Basal-Type Breast Cancer Stem Cells Over-Express Chromosomal Passenger Complex Proteins

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Line
2.2. Cancer Stem Cells Isolation
2.3. RNA Analyses
2.4. Xenotransplant Assays
2.5. BIRC5 Overexpression and Inhibition
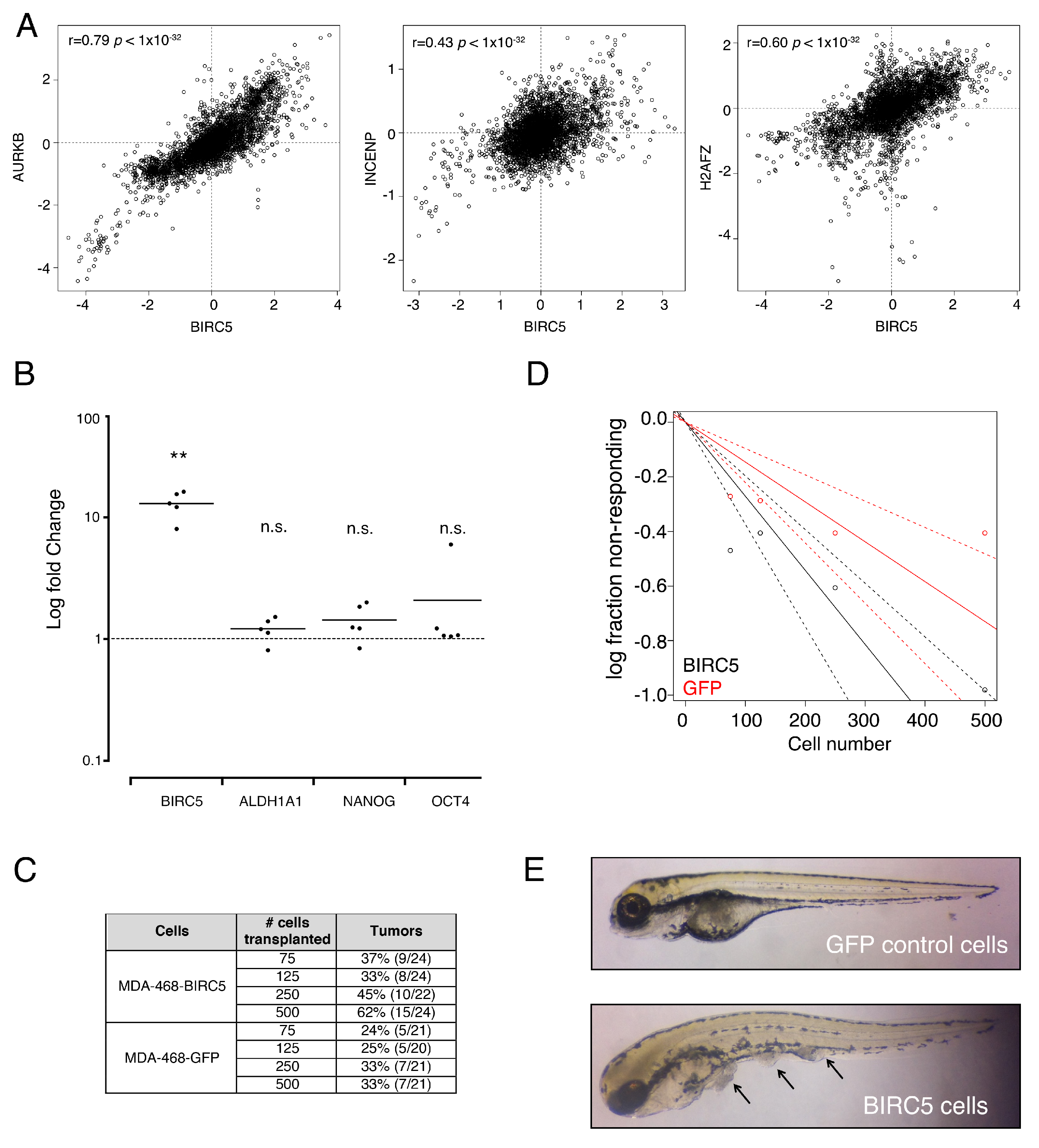
3. Results
3.1. Isolation of Breast Cancer Stem Cells
3.2. Breast Cancer Stem Cells Transcriptome Analysis
3.3. Genes from a Chromosomal Passenger Proteins Module (CPPM) are Overexpressed in CSCs
3.4. Genes from a CPPM Have Prognostic Significance in Breast Cancer Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Perou, C.M.; Borresen-Dale, A.L. Systems biology and genomics of breast cancer. Cold Spring Harb Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.; Ma, J.; Zou, Z.; Jemal, A. Cancer statistics, 2014. CA Cancer J. Clin. 2014, 64, 9–29. [Google Scholar] [CrossRef] [PubMed]
- Bertucci, F.; Finetti, P.; Birnbaum, D. Basal breast cancer: A complex and deadly molecular subtype. Curr. Mol. Med. 2012, 12, 96–110. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Wang, X.; Chen, G.Y.; Dalerba, P.; Gurney, A.; Hoey, T.; Sherlock, G.; Lewicki, J.; Shedden, K.; Clarke, M.F. The prognostic role of a gene signature from tumorigenic breast-cancer cells. N Engl. J. Med. 2007, 356, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Stingl, J.; Eirew, P.; Ricketson, I.; Shackleton, M.; Vaillant, F.; Choi, D.; Li, H.I.; Eaves, C.J. Purification and unique properties of mammary epithelial stem cells. Nature 2006, 439, 993–997. [Google Scholar] [CrossRef] [PubMed]
- Schwarz-Cruz, Y.C.A.; Espinosa, M.; Maldonado, V.; Melendez-Zajgla, J. Advances in the knowledge of breast Cancer Stem Cells. A Review. Histol. Histopathol. 2016, 31, 601. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. Bmc Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Leng, N.; Dawson, J.A.; Thomson, J.A.; Ruotti, V.; Rissman, A.I.; Smits, B.M.; Haag, J.D.; Gould, M.N.; Stewart, R.M.; Kendziorski, C. EBSeq: An empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics 2013, 29, 1035–1043. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U S A 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Okamura, Y.; Aoki, Y.; Obayashi, T.; Tadaka, S.; Ito, S.; Narise, T.; Kinoshita, K. COXPRESdb in 2015: Coexpression database for animal species by DNA-microarray and RNAseq-based expression data with multiple quality assessment systems. Nucleic Acids Res. 2015, 43, D82–D86. [Google Scholar] [CrossRef] [PubMed]
