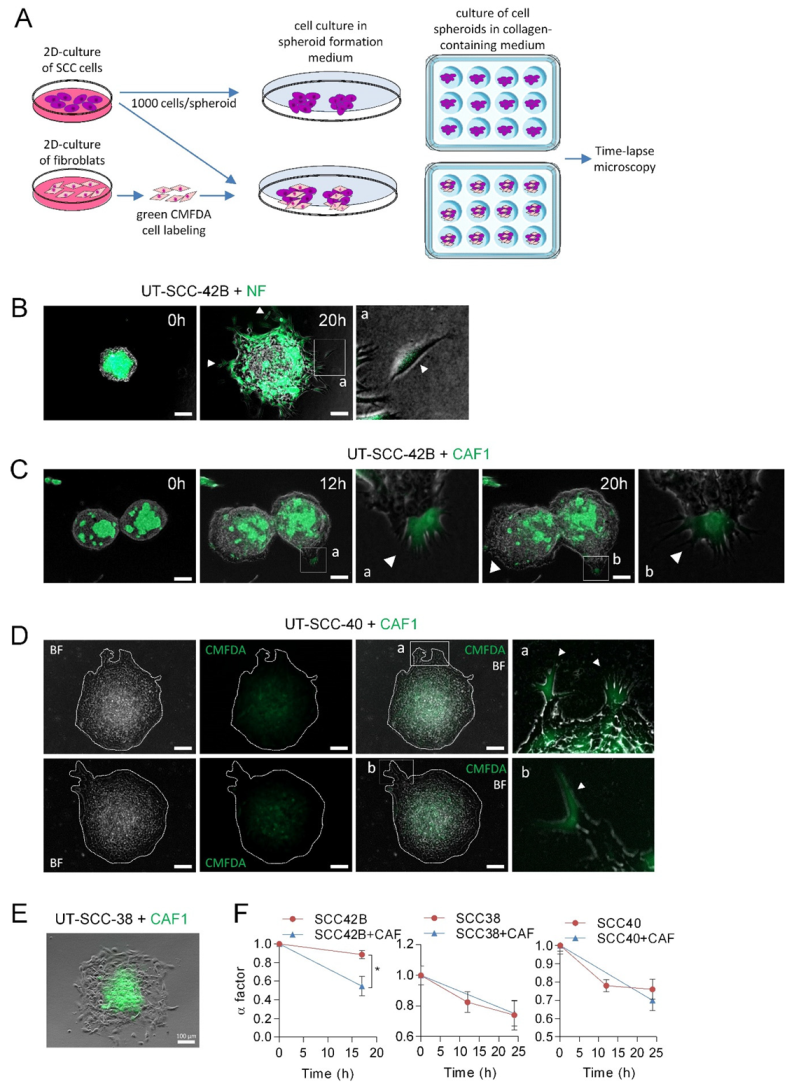
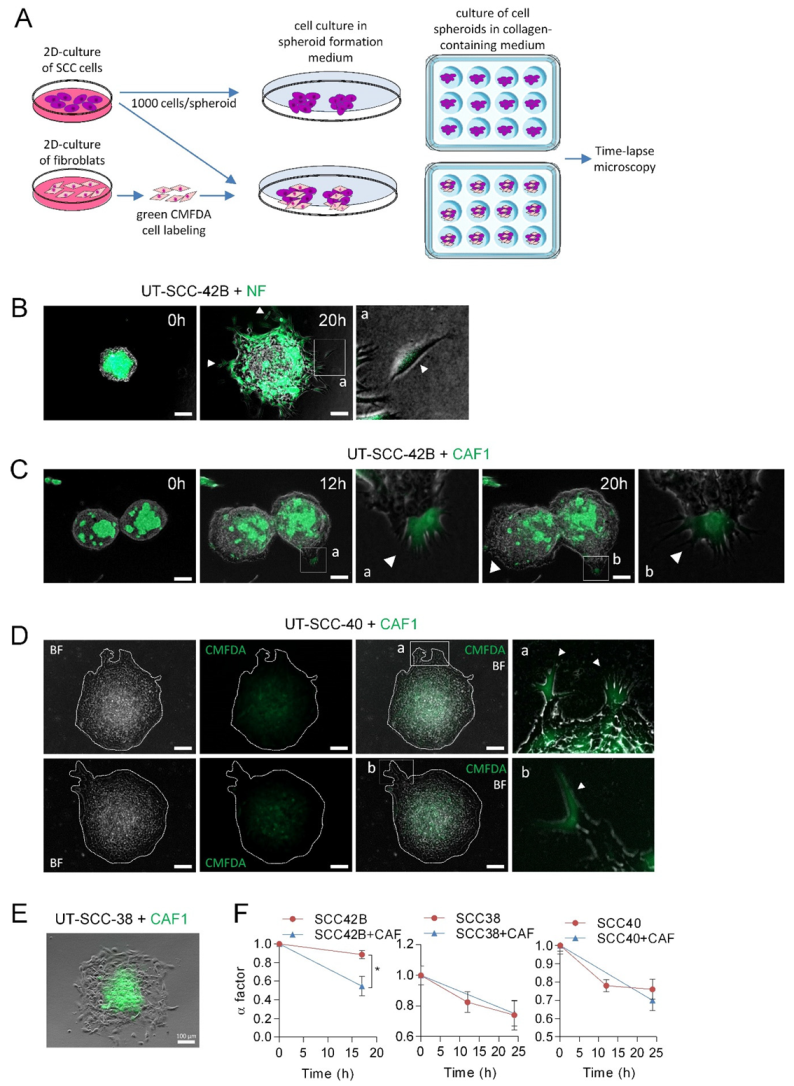
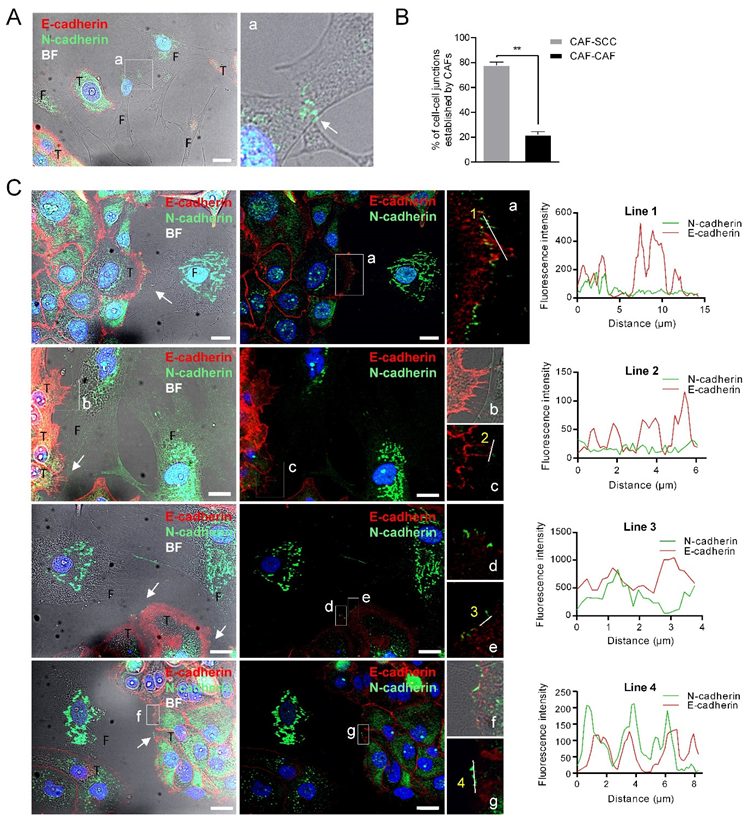
3.3. CAFs as Leaders of Collective Migration of SCC Cells
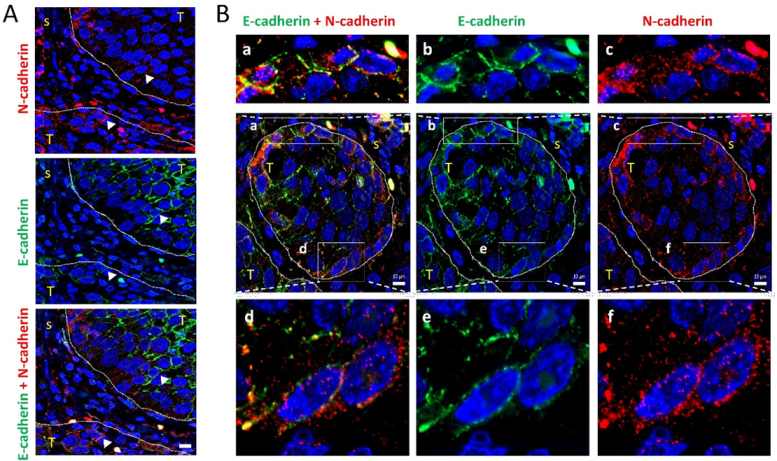
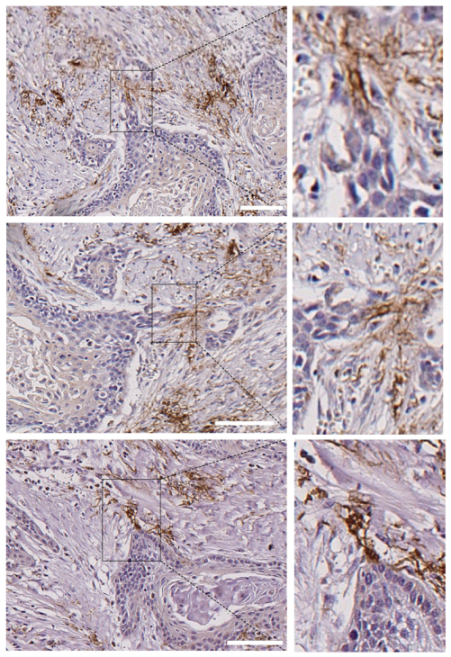
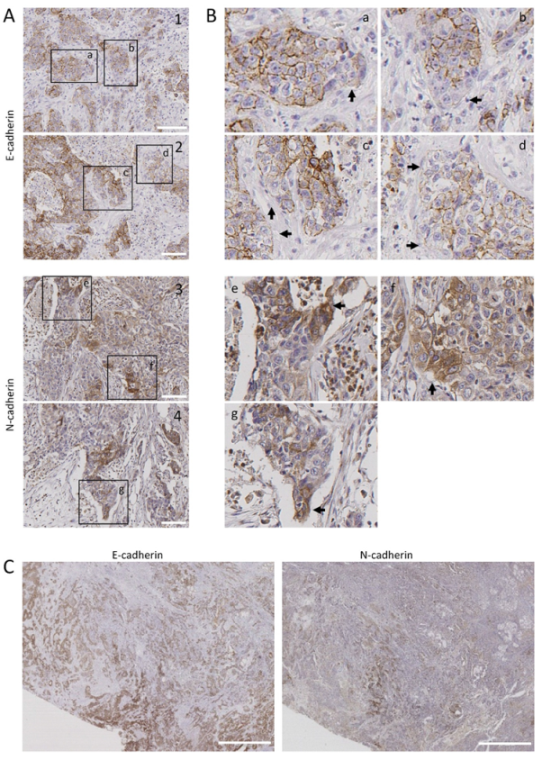
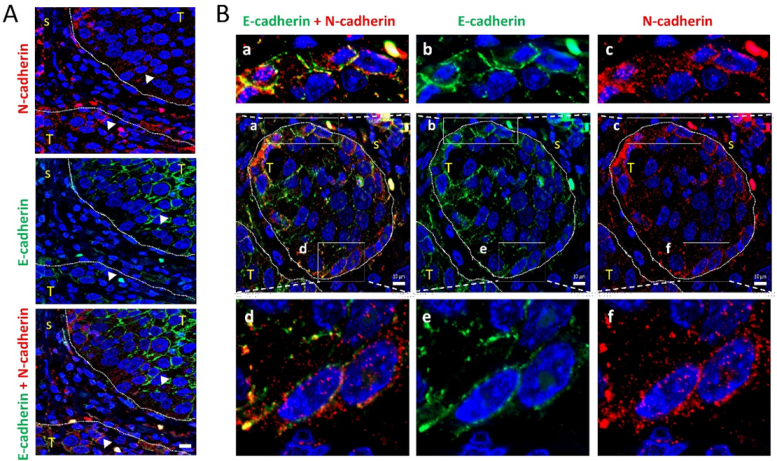
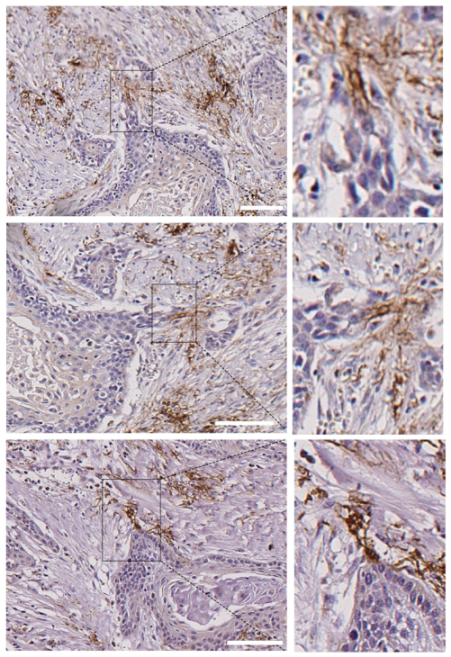
3.4. Leader-Like Positions of Collective Migration of N-Cadherin-Expressing Tumor Cells and CAFs in Human SCC Tissues
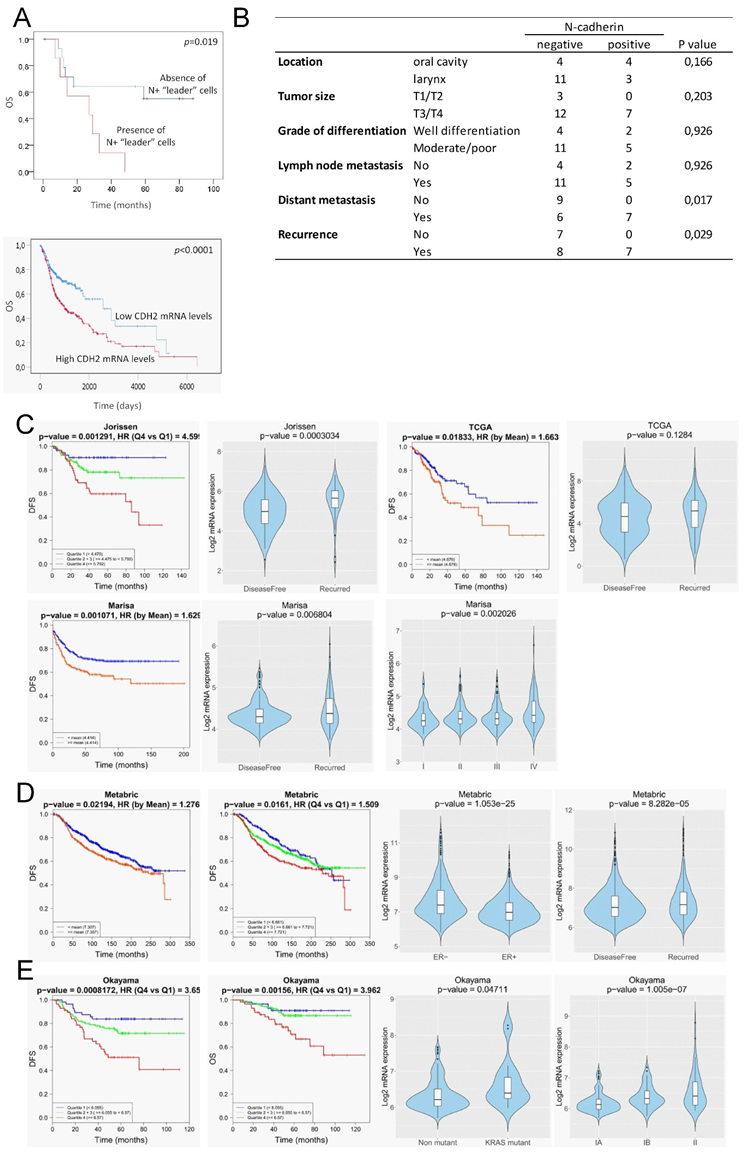
3.5. Clinical Outcome of Patients with Head and Neck SCC Containing “Leader” N-Cadherin-Expressing Cells
3.6. In Silico Analysis of N-Cadherin Aberrant Expression and Clinical Outcome in Patients with Breast, Colorectal and Lung Cancer
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Friedl, P.; Sahai, E.; Weiss, S.; Yamada, K.M. New dimensions in cell migration. Nat. Rev. Mol. Cell Biol. 2012, 13, 743–747. [Google Scholar] [CrossRef]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging Biological Principles of Metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Pascalis, C.; Etienne-Manneville, S. Single and collective cell migration: The mechanics of adhesions. Mol. Biol. Cell 2017, 28, 1833–1846. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [Green Version]
- Nieto, M.A.; Huang, R.Y.-J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savagner, P. Epithelial–Mesenchymal Transitions. Curr. Top. Dev. Biol. 2015, 112, 273–300. [Google Scholar]
