Cyclin-Dependent Kinase 18 Controls Trafficking of Aquaporin-2 and Its Abundance through Ubiquitin Ligase STUB1, Which Functions as an AKAP


 , and
, and 
Abstract
1. Introduction
2. Materials and Methods
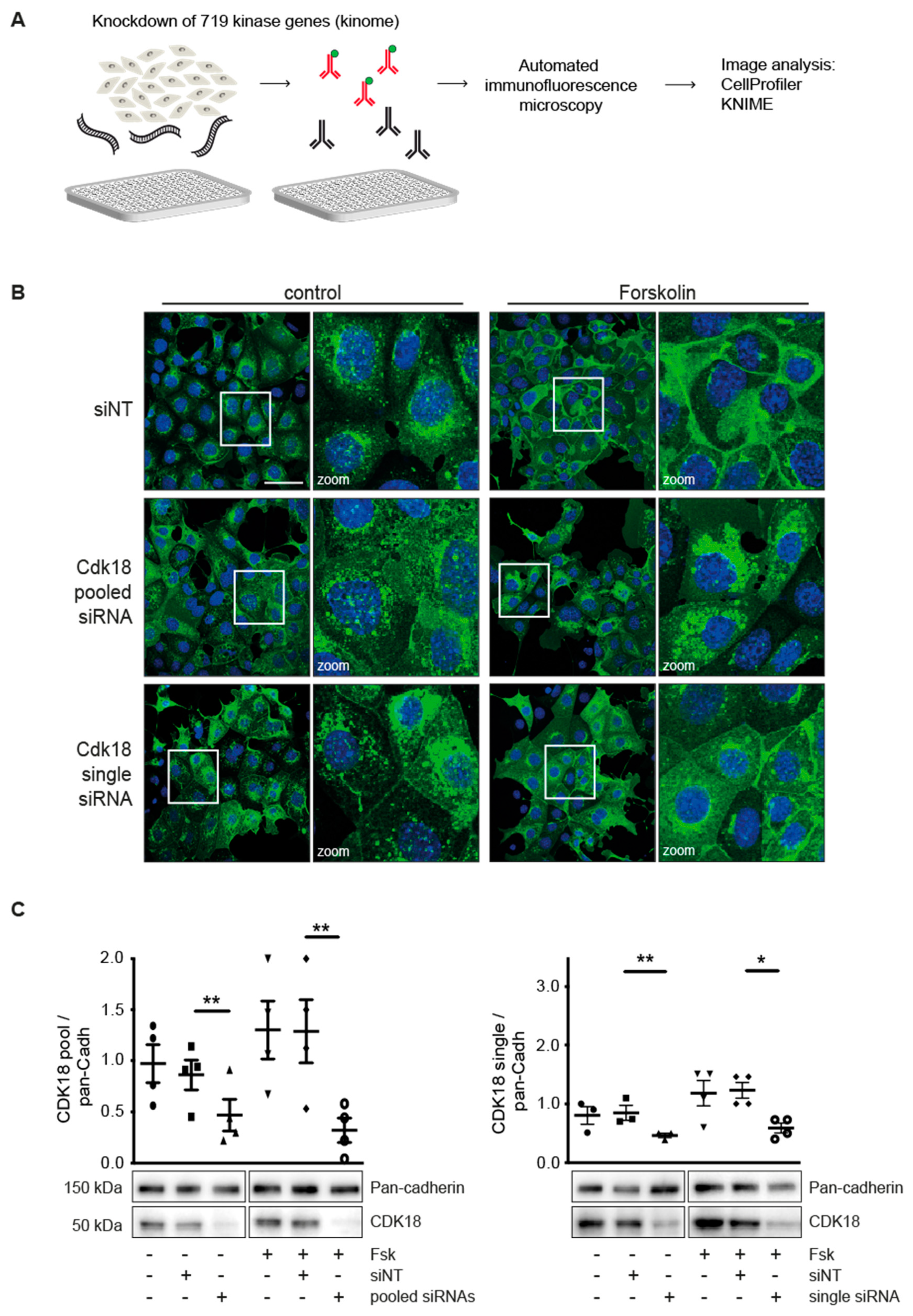
2.1. Kinome Screening and Automated Immunofluorescence Microscopy
2.2. Cell Culture, Transfection and