Modulation of the Pol II CTD Phosphorylation Code by Rac1 and Cdc42 Small GTPases in Cultured Human Cancer Cells and Its Implication for Developing a Synthetic-Lethal Cancer Therapy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Human Cancer Cell Cultures
2.2. siRNA Transfection
2.3. Inhibitor Treatments
2.4. Western Blot
2.5. Quantitative Analysis of Western Blots and Cell Counting
3. Results
3.1. Treatments by Rho/Rac/Cdc42 Inhibitors Decreased the Pol II CTD Ser2 and Ser5 Phosphorylation Levels
3.2. Rac1 and Cdc42 Knockdown by siRNA Decreased CTD Ser2P and Ser5P Levels and Differentially Affected Levels of CTD Phosphatases and Kinases
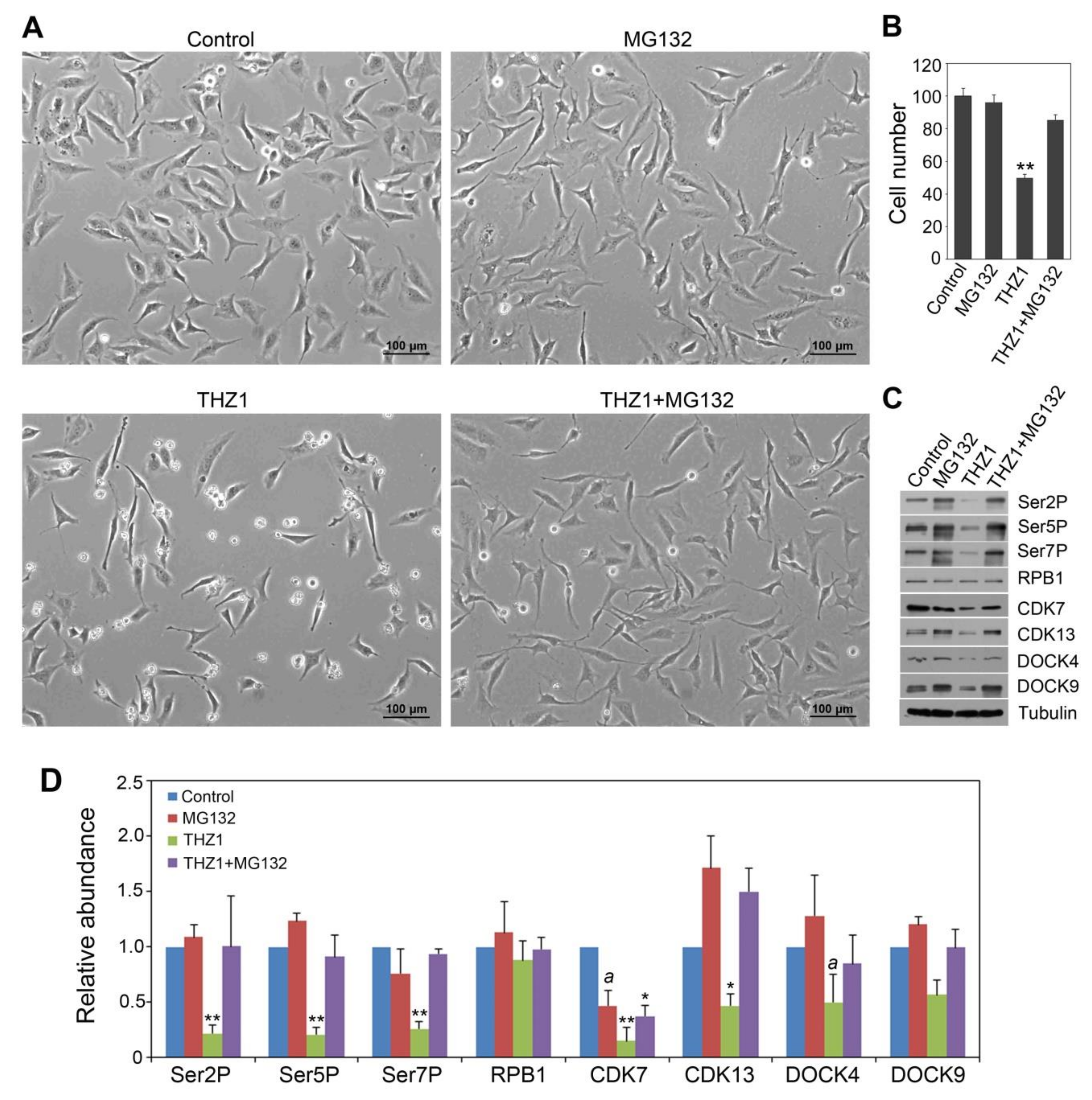
3.3. Effects of THZ1 in CTD Code Modulation and Accumulation of CDK and DOCK Proteins Can Be Reversed by MG132 Treatment
3.4. Serum Deprivation and Torin1 Treatments Enhanced the THZ1 Effect
4. Discussion
4.1. Modulation of the Pol II CTD Phosphorylation Code by Rho Family GTPases Is Likely Conserved in Cultured Human Cells via Regulating CTD Kinases and Phosphatases
4.2. A Hypothetical Model for the Rho-Pol II signaling shortcut in transcriptional control in cultured human cells
4.3. Potential for Developing a Synthetic-Lethal Cancer Therapy
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Jaffe, A.B.; Hall, A. Rho GTPases: Biochemistry and biology. Annu. Rev. Cell Dev. Biol. 2005, 21, 247–269. [Google Scholar] [CrossRef]
- Lin, Y.; Zheng, Y. Approaches of targeting Rho GTPases in cancer drug discovery. Expert Opin. Drug Discov. 2015, 10, 991–1010. [Google Scholar] [CrossRef] [PubMed]
- Porter, A.P.; Papaioannou, A.; Malliri, A. Deregulation of Rho GTPases in cancer. Small GTPases 2016, 7, 123–138. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, G.A.; Der, C.J.; Rossman, K.L. RAS isoforms and mutations in cancer at a glance. J. Cell Sci. 2016, 129, 1287–1292. [Google Scholar] [CrossRef] [PubMed]
- Bustelo, X.R.; Crespo, P.; Fernandez-Pisonero, I.; Rodriguez-Fdez, S. RAS GTPase-dependent pathways in developmental diseases: Old guys, new lads, and current challenges. Curr. Opin. Cell Biol. 2018, 55, 42–51. [Google Scholar] [CrossRef]
- Zheng, Z.L. Ras and Rho GTPase regulation of Pol II transcription: A shortcut model revisited. Transcription 2017, 8, 268–274. [Google Scholar] [CrossRef][Green Version]
- Chang, Y.W.; Howard, S.C.; Herman, P.K. The Ras/PKA signaling pathway directly targets the Srb9 protein, a component of the general RNA polymerase II transcription apparatus. Mol. Cell 2004, 15, 107–116. [Google Scholar] [CrossRef]
- Zhang, B.; Yang, G.; Chen, Y.; Zhao, Y.; Gao, P.; Liu, B.; Wang, H.; Zheng, Z.L. C-terminal domain (CTD) phosphatase links Rho GTPase signaling to Pol II CTD phosphorylation in Arabidopsis and yeast. Proc. Natl. Acad. Sci. USA 2016, 113, E8197–E8206. [Google Scholar] [CrossRef]