- Wheelock, M.J.; Shintani, Y.; Maeda, M.; Fukumoto, Y.; Johnson, K.R. Cadherin switching. J. Cell Sci. 2008, 121, 727–735. [Google Scholar] [CrossRef] [Green Version]
- Friedl, P.; Mayor, R. Tuning Collective Cell Migration by Cell–Cell Junction Regulation. Cold Spring Harb. Perspect. Biol. 2017, 9, a029199. [Google Scholar] [CrossRef] [Green Version]
- Friedl, P.; Gilmour, D. Collective cell migration in morphogenesis, regeneration and cancer. Nat. Rev. Mol. Cell Biol. 2009, 10, 445–457. [Google Scholar] [CrossRef]
- Haeger, A.; Wolf, K.; Zegers, M.M.; Friedl, P. Collective cell migration: Guidance principles and hierarchies. Trends Cell Biol. 2015, 25, 556–566. [Google Scholar] [CrossRef]
- Campbell, K.; Casanova, J. A role for E-cadherin in ensuring cohesive migration of a heterogeneous population of non-epithelial cells. Nat. Commun. 2015, 6, 7998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaggioli, C.; Hooper, S.; Hidalgo-Carcedo, C.; Grosse, R.; Marshall, J.F.; Harrington, K.; Sahai, E. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat. Cell Biol. 2007, 9, 1392–1400. [Google Scholar] [CrossRef] [PubMed]
- Labernadie, A.; Kato, T.; Brugués, A.; Serra-Picamal, X.; Derzsi, S.; Arwert, E.; Weston, A.; González-Tarragó, V.; Elosegui-Artola, A.; Albertazzi, L.; et al. A mechanically active heterotypic E-cadherin/N-cadherin adhesion enables fibroblasts to drive cancer cell invasion. Nat. Cell Biol. 2017, 19, 224–237. [Google Scholar] [CrossRef] [PubMed]
- Friedl, P.; Noble, P.B.; Walton, P.A.; Laird, D.W.; Chauvin, P.J.; Tabah, R.J.; Black, M.; Zänker, K.S. Migration of coordinated cell clusters in mesenchymal and epithelial cancer explants in vitro. Cancer Res. 1995, 55, 4557–4560. [Google Scholar] [PubMed]