Generation of Expression Plasmids
2.3. Laser Scanning Microscopy
2.4. Cell Lysis, Immunoprecipitation, cAMP Pull-Down, Western Blotting and RII Overlay
2.5. Kinase and pNPP Phosphatase Activity Assays
2.6. Synthetic and Spot-Synthesised Peptides
2.7. Sample Preparation for Mass Spectrometry
2.8. Statistical Analysis
3. Results
3.1. Kinome Knockdown Identifies Candidates Controlling the Redistribution of AQP2 to the Plasma Membrane
3.2. Knockdown of CDK18 Inhibits the cAMP-Induced Redistribution of AQP2
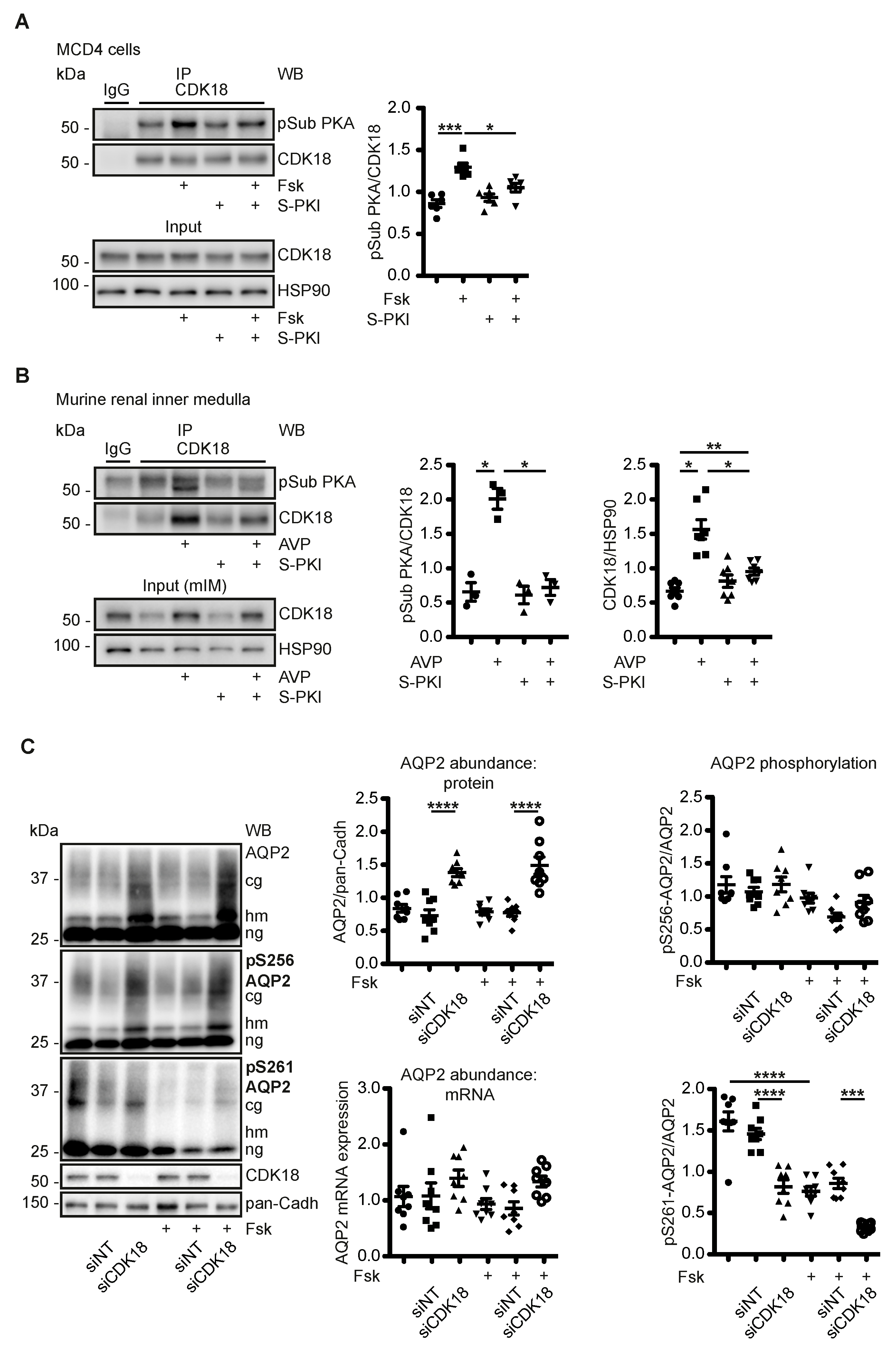