- Hajheidari, M.; Koncz, C.; Eick, D. Emerging roles for RNA polymerase II CTD in Arabidopsis. Trends Plant Sci. 2013, 18, 633–643. [Google Scholar] [CrossRef]
- Aristizabal, M.J.; Kobor, M.S. A single flexible RNAPII-CTD integrates many different transcriptional programs. Transcription 2016, 7, 50–56. [Google Scholar] [CrossRef][Green Version]
- Jeronimo, C.; Collin, P.; Robert, F. The RNA Polymerase II CTD: The Increasing Complexity of a Low-Complexity Protein Domain. J. Mol. Biol. 2016, 428, 2607–2622. [Google Scholar] [CrossRef] [PubMed]
- Zaborowska, J.; Egloff, S.; Murphy, S. The pol II CTD: New twists in the tail. Nat. Struct. Mol. Biol. 2016, 23, 771–777. [Google Scholar] [CrossRef] [PubMed]
- Jansen, B.; Schlagbauer-Wadl, H.; Kahr, H.; Heere-Ress, E.; Mayer, B.X.; Eichler, H.; Pehamberger, H.; Gana-Weisz, M.; Ben-David, E.; Kloog, Y.; et al. Novel Ras antagonist blocks human melanoma growth. Proc. Natl. Acad. Sci. USA 1999, 96, 14019–14024. [Google Scholar] [CrossRef] [PubMed]
- Shang, X.; Marchioni, F.; Evelyn, C.R.; Sipes, N.; Zhou, X.; Seibel, W.; Wortman, M.; Zheng, Y. Small-molecule inhibitors targeting G-protein-coupled Rho guanine nucleotide exchange factors. Proc. Natl. Acad. Sci. USA 2013, 110, 3155–3160. [Google Scholar] [CrossRef] [PubMed]
- Montalvo-Ortiz, B.L.; Castillo-Pichardo, L.; Hernandez, E.; Humphries-Bickley, T.; De la Mota-Peynado, A.; Cubano, L.A.; Vlaar, C.P.; Dharmawardhane, S. Characterization of EHop-016, novel small molecule inhibitor of Rac GTPase. J. Biol. Chem. 2012, 287, 13228–13238. [Google Scholar] [CrossRef] [PubMed]
- Surviladze, Z.; Waller, A.; Strouse, J.J.; Bologa, C.; Ursu, O.; Salas, V.; Parkinson, J.F.; Phillips, G.K.; Romero, E.; Wandinger-Ness, A.; et al. A Potent and Selective Inhibitor of Cdc42 GTPase. In Probe Reports from the NIH Molecular Libraries Program. In Probe Reports from the NIH Molecular Libraries Program; National Center for Biotechnology Information (US): Bethesda, MD, USA,, 2010. [Google Scholar]
- Czuchra, A.; Wu, X.; Meyer, H.; van Hengel, J.; Schroeder, T.; Geffers, R.; Rottner, K.; Brakebusch, C. Cdc42 is not essential for filopodium formation, directed migration, cell polarization, and mitosis in fibroblastoid cells. Mol. Biol. Cell 2005, 16, 4473–4484. [Google Scholar] [CrossRef]
- Lin, P.S.; Dubois, M.F.; Dahmus, M.E. TFIIF-associating carboxyl-terminal domain phosphatase dephosphorylates phosphoserines 2 and 5 of RNA polymerase II. J. Biol. Chem. 2002, 277, 45949–45956. [Google Scholar] [CrossRef]
- Egloff, S.; Zaborowska, J.; Laitem, C.; Kiss, T.; Murphy, S. Ser7 phosphorylation of the CTD recruits the RPAP2 Ser5 phosphatase to snRNA genes. Mol. Cell 2012, 45, 111–122. [Google Scholar] [CrossRef]
- Akhtar, M.S.; Heidemann, M.; Tietjen, J.R.; Zhang, D.W.; Chapman, R.D.; Eick, D.; Ansari, A.Z. TFIIH kinase places bivalent marks on the carboxy-terminal domain of RNA polymerase II. Mol. Cell 2009, 34, 387–393. [Google Scholar] [CrossRef]