- Talmi, Y.P.; Takes, R.P.; Alon, E.E.; Nixon, I.J.; López, F.; de Bree, R.; Rodrigo, J.P.; Shaha, A.R.; Halmos, G.B.; Rinaldo, A.; et al. Prognostic value of lymph node ratio in head and neck squamous cell carcinoma. Head Neck 2018, 40, 1082–1090. [Google Scholar] [CrossRef]
- Sáenz-de-Santa-María, I.; Bernardo-Castiñeira, C.; Enciso, E.; García-Moreno, I.; Chiara, J.L.; Suarez, C.; Chiara, M.D. Control of long-distance cell-to-cell communication and autophagosome transfer in squamous cell carcinoma via tunneling nanotubes. Oncotarget 2017, 8, 20939–20960. [Google Scholar] [CrossRef] [Green Version]
- Anon, E.; Serra-Picamal, X.; Hersen, P.; Gauthier, N.C.; Sheetz, M.P.; Trepat, X.; Ladoux, B. Cell crawling mediates collective cell migration to close undamaged epithelial gaps. Proc. Natl. Acad. Sci. USA 2012, 109, 10891–10896. [Google Scholar] [CrossRef] [Green Version]
- Cortazar, A.R.; Torrano, V.; Martín-Martín, N.; Caro-Maldonado, A.; Camacho, L.; Hermanova, I.; Guruceaga, E.; Lorenzo-Martín, L.F.; Caloto, R.; Gomis, R.R.; et al. CANCERTOOL: A Visualization and Representation Interface to Exploit Cancer Datasets. Cancer Res. 2018, 78, 6320–6328. [Google Scholar] [CrossRef] [Green Version]
- Canel, M.; Secades, P.; Garzón-Arango, M.; Allonca, E.; Suarez, C.; Serrels, A.; Frame, M.C.; Brunton, V.; Chiara, M.-D. Involvement of focal adhesion kinase in cellular invasion of head and neck squamous cell carcinomas via regulation of MMP-2 expression. Br. J. Cancer 2008, 98, 1274–1284. [Google Scholar] [CrossRef] [Green Version]