3.3. AVP Induces PKA-Dependent Increases of CDK18 Phosphorylation and Abundance
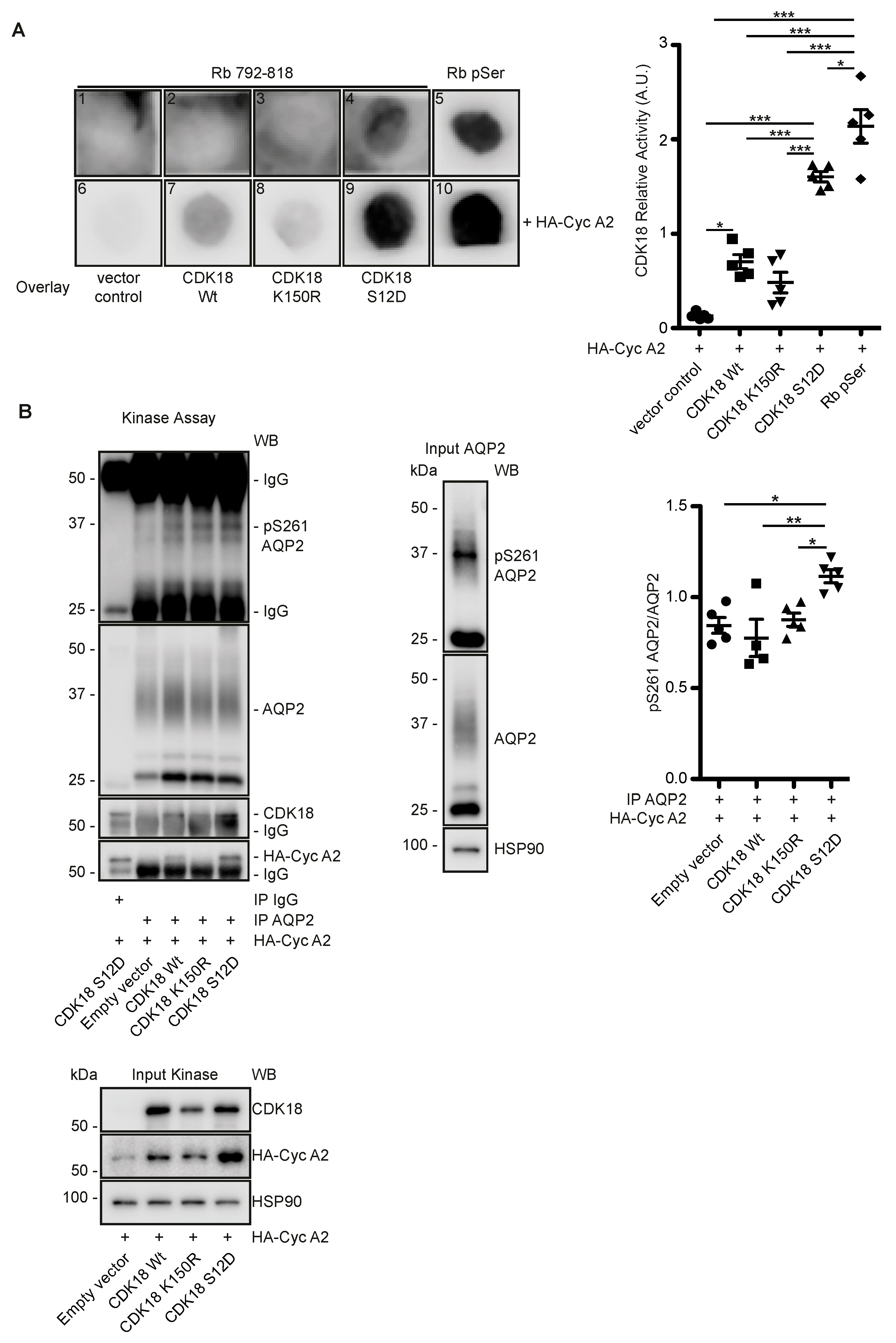
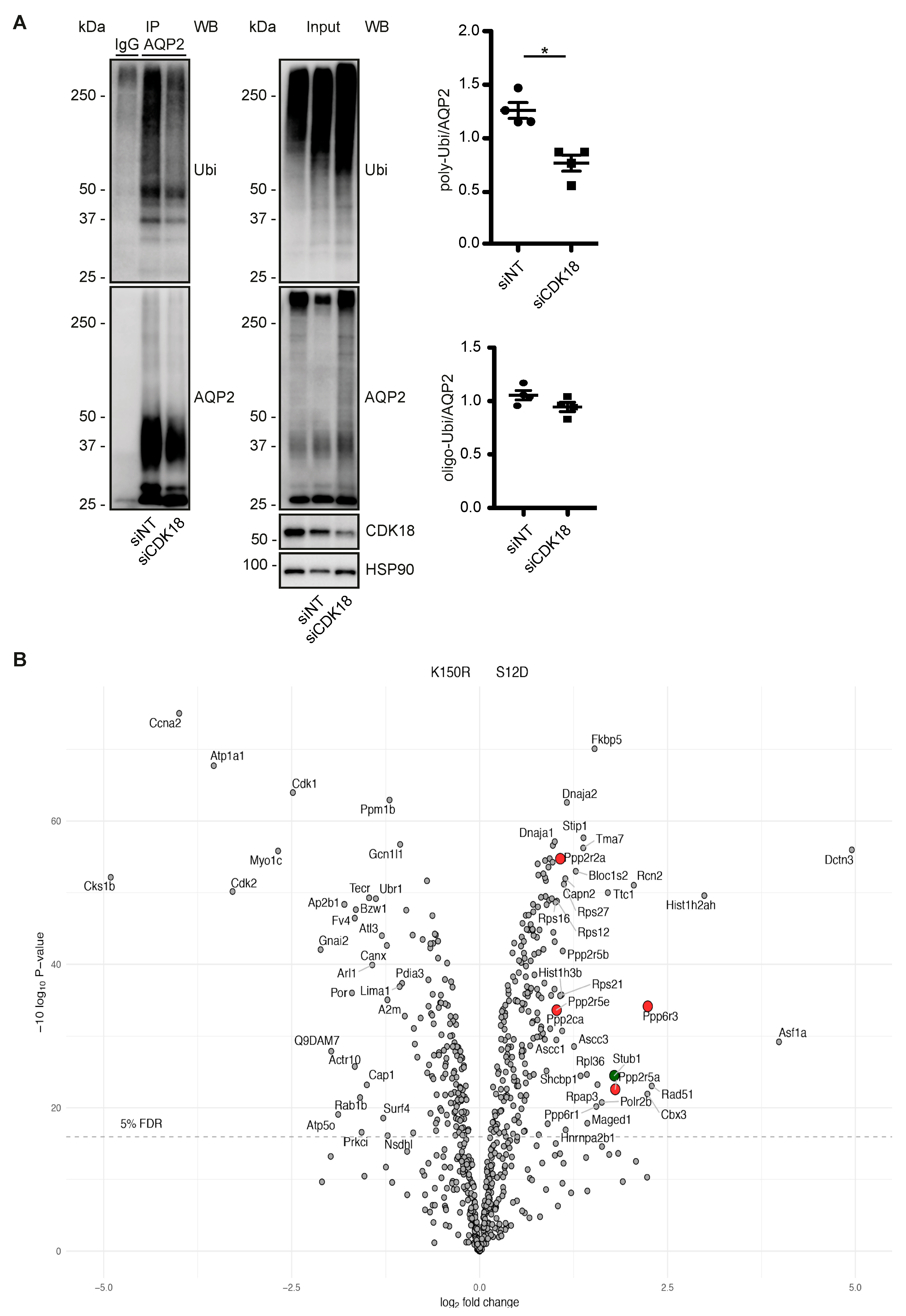