- Ringner, M.; Fredlund, E.; Hakkinen, J.; Borg, A.; Staaf, J. GOBO: Gene expression-based outcome for breast cancer online. PLoS ONE 2011, 6, e17911. [Google Scholar] [CrossRef] [PubMed]
- Varley, K.E.; Gertz, J.; Roberts, B.S.; Davis, N.S.; Bowling, K.M.; Kirby, M.K.; Nesmith, A.S.; Oliver, P.G.; Grizzle, W.E.; Forero, A.; et al. Recurrent read-through fusion transcripts in breast cancer. Breast Cancer Res. Treat. 2014, 146, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas, N. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Gyorffy, B.; Lanczky, A.; Eklund, A.C.; Denkert, C.; Budczies, J.; Li, Q.; Szallasi, Z. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res. Treat. 2010, 123, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Mihaly, Z.; Kormos, M.; Lanczky, A.; Dank, M.; Budczies, J.; Szasz, M.A.; Gyorffy, B. A meta-analysis of gene expression-based biomarkers predicting outcome after tamoxifen treatment in breast cancer. Breast Cancer Res. Treat. 2013, 140, 219–232. [Google Scholar] [CrossRef]
- Bharadwaj, R.; Yu, H. The spindle checkpoint, aneuploidy, and cancer. Oncogene 2004, 23, 2016–2027. [Google Scholar] [CrossRef]
- Sansregret, L.; Swanton, C. The Role of Aneuploidy in Cancer Evolution. Cold Spring Harb Perspect. Med. 2017, 7. [Google Scholar] [CrossRef]
- Hu, Y.; Smyth, G.K. ELDA: Extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J. Immunol. Methods 2009, 347, 70–78. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Invest. 2011, 121, 2750–2767. [Google Scholar] [CrossRef]
- Asiedu, M.K.; Ingle, J.N.; Behrens, M.D.; Radisky, D.C.; Knutson, K.L. TGFbeta/TNF(alpha)-mediated epithelial-mesenchymal transition generates breast cancer stem cells with a claudin-low phenotype. Cancer Res. 2011, 71, 4707–4719. [Google Scholar] [CrossRef] [PubMed]
- Memmi, E.M.; Sanarico, A.G.; Giacobbe, A.; Peschiaroli, A.; Frezza, V.; Cicalese, A.; Pisati, F.; Tosoni, D.; Zhou, H.; Tonon, G.; et al. p63 sustains self-renewal of mammary cancer stem cells through regulation of Sonic Hedgehog signaling. Proc. Natl. Acad. Sci. 2015, 112, 3499–3504. [Google Scholar] [CrossRef] [PubMed]
- Shostak, K.; Chariot, A. NF-kappaB, stem cells and breast cancer: The links get stronger. Breast Cancer Res. 2011, 13, 214. [Google Scholar] [CrossRef]
- Vermeulen, L.; De Sousa, E.M.F.; van der Heijden, M.; Cameron, K.; de Jong, J.H.; Borovski, T.; Tuynman, J.B.; Todaro, M.; Merz, C.; Rodermond, H.; et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat. Cell Biol. 2010, 12, 468–476. [Google Scholar] [CrossRef]
