Functional Alterations in Ciliogenesis-Associated Kinase 1 (CILK1) that Result from Mutations Linked to Juvenile Myoclonic Epilepsy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Plasmids and Antibodies
2.2. Cell Culture and Transfection
2.3. Immunoprecipitation and Immunoblotting
2.4. Confocal Immunofluorescence Microscopy and Imaging
2.5. Statistical Analysis
3. Results
3.1. CILK1 Variants Lack Restrictive Effects on Cilia Length
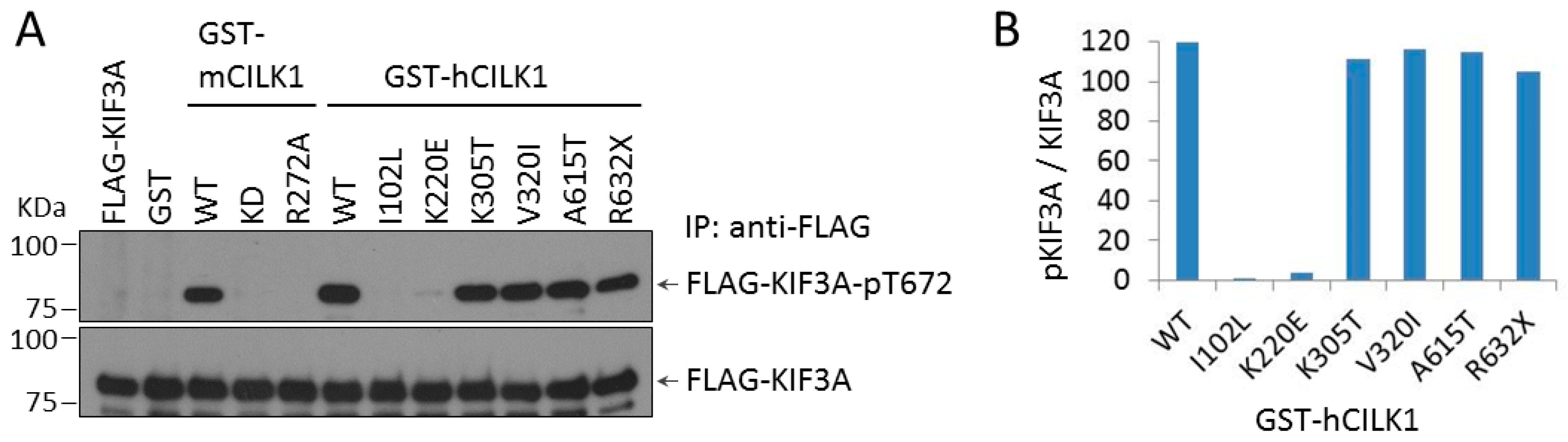
3.2. Phosphorylation of KIF3A by CILK1 Variants Found in Human JME
3.3. CILK1 Variants Gain the Ability to Promote Ciliogenesis
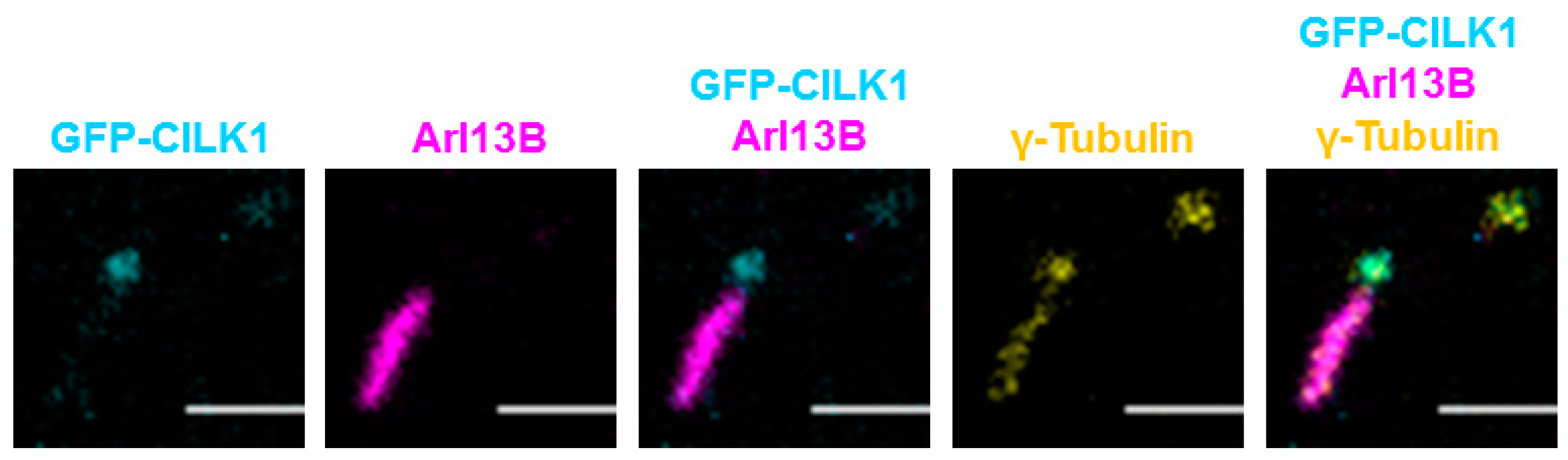
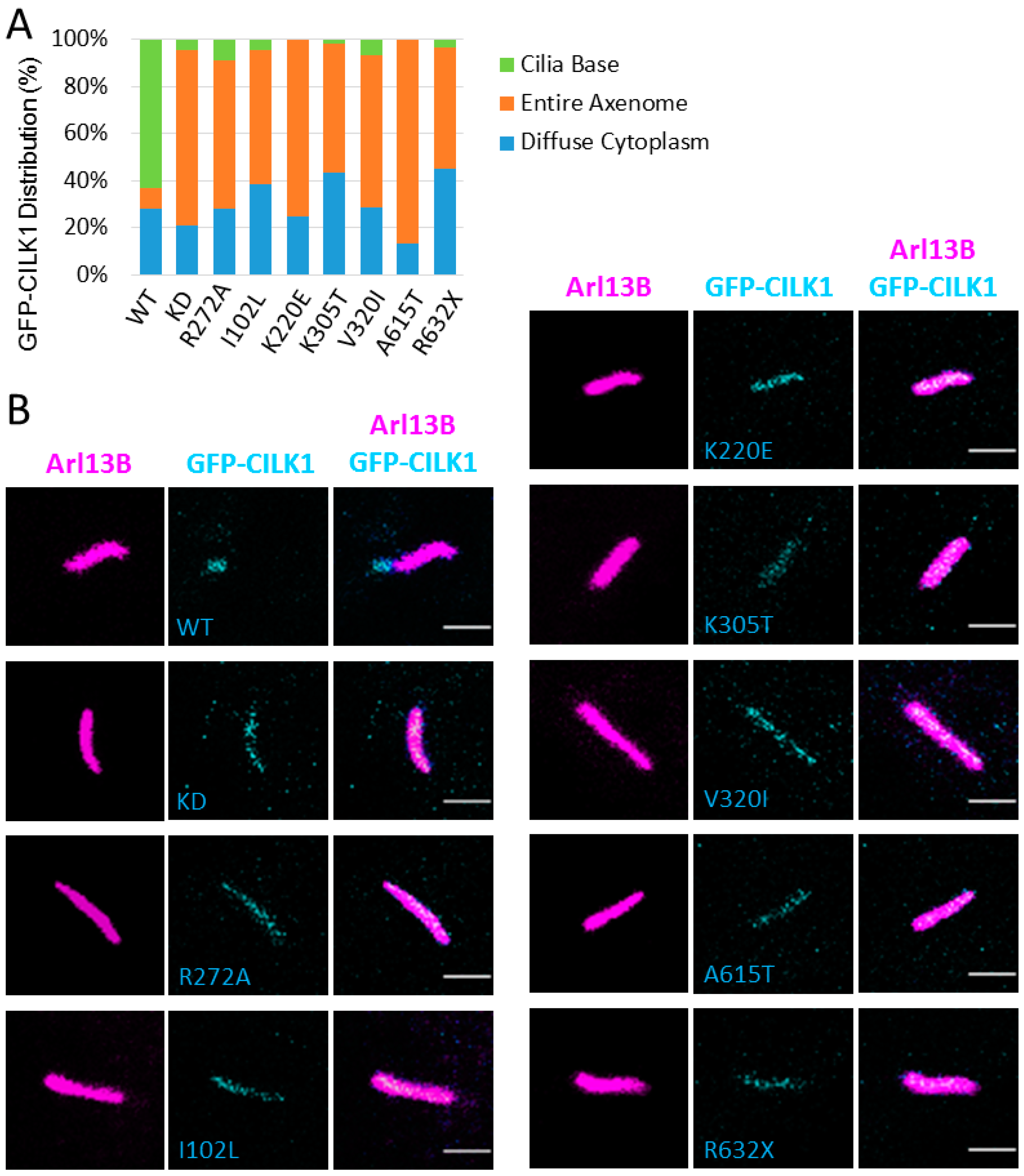
3.4. CILK1 Variants Are Mis-Localized Along the Axoneme of the Primary Cilium
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Malicki, J.; Johnson, C. The Cilium: Cellular Antenna and Central Processing Unit. Trends Cell Biol. 2016, 27, 126–140. [Google Scholar] [CrossRef] [PubMed]
- Reiter, J.F.; Leroux, M. Genes and molecular pathways underpinning ciliopathies. Nat. Rev. Mol. Cell Biol. 2017, 18, 533–547. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Gailey, C.D.; Wang, E.J.; Brautigan, D.L. Ciliogenesis associated kinase 1: Targets and functions in various organ systems. FEBS Lett. 2019, 593, 2990–3002. [Google Scholar] [CrossRef] [PubMed]
- Abe, S.; Yagi, T.; Ishiyama, S.; Hiroe, M.; Marumo, F.; Ikawa, Y. Molecular cloning of a novel serine/threonine kinase, MRK, possibly involved in cardiac development. Oncogene 1995, 11, 2187–2195. [Google Scholar]
- Togawa, K.; Yan, Y.X.; Inomoto, T.; Slaugenhaupt, S.; Rustgi, A.K. Intestinal cell kinase (ICK) localizes to the crypt region and requires a dual phosphorylation site found in map kinases. J. Cell. Physiol. 2000, 183, 129–139. [Google Scholar] [CrossRef]