- Richardson, A.M.; Havel, L.S.; Koyen, A.E.; Konen, J.M.; Shupe, J.; Wiles, W., IV; Martin, W.D.; Grossniklaus, H.E.; Sica, G.; Gilbert-Ross, M. Vimentin Is Required for Lung Adenocarcinoma Metastasis via Heterotypic Tumor Cell–Cancer-Associated Fibroblast Interactions during Collective Invasion. Clin. Cancer Res. 2018, 24, 420–432. [Google Scholar] [CrossRef] [Green Version]
- Roepman, P.; Schlicker, A.; Tabernero, J.; Majewski, I.; Tian, S.; Moreno, V.; Snel, M.H.; Chresta, C.M.; Rosenberg, R.; Nitsche, U.; et al. Colorectal cancer intrinsic subtypes predict chemotherapy benefit, deficient mismatch repair and epithelial-to-mesenchymal transition. Int. J. Cancer 2014, 134, 552–562. [Google Scholar] [CrossRef] [PubMed]
- Marisa, L.; de Reyniès, A.; Duval, A.; Selves, J.; Gaub, M.P.; Vescovo, L.; Etienne-Grimaldi, M.-C.; Schiappa, R.; Guenot, D.; Ayadi, M.; et al. Gene Expression Classification of Colon Cancer into Molecular Subtypes: Characterization, Validation, and Prognostic Value. PLoS Med. 2013, 10, e1001453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laibe, S.; Lagarde, A.; Ferrari, A.; Monges, G.; Birnbaum, D. Olschwang, and the COL2 Project, S. A Seven-Gene Signature Aggregates a Subgroup of Stage II Colon Cancers with Stage III. Omi. A J. Integr. Biol. 2012, 16, 560–565. [Google Scholar] [CrossRef] [PubMed]
- Kemper, K.; Versloot, M.; Cameron, K.; Colak, S.; e Melo, F.d.S.; de Jong, J.H.; Bleackley, J.; Vermeulen, L.; Versteeg, R.; Koster, J.; et al. Mutations in the Ras-Raf Axis Underlie the Prognostic Value of CD133 in Colorectal Cancer. Clin. Cancer Res. 2012, 18, 3132–3141. [Google Scholar] [CrossRef] [Green Version]