- Tam, W.L.; Lu, H.; Buikhuisen, J.; Soh, B.S.; Lim, E.; Reinhardt, F.; Wu, Z.J.; Krall, J.A.; Bierie, B.; Guo, W.; et al. Protein kinase C alpha is a central signaling node and therapeutic target for breast cancer stem cells. Cancer Cell 2013, 24, 347–364. [Google Scholar] [CrossRef] [PubMed]
- Ceppi, P.; Hadji, A.; Kohlhapp, F.J.; Pattanayak, A.; Hau, A.; Liu, X.; Liu, H.; Murmann, A.E.; Peter, M.E. CD95 and CD95L promote and protect cancer stem cells. Nat. Commun 2014, 5, 5238. [Google Scholar] [CrossRef] [PubMed]
- Pathak, S.; Multani, A.S. Aneuploidy, stem cells and cancer. EXS 2006, 96, 49–64. [Google Scholar]
- Carmena, M.; Wheelock, M.; Funabiki, H.; Earnshaw, W.C. The chromosomal passenger complex (CPC): From easy rider to the godfather of mitosis. Nat. Rev. Mol Cell Biol 2012, 13, 789–803. [Google Scholar] [CrossRef] [PubMed]
- Ling, X.; Marini, F.; Konopleva, M.; Schober, W.; Shi, Y.; Burks, J.; Clise-Dwyer, K.; Wang, R.Y.; Zhang, W.; Yuan, X.; et al. Mesenchymal Stem Cells Overexpressing IFN-beta Inhibit Breast Cancer Growth and Metastases through Stat3 Signaling in a Syngeneic Tumor Model. Cancer Microenviron 2010, 3, 83–95. [Google Scholar] [CrossRef]
- Shen, C.J.; Chan, T.F.; Chen, C.C.; Hsu, Y.C.; Long, C.Y.; Lai, C.S. Human umbilical cord matrix-derived stem cells expressing interferon-beta gene inhibit breast cancer cells via apoptosis. Oncotarget 2016, 7, 34172–34179. [Google Scholar] [CrossRef]
- Choi, H.S.; Kim, D.A.; Chung, H.; Park, I.H.; Kim, B.H.; Oh, E.S.; Kang, D.H. Screening of breast cancer stem cell inhibitors using a protein kinase inhibitor library. Cancer Cell Int. 2017, 17, 25. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.S.; Kim, J.H.; Kim, S.L.; Lee, D.S. Disruption of the NF-kappaB/IL-8 Signaling Axis by Sulconazole Inhibits Human Breast Cancer Stem Cell Formation. Cells 2019, 8, 1007. [Google Scholar] [CrossRef] [PubMed]
- Charpentier, M.S.; Whipple, R.A.; Vitolo, M.I.; Boggs, A.E.; Slovic, J.; Thompson, K.N.; Bhandary, L.; Martin, S.S. Curcumin targets breast cancer stem-like cells with microtentacles that persist in mammospheres and promote reattachment. Cancer Res. 2014, 74, 1250–1260. [Google Scholar] [CrossRef] [PubMed]
- Wolf, J.; Dewi, D.L.; Fredebohm, J.; Muller-Decker, K.; Flechtenmacher, C.; Hoheisel, J.D.; Boettcher, M. A mammosphere formation RNAi screen reveals that ATG4A promotes a breast cancer stem-like phenotype. Breast Cancer Res. 2013, 15, R109. [Google Scholar] [CrossRef]