- Matsushime, H.; Jinno, A.; Takagi, N.; Shibuya, M. A novel mammalian protein kinase gene (mak) is highly expressed in testicular germ cells at and after meiosis. Mol. Cell. Biol. 1990, 10, 2261–2268. [Google Scholar] [CrossRef]
- Bladt, F.; Birchmeier, C. Characterization and expression analysis of the murine rck gene: A protein kinase with a potential function in sensory cells. Differentiation 1993, 53, 115–122. [Google Scholar] [CrossRef]
- Miyata, Y.; Akashi, M.; Nishida, E. Molecular cloning and characterization of a novel member of the MAP kinase superfamily. Genes Cells 1999, 4, 299–309. [Google Scholar] [CrossRef]
- Fu, Z.; Schroeder, M.J.; Shabanowitz, J.; Kaldis, P.; Togawa, K.; Rustgi, A.K.; Hunt, N.F.; Sturgill, T.W. Activation of a Nuclear Cdc2-Related Kinase within a Mitogen-Activated Protein Kinase-Like TDY Motif by Autophosphorylation and Cyclin-Dependent Protein Kinase-Activating Kinase. Mol. Cell. Biol. 2005, 25, 6047–6064. [Google Scholar] [CrossRef]
- Fu, Z.; Larson, K.A.; Chitta, R.K.; Parker, S.A.; Turk, B.E.; Lawrence, M.W.; Kaldis, P.; Galaktionov, K.; Cohn, S.M.; Shabanowitz, J.; et al. Identification of Yin-Yang Regulators and a Phosphorylation Consensus for Male Germ Cell-Associated Kinase (MAK)-Related Kinase. Mol. Cell. Biol. 2006, 26, 8639–8654. [Google Scholar] [CrossRef]
- Bosakova, M.K.; Nita, A.; Gregor, T.; Varecha, M.; Gudernova, I.; Fafilek, B.; Barta, T.; Basheer, N.; Abraham, S.P.; Balek, L.; et al. Fibroblast growth factor receptor influences primary cilium length through an interaction with intestinal cell kinase. Proc. Natl. Acad. Sci. USA 2019, 116, 4316–4325. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.S.; Wang, E.; Gailey, C.D.; Brautigan, D.L.; Allen, B.L.; Fu, Z. Ciliopathy-Associated Protein Kinase ICK Requires Its Non-Catalytic Carboxyl-Terminal Domain for Regulation of Ciliogenesis. Cells 2019, 8, 677. [Google Scholar] [CrossRef] [PubMed]