3.4. CDK18 Controls the Phosphorylation of AQP2 at S261, Its Ubiquitination and Abundance
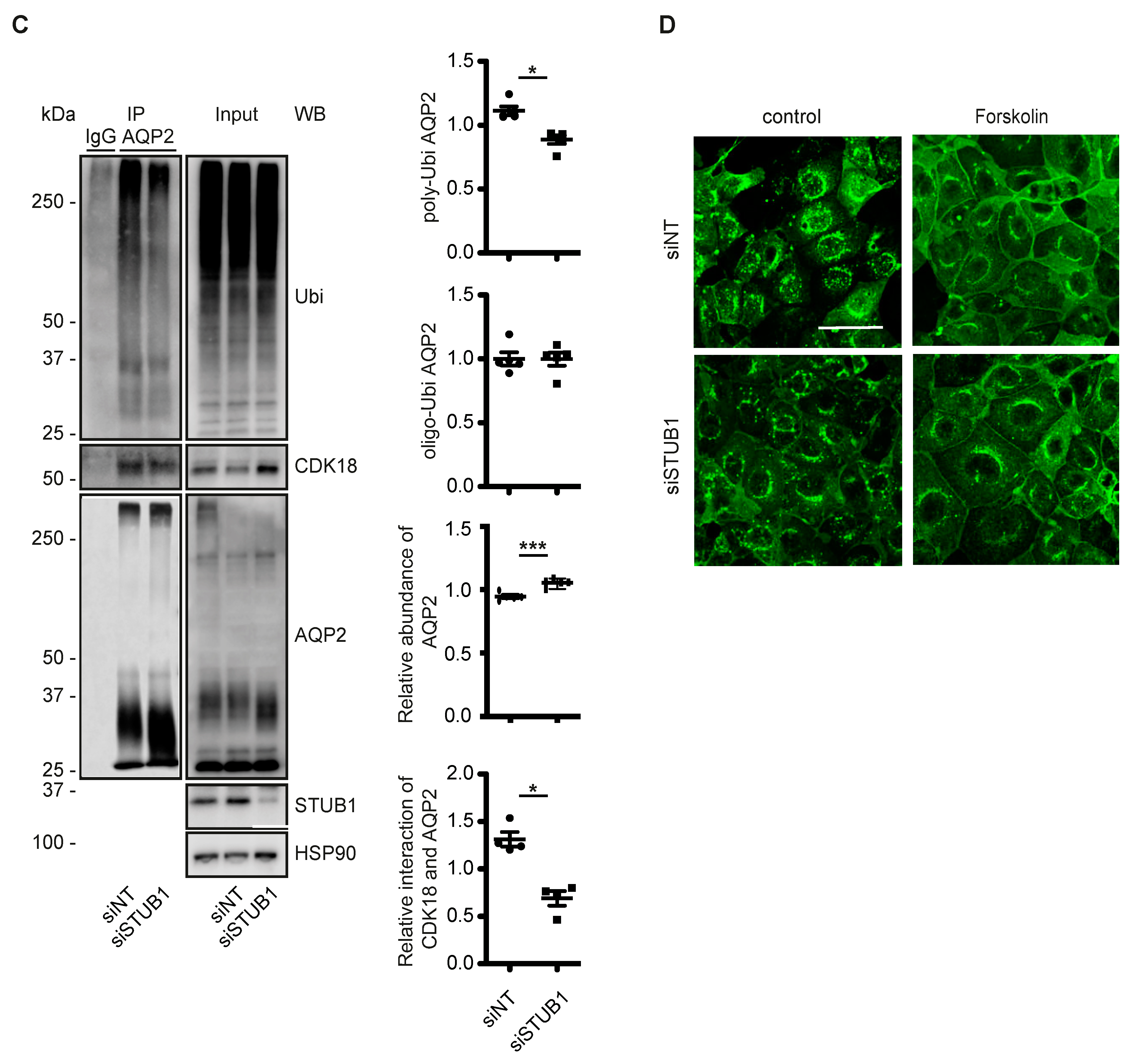
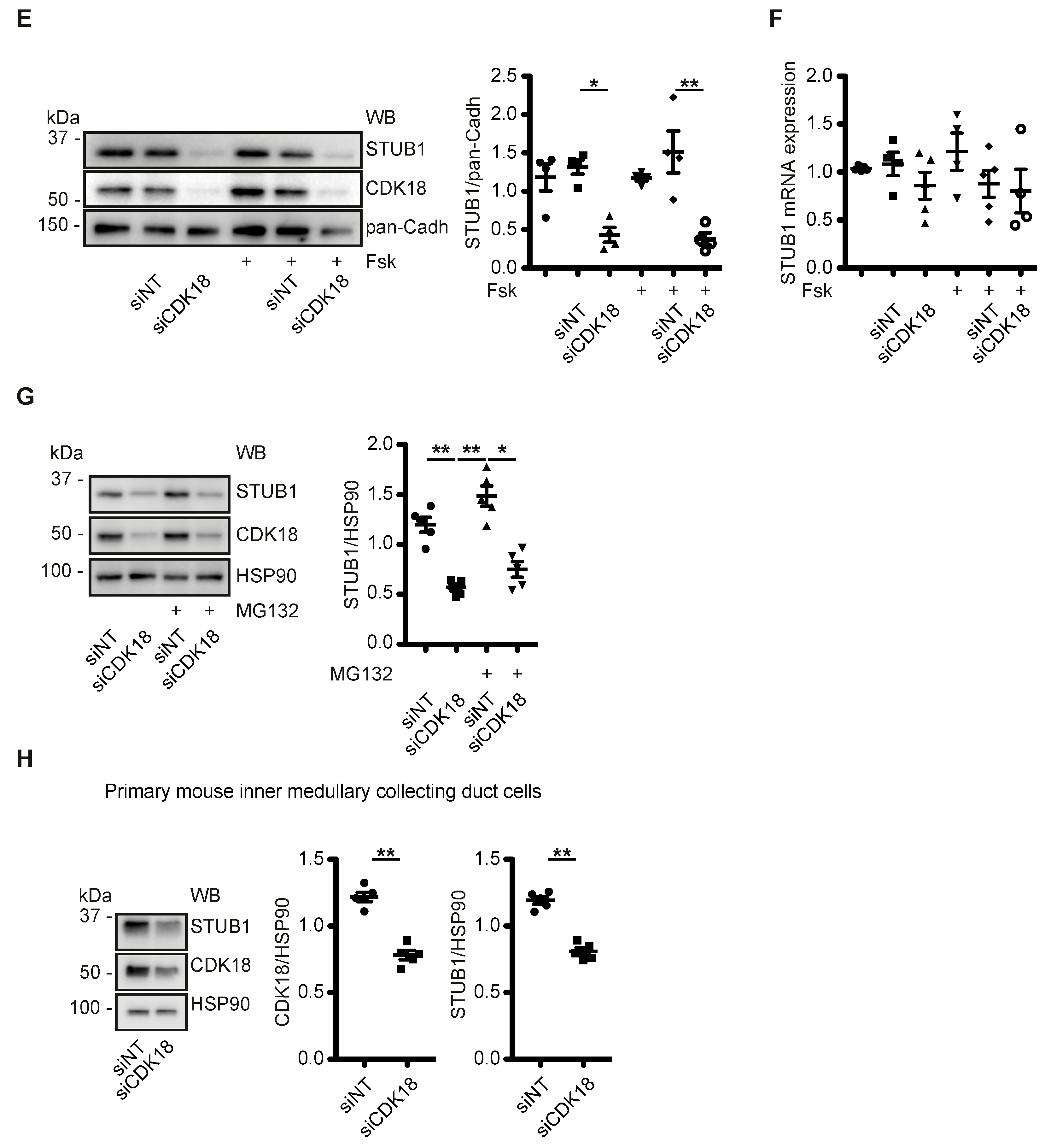