- Jorissen, R.N.; Gibbs, P.; Christie, M.; Prakash, S.; Lipton, L.; Desai, J.; Ørntoft, T.F. Metastasis-Associated Gene Expression Changes Predict Poor Outcomes in Patients with Dukes Stage B and C Colorectal Cancer. Clin. Cancer Res. 2009, 15, 7642–7651. [Google Scholar] [CrossRef] [Green Version]
- Ivshina, A.V.; George, J.; Senko, O.; Mow, B.; Putti, T.C.; Smeds, J.; Lindahl, T.; Pawitan, Y.; Hall, P.; Nordgren, H.; et al. Genetic Reclassification of Histologic Grade Delineates New Clinical Subtypes of Breast Cancer. Cancer Res. 2006, 66, 10292–10301. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Klijn, J.G.; Zhang, Y.; Sieuwerts, A.M.; Look, M.P.; Yang, F.; Talantov, D.; Timmermans, M.; Meijer-van Gelder, M.E.; Yu, J.; et al. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet 2005, 365, 671–679. [Google Scholar] [CrossRef]
- Pawitan, Y.; Bjöhle, J.; Amler, L.; Borg, A.L.; Egyhazi, S.; Hall, P.; Han, X.; Holmberg, L.; Huang, F.; Klaar, S.; et al. Gene expression profiling spares early breast cancer patients from adjuvant therapy: Derived and validated in two population-based cohorts. Breast Cancer Res. 2005, 7, R953. [Google Scholar] [CrossRef]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef]
- Lu, X.; Lu, X.; Wang, Z.C.; Iglehart, J.D.; Zhang, X.; Richardson, A.L. Predicting features of breast cancer with gene expression patterns. Breast Cancer Res. Treat. 2008, 108, 191–201. [Google Scholar] [CrossRef]