- Chaya, T.; Omori, Y.; Kuwahara, R.; Furukawa, T. ICK is essential for cell type-specific ciliogenesis and the regulation of ciliary transport. EMBO J. 2014, 33, 1227–1242. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Park, S.H.; Wu, D.; Xu, W.; Guillot, S.J.; Jin, L.; Li, X.; Wang, Y.; Lin, C.-S.; Fu, Z. An essential role of intestinal cell kinase in lung development is linked to the perinatal lethality of human ECO syndrome. FEBS Lett. 2017, 591, 1247–1257. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Chapman, J.R.; Wang, L.; Harris, T.E.; Shabanowitz, J.; Hunt, D.F.; Fu, Z. Intestinal Cell Kinase (ICK) Promotes Activation of mTOR Complex 1 (mTORC1) through Phosphorylation of Raptor Thr-908. J. Biol. Chem. 2012, 287, 12510–12519. [Google Scholar] [CrossRef] [PubMed]
- Burghoorn, J.; Dekkers, M.P.J.; Rademakers, S.; De Jong, T.; Willemsen, R.; Jansen, G. Mutation of the MAP kinase DYF-5 affects docking and undocking of kinesin-2 motors and reduces their speed in the cilia of Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2007, 104, 7157–7162. [Google Scholar] [CrossRef]
- Berman, S.A.; Wilson, N.F.; Haas, N.A.; Lefebvre, P.A. A novel MAP kinase regulates flagellar length in Chlamydomonas. Curr. Biol. 2003, 13, 1145–1149. [Google Scholar] [CrossRef]
- Bengs, F.; Scholz, A.; Kühn, D.; Wiese, M. LmxMPK9, a mitogen-activated protein kinase homologue affects flagellar length in Leishmania mexicana. Mol. Microbiol. 2005, 55, 1606–1615. [Google Scholar] [CrossRef]
- Jiang, Y.Y.; Maier, W.; Baumeister, R.; Minevich, G.; Joachimiak, E.; Wloga, D.; Ruan, Z.; Kannan, N.; Bocarro, S.; Bahraini, A.; et al. LF4/MOK and a CDK-related kinase regulate the number and length of cilia in Tetrahymena. PLOS Genet. 2019, 15, e1008099. [Google Scholar] [CrossRef]
- Broekhuis, J.R.; Verhey, K.J.; Jansen, G. Regulation of Cilium Length and Intraflagellar Transport by the RCK-Kinases ICK and MOK in Renal Epithelial Cells. PLoS ONE 2014, 9, e108470. [Google Scholar] [CrossRef]