- Liang, Y.; Zhong, Z.; Huang, Y.; Deng, W.; Cao, J.; Tsao, G.; Liu, Q.; Pei, D.; Kang, T.; Zeng, Y.X. Stem-like cancer cells are inducible by increasing genomic instability in cancer cells. J. Biol. Chem. 2010, 285, 4931–4940. [Google Scholar] [CrossRef]
- Nguyen, H.G.; Ravid, K. Tetraploidy/aneuploidy and stem cells in cancer promotion: The role of chromosome passenger proteins. J. Cell Physiol. 2006, 208, 12–22. [Google Scholar] [CrossRef]
- Siddharth, S.; Das, S.; Nayak, A.; Kundu, C.N. SURVIVIN as a marker for quiescent-breast cancer stem cells-An intermediate, adherent, pre-requisite phase of breast cancer metastasis. Clin. Exp. Metastasis 2016, 33, 661–675. [Google Scholar] [CrossRef]
- Yu, C.J.; Ou, J.H.; Wang, M.L.; Jialielihan, N.; Liu, Y.H. Elevated survivin mediated multidrug resistance and reduced apoptosis in breast cancer stem cells. J. Buon. 2015, 20, 1287–1294. [Google Scholar]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Kaklamani, V. A genetic signature can predict prognosis and response to therapy in breast cancer: Oncotype DX. Expert Rev. Mol. Diagn 2006, 6, 803–809. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schwarz-Cruz y Celis, A.; Ceballos-Cancino, G.; Vazquez-Santillan, K.; Espinosa, M.; Zampedri, C.; Bahena, I.; Ruiz, V.; Maldonado, V.; Melendez-Zajgla, J. Basal-Type Breast Cancer Stem Cells Over-Express Chromosomal Passenger Complex Proteins. Cells 2020, 9, 709. https://doi.org/10.3390/cells9030709
Schwarz-Cruz y Celis A, Ceballos-Cancino G, Vazquez-Santillan K, Espinosa M, Zampedri C, Bahena I, Ruiz V, Maldonado V, Melendez-Zajgla J. Basal-Type Breast Cancer Stem Cells Over-Express Chromosomal Passenger Complex Proteins. Cells. 2020; 9(3):709. https://doi.org/10.3390/cells9030709
Chicago/Turabian StyleSchwarz-Cruz y Celis, Angela, Gisela Ceballos-Cancino, Karla Vazquez-Santillan, Magali Espinosa, Cecilia Zampedri, Ivan Bahena, Victor Ruiz, Vilma Maldonado, and Jorge Melendez-Zajgla. 2020. "Basal-Type Breast Cancer Stem Cells Over-Express Chromosomal Passenger Complex Proteins" Cells 9, no. 3: 709. https://doi.org/10.3390/cells9030709
APA StyleSchwarz-Cruz y Celis, A., Ceballos-Cancino, G., Vazquez-Santillan, K., Espinosa, M., Zampedri, C., Bahena, I., Ruiz, V., Maldonado, V., & Melendez-Zajgla, J. (2020). Basal-Type Breast Cancer Stem Cells Over-Express Chromosomal Passenger Complex Proteins. Cells, 9(3), 709. https://doi.org/10.3390/cells9030709