- Greifenberg, A.K.; Honig, D.; Pilarova, K.; Duster, R.; Bartholomeeusen, K.; Bosken, C.A.; Anand, K.; Blazek, D.; Geyer, M. Structural and Functional Analysis of the Cdk13/Cyclin K Complex. Cell Rep. 2016, 14, 320–331. [Google Scholar] [CrossRef]
- Kwiatkowski, N.; Zhang, T.; Rahl, P.B.; Abraham, B.J.; Reddy, J.; Ficarro, S.B.; Dastur, A.; Amzallag, A.; Ramaswamy, S.; Tesar, B.; et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature 2014, 511, 616–620. [Google Scholar] [CrossRef] [PubMed]
- Chipumuro, E.; Marco, E.; Christensen, C.L.; Kwiatkowski, N.; Zhang, T.; Hatheway, C.M.; Abraham, B.J.; Sharma, B.; Yeung, C.; Altabef, A.; et al. CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell 2014, 159, 1126–1139. [Google Scholar] [CrossRef] [PubMed]
- Christensen, C.L.; Kwiatkowski, N.; Abraham, B.J.; Carretero, J.; Al-Shahrour, F.; Zhang, T.; Chipumuro, E.; Herter-Sprie, G.S.; Akbay, E.A.; Altabef, A.; et al. Targeting transcriptional addictions in small cell lung cancer with a covalent CDK7 inhibitor. Cancer Cell 2014, 26, 909–922. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, T.; Kwiatkowski, N.; Abraham, B.J.; Lee, T.I.; Xie, S.; Yuzugullu, H.; Von, T.; Li, H.; Lin, Z.; et al. CDK7-dependent transcriptional addiction in triple-negative breast cancer. Cell 2015, 163, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Hur, J.Y.; Kim, H.R.; Lee, J.Y.; Park, S.; Hwang, J.A.; Kim, W.S.; Yoon, S.; Choi, C.M.; Rho, J.K.; Lee, J.C. CDK7 inhibition as a promising therapeutic strategy for lung squamous cell carcinomas with a SOX2 amplification. Cell. Oncol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Meller, N.; Irani-Tehrani, M.; Kiosses, W.B.; Del Pozo, M.A.; Schwartz, M.A. Zizimin1, a novel Cdc42 activator, reveals a new GEF domain for Rho proteins. Nat. Cell Biol. 2002, 4, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Kwofie, M.A.; Skowronski, J. Specific recognition of Rac2 and Cdc42 by DOCK2 and DOCK9 guanine nucleotide exchange factors. J. Biol. Chem. 2008, 283, 3088–3096. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, Z.; Roe, S.M.; Marshall, C.J.; Barford, D. Activation of Rho GTPases by DOCK exchange factors is mediated by a nucleotide sensor. Science 2009, 325, 1398–1402. [Google Scholar] [CrossRef]
- Abraham, S.; Scarcia, M.; Bagshaw, R.D.; McMahon, K.; Grant, G.; Harvey, T.; Yeo, M.; Esteves, F.O.G.; Thygesen, H.H.; Jones, P.F.; et al. A Rac/Cdc42 exchange factor complex promotes formation of lateral filopodia and blood vessel lumen morphogenesis. Nat. Commun. 2015, 6, 7286. [Google Scholar] [CrossRef]
- Yajnik, V.; Paulding, C.; Sordella, R.; McClatchey, A.I.; Saito, M.; Wahrer, D.C.; Reynolds, P.; Bell, D.W.; Lake, R.; van den Heuvel, S.; et al. DOCK4, a GTPase activator, is disrupted during tumorigenesis. Cell 2003, 112, 673–684. [Google Scholar] [CrossRef]