- Wilkerson, M.D.; Yin, X.; Walter, V.; Zhao, N.; Cabanski, C.R.; Hayward, M.C.; Miller, C.R.; Socinski, M.A.; Parsons, A.M.; Thorne, L.B.; et al. Differential Pathogenesis of Lung Adenocarcinoma Subtypes Involving Sequence Mutations, Copy Number, Chromosomal Instability, and Methylation. PLoS ONE 2012, 7, e36530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shedden, K.; Taylor, J.M.; Enkemann, S.A.; Tsao, M.S.; Yeatman, T.J.; Gerald, W.L.; Eschrich, S.; Jurisica, I.; Venkatraman, S.E.; Meyerson, M.; et al. Gene expression–based survival prediction in lung adenocarcinoma: A multi-site, blinded validation study. Nat. Med. 2008, 14, 822–827. [Google Scholar] [PubMed]
- Okayama, H.; Kohno, T.; Ishii, Y.; Shimada, Y.; Shiraishi, K.; Iwakawa, R.; Furuta, K.; Tsuta, K.; Shibata, T.; Yamamoto, S.; et al. dentification of Genes Upregulated in ALK-Positive and EGFR/KRAS/ALK-Negative Lung Adenocarcinomas. Cancer Res. 2012, 72, 100–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chitale, D.; Gong, Y.; Taylor, B.S.; Broderick, S.; Brennan, C.; Somwar, R.; Golas, B.; Wang, L.; Motoi, N.; Szoke, J.; et al. An integrated genomic analysis of lung cancer reveals loss of DUSP4 in EGFR-mutant tumors. Oncogene 2009, 28, 2773–2783. [Google Scholar] [CrossRef] [Green Version]
- Harrison, O.J.; Bahna, F.; Katsamba, P.S.; Jin, X.; Brasch, J.; Vendome, J.; Ahlsen, G.; Carroll, K.J.; Price, S.R.; Honig, B.; et al. Two-step adhesive binding by classical cadherins. Nat. Struct. Mol. Biol. 2010, 17, 348–357. [Google Scholar] [CrossRef]