- Omori, Y.; Chaya, T.; Katoh, K.; Kajimura, N.; Sato, S.; Muraoka, K.; Ueno, S.; Koyasu, T.; Kondo, M.; Furukawa, T. Negative regulation of ciliary length by ciliary male germ cell-associated kinase (Mak) is required for retinal photoreceptor survival. Proc. Natl. Acad. Sci. USA 2010, 107, 22671–22676. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.; Song, J.; Shin, J.-O.; Lee, H.; Kim, H.-K.; Eggenschwiller, J.T.; Bok, J.; Ko, H.W. Intestinal cell kinase, a protein associated with endocrine-cerebro-osteodysplasia syndrome, is a key regulator of cilia length and Hedgehog signaling. Proc. Natl. Acad. Sci. USA 2014, 111, 8541–8546. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.P.; Bosakova, M.K.; Varecha, M.; Balek, L.; Barta, T.; Trantírek, L.; Jelinkova, I.; Duran, I.; Vesela, I.; Forlenza, K.N.; et al. An inactivating mutation in intestinal cell kinase, ICK, impairs hedgehog signalling and causes short rib-polydactyly syndrome. Hum. Mol. Genet. 2016, 25, 3998–4011. [Google Scholar] [CrossRef] [PubMed]
- Lahiry, P.; Wang, J.; Robinson, J.F.; Turowec, J.P.; Litchfield, D.W.; Lanktree, M.; Gloor, G.B.; Puffenberger, E.; Strauss, K.A.; Martens, M.B.; et al. A Multiplex Human Syndrome Implicates a Key Role for Intestinal Cell Kinase in Development of Central Nervous, Skeletal, and Endocrine Systems. Am. J. Hum. Genet. 2009, 84, 134–147. [Google Scholar] [CrossRef]
- Oud, M.M.; Bonnard, C.; Mans, D.A.; Altunoğlu, U.; Tohari, S.; Ng, A.Y.J.; Eskin, A.; Lee, H.; Rupar, C.A.; De Wagenaar, N.P.; et al. A novel ICK mutation causes ciliary disruption and lethal endocrine-cerebro-osteodysplasia syndrome. Cilia 2016, 5, 8. [Google Scholar] [CrossRef]
- Ding, M.; Jin, L.; Xie, L.; Park, S.H.; Tong, Y.; Wu, D.; Chhabra, A.B.; Fu, Z.; Liang, H. A Murine Model for Human ECO Syndrome Reveals a Critical Role of Intestinal Cell Kinase in Skeletal Development. Calcif. Tissue Int. 2017, 102, 348–357. [Google Scholar] [CrossRef]