- Upadhyay, G.; Goessling, W.; North, T.E.; Xavier, R.; Zon, L.I.; Yajnik, V. Molecular association between beta-catenin degradation complex and Rac guanine exchange factor DOCK4 is essential for Wnt/beta-catenin signaling. Oncogene 2008, 27, 5845–5855. [Google Scholar] [CrossRef] [PubMed]
- Kuo, K.T.; Guan, B.; Feng, Y.; Mao, T.L.; Chen, X.; Jinawath, N.; Wang, Y.; Kurman, R.J.; Shih Ie, M.; Wang, T.L. Analysis of DNA copy number alterations in ovarian serous tumors identifies new molecular genetic changes in low-grade and high-grade carcinomas. Cancer Res. 2009, 69, 4036–4042. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.R.; Tai, Y.; Jin, Y.; Hammell, M.C.; Wilkinson, J.E.; Roe, J.S.; Vakoc, C.R.; Van Aelst, L. TGF-beta/Smad signaling through DOCK4 facilitates lung adenocarcinoma metastasis. Genes Dev. 2015, 29, 250–261. [Google Scholar] [CrossRef] [PubMed]
- Sundaravel, S.; Kuo, W.L.; Jeong, J.J.; Choudhary, G.S.; Gordon-Mitchell, S.; Liu, H.; Bhagat, T.D.; McGraw, K.L.; Gurbuxani, S.; List, A.F.; et al. Loss of Function of DOCK4 in Myelodysplastic Syndromes Stem Cells is Restored by Inhibitors of DOCK4 Signaling Networks. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 5638–5649. [Google Scholar] [CrossRef] [PubMed]
- Hirata, E.; Yukinaga, H.; Kamioka, Y.; Arakawa, Y.; Miyamoto, S.; Okada, T.; Sahai, E.; Matsuda, M. In vivo fluorescence resonance energy transfer imaging reveals differential activation of Rho-family GTPases in glioblastoma cell invasion. J. Cell Sci. 2012, 125, 858–868. [Google Scholar] [CrossRef]
- Egnuni, T.; Speirs, V.; Chakrabarty, A.; Wurdak, H.; Lorger, M.; Short, S.; Mavria, G. PP17. Rho GTPase signalling in cancer cell invaision and angiogenesis in brain tumors. NeuroOncology 2017, 19, i5. [Google Scholar] [CrossRef][Green Version]
- Zhao, J.; Zhai, B.; Gygi, S.P.; Goldberg, A.L. mTOR inhibition activates overall protein degradation by the ubiquitin proteasome system as well as by autophagy. Proc. Natl. Acad. Sci. USA 2015, 112, 15790–15797. [Google Scholar] [CrossRef]
- Abdellatif, M.; Packer, S.E.; Michael, L.H.; Zhang, D.; Charng, M.J.; Schneider, M.D. A Ras-dependent pathway regulates RNA polymerase II phosphorylation in cardiac myocytes: Implications for cardiac hypertrophy. Mol. Cell. Biol. 1998, 18, 6729–6736. [Google Scholar] [CrossRef]
- Schuller, R.; Forne, I.; Straub, T.; Schreieck, A.; Texier, Y.; Shah, N.; Decker, T.M.; Cramer, P.; Imhof, A.; Eick, D. Heptad-Specific Phosphorylation of RNA Polymerase II CTD. Mol. Cell 2016, 61, 305–314. [Google Scholar] [CrossRef]
- Suh, H.; Ficarro, S.B.; Kang, U.B.; Chun, Y.; Marto, J.A.; Buratowski, S. Direct Analysis of Phosphorylation Sites on the Rpb1 C-Terminal Domain of RNA Polymerase II. Mol. Cell 2016, 61, 297–304. [Google Scholar] [CrossRef]