3.5. CDK18 Controls AQP2 Ubiquitination and Abundance through the E3 Ubiquitin Ligase STUB1
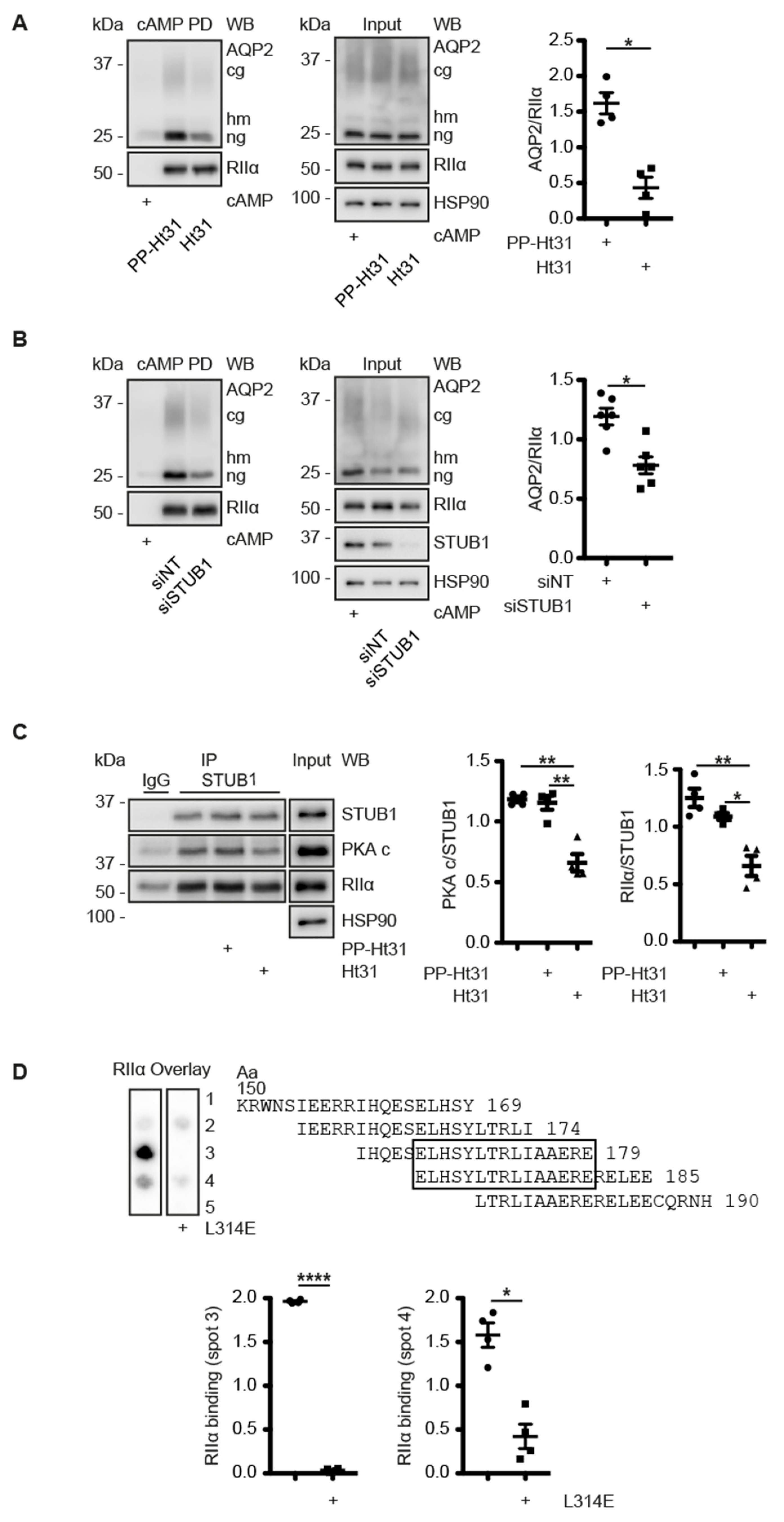
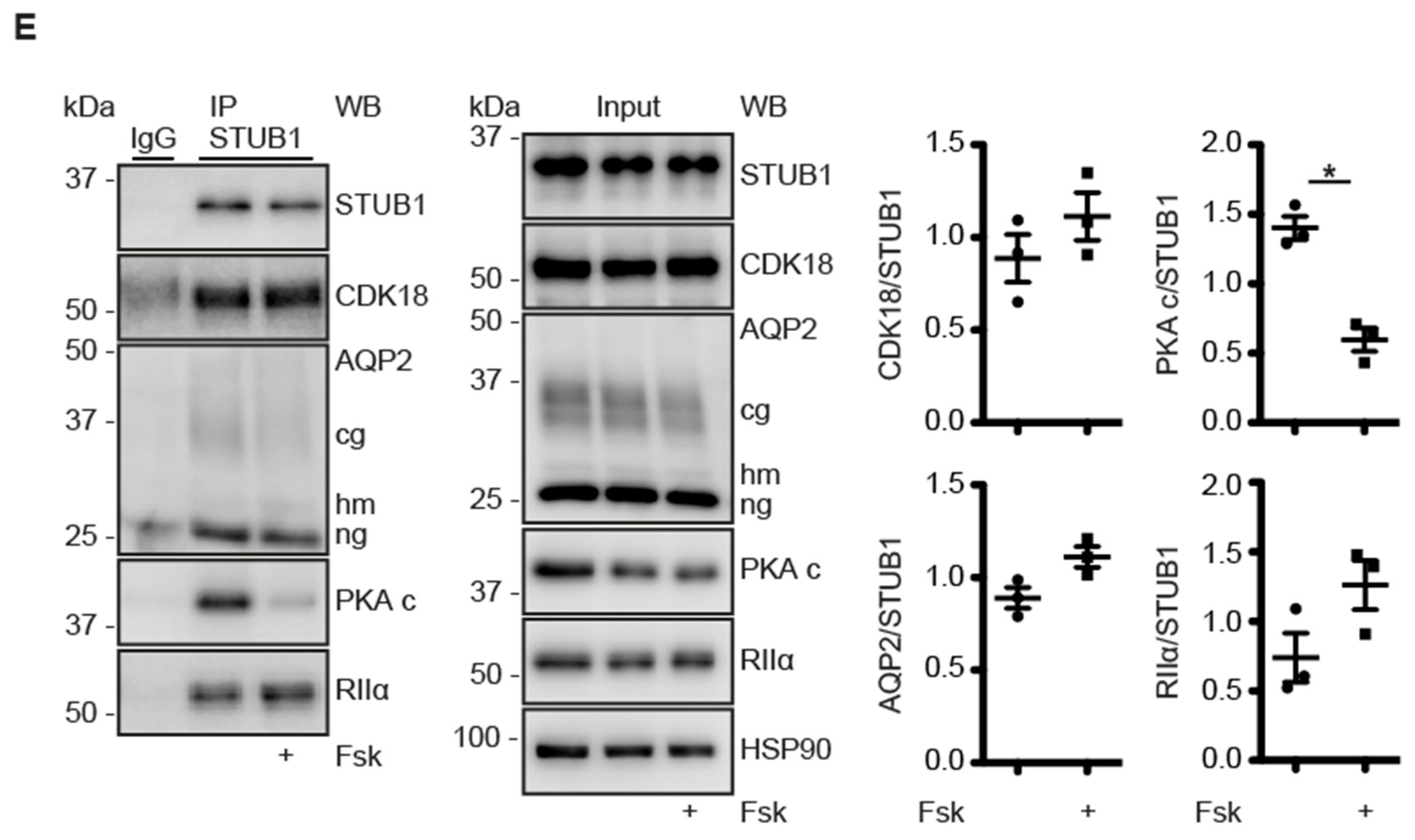