- Carey, S.P.; Starchenko, A.; McGregor, A.L.; Reinhart-King, C.A. Leading malignant cells initiate collective epithelial cell invasion in a three-dimensional heterotypic tumor spheroid model. Clin. Exp. Metastasis 2013, 30, 615–630. [Google Scholar] [CrossRef] [Green Version]
- Shih, W.; Yamada, S. N-cadherin as a key regulator of collective cell migration in a 3D environment. Cell Adh. Migr. 2012, 6, 513–517. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Yu, T.; Zhang, Q.; Fu, Q.; Hu, Y.; Xiang, M.; Peng, H.; Zheng, T.; Lu, L.; Shi, H. Upregulated N-cadherin expression is associated with poor prognosis in epithelial-derived solid tumours: A meta-analysis. Eur. J. Clin. Investig. 2018, 48, e12903. [Google Scholar] [CrossRef]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saénz-de-Santa-María, I.; Celada, L.; Chiara, M.-D. The Leader Position of Mesenchymal Cells Expressing N-Cadherin in the Collective Migration of Epithelial Cancer. Cells 2020, 9, 731. https://doi.org/10.3390/cells9030731
Saénz-de-Santa-María I, Celada L, Chiara M-D. The Leader Position of Mesenchymal Cells Expressing N-Cadherin in the Collective Migration of Epithelial Cancer. Cells. 2020; 9(3):731. https://doi.org/10.3390/cells9030731
Chicago/Turabian StyleSaénz-de-Santa-María, Inés, Lucía Celada, and María-Dolores Chiara. 2020. "The Leader Position of Mesenchymal Cells Expressing N-Cadherin in the Collective Migration of Epithelial Cancer" Cells 9, no. 3: 731. https://doi.org/10.3390/cells9030731
APA StyleSaénz-de-Santa-María, I., Celada, L., & Chiara, M.-D. (2020). The Leader Position of Mesenchymal Cells Expressing N-Cadherin in the Collective Migration of Epithelial Cancer. Cells, 9(3), 731. https://doi.org/10.3390/cells9030731