- Engelke, M.; Waas, B.; Kearns, S.E.; Suber, A.; Boss, A.; Allen, B.L.; Verhey, K.J. Acute Inhibition of Heterotrimeric Kinesin-2 Function Reveals Mechanisms of Intraflagellar Transport in Mammalian Cilia. Curr. Biol. 2019, 29, 1137–1148.e4. [Google Scholar] [CrossRef]
- Bailey, J.N.; De Nijs, L.; Bai, N.; Suzuki, T.; Miyamoto, H.; Tanaka, M.; Patterson, C.; Lin, Y.-C.; Medina, M.T.; Alonso, M.E.; et al. Variant Intestinal-Cell Kinase in Juvenile Myoclonic Epilepsy. New Engl. J. Med. 2018, 378, 1018–1028. [Google Scholar] [CrossRef]
- Sambrook, J.; Russell, D.W. Calcium-phosphate-mediated Transfection of Eukaryotic Cells with Plasmid DNAs. Cold Spring Harb. Protoc. 2006, 2006. [Google Scholar] [CrossRef]
- Dummer, A.; Poelma, C.; DeRuiter, M.C.; Goumans, M.J.; Hierck, B. Measuring the primary cilium length: Improved method for unbiased high-throughput analysis. Cilia 2016, 5, 7. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, E.J.; Gailey, C.D.; Brautigan, D.L.; Fu, Z. Functional Alterations in Ciliogenesis-Associated Kinase 1 (CILK1) that Result from Mutations Linked to Juvenile Myoclonic Epilepsy. Cells 2020, 9, 694. https://doi.org/10.3390/cells9030694
Wang EJ, Gailey CD, Brautigan DL, Fu Z. Functional Alterations in Ciliogenesis-Associated Kinase 1 (CILK1) that Result from Mutations Linked to Juvenile Myoclonic Epilepsy. Cells. 2020; 9(3):694. https://doi.org/10.3390/cells9030694
Chicago/Turabian StyleWang, Eric J., Casey D. Gailey, David L. Brautigan, and Zheng Fu. 2020. "Functional Alterations in Ciliogenesis-Associated Kinase 1 (CILK1) that Result from Mutations Linked to Juvenile Myoclonic Epilepsy" Cells 9, no. 3: 694. https://doi.org/10.3390/cells9030694
APA StyleWang, E. J., Gailey, C. D., Brautigan, D. L., & Fu, Z. (2020). Functional Alterations in Ciliogenesis-Associated Kinase 1 (CILK1) that Result from Mutations Linked to Juvenile Myoclonic Epilepsy. Cells, 9(3), 694. https://doi.org/10.3390/cells9030694