- Wang, H.; Jo, Y.J.; Sun, T.Y.; Namgoong, S.; Cui, X.S.; Oh, J.S.; Kim, N.H. Inhibition of CDK7 bypasses spindle assembly checkpoint via premature cyclin B degradation during oocyte meiosis. Biochim. Biophys. Acta 2016, 1863, 2993–3000. [Google Scholar] [CrossRef] [PubMed]
- Greenall, S.A.; Lim, Y.C.; Mitchell, C.B.; Ensbey, K.S.; Stringer, B.W.; Wilding, A.L.; O’Neill, G.M.; McDonald, K.L.; Gough, D.J.; Day, B.W.; et al. Cyclin-dependent kinase 7 is a therapeutic target in high-grade glioma. Oncogenesis 2017, 6, e336. [Google Scholar] [CrossRef] [PubMed]
- Cayrol, F.; Praditsuktavorn, P.; Fernando, T.M.; Kwiatkowski, N.; Marullo, R.; Calvo-Vidal, M.N.; Phillip, J.; Pera, B.; Yang, S.N.; Takpradit, K.; et al. THZ1 targeting CDK7 suppresses STAT transcriptional activity and sensitizes T-cell lymphomas to BCL2 inhibitors. Nat. Commun. 2017, 8, 14290. [Google Scholar] [CrossRef] [PubMed]
- Kalan, S.; Amat, R.; Schachter, M.M.; Kwiatkowski, N.; Abraham, B.J.; Liang, Y.; Zhang, T.; Olson, C.M.; Larochelle, S.; Young, R.A.; et al. Activation of the p53 Transcriptional Program Sensitizes Cancer Cells to Cdk7 Inhibitors. Cell Rep. 2017, 21, 467–481. [Google Scholar] [CrossRef] [PubMed]
- Rusan, M.; Li, K.; Li, Y.; Christensen, C.L.; Abraham, B.J.; Kwiatkowski, N.; Buczkowski, K.A.; Bockorny, B.; Chen, T.; Li, S.; et al. Suppression of Adaptive Responses to Targeted Cancer Therapy by Transcriptional Repression. Cancer Discov. 2018, 8, 59–73. [Google Scholar] [CrossRef]
- Liu, F.; Jiang, W.; Sui, Y.; Meng, W.; Hou, L.; Li, T.; Li, M.; Zhang, L.; Mo, J.; Wang, J.; et al. CDK7 inhibition suppresses aberrant hedgehog pathway and overcomes resistance to smoothened antagonists. Proc. Natl. Acad. Sci. USA 2019. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, B.; Zhong, X.; Sauane, M.; Zhao, Y.; Zheng, Z.-L. Modulation of the Pol II CTD Phosphorylation Code by Rac1 and Cdc42 Small GTPases in Cultured Human Cancer Cells and Its Implication for Developing a Synthetic-Lethal Cancer Therapy. Cells 2020, 9, 621. https://doi.org/10.3390/cells9030621
Zhang B, Zhong X, Sauane M, Zhao Y, Zheng Z-L. Modulation of the Pol II CTD Phosphorylation Code by Rac1 and Cdc42 Small GTPases in Cultured Human Cancer Cells and Its Implication for Developing a Synthetic-Lethal Cancer Therapy. Cells. 2020; 9(3):621. https://doi.org/10.3390/cells9030621
Chicago/Turabian StyleZhang, Bo, Xuelin Zhong, Moira Sauane, Yihong Zhao, and Zhi-Liang Zheng. 2020. "Modulation of the Pol II CTD Phosphorylation Code by Rac1 and Cdc42 Small GTPases in Cultured Human Cancer Cells and Its Implication for Developing a Synthetic-Lethal Cancer Therapy" Cells 9, no. 3: 621. https://doi.org/10.3390/cells9030621
APA StyleZhang, B., Zhong, X., Sauane, M., Zhao, Y., & Zheng, Z.-L. (2020). Modulation of the Pol II CTD Phosphorylation Code by Rac1 and Cdc42 Small GTPases in Cultured Human Cancer Cells and Its Implication for Developing a Synthetic-Lethal Cancer Therapy. Cells, 9(3), 621. https://doi.org/10.3390/cells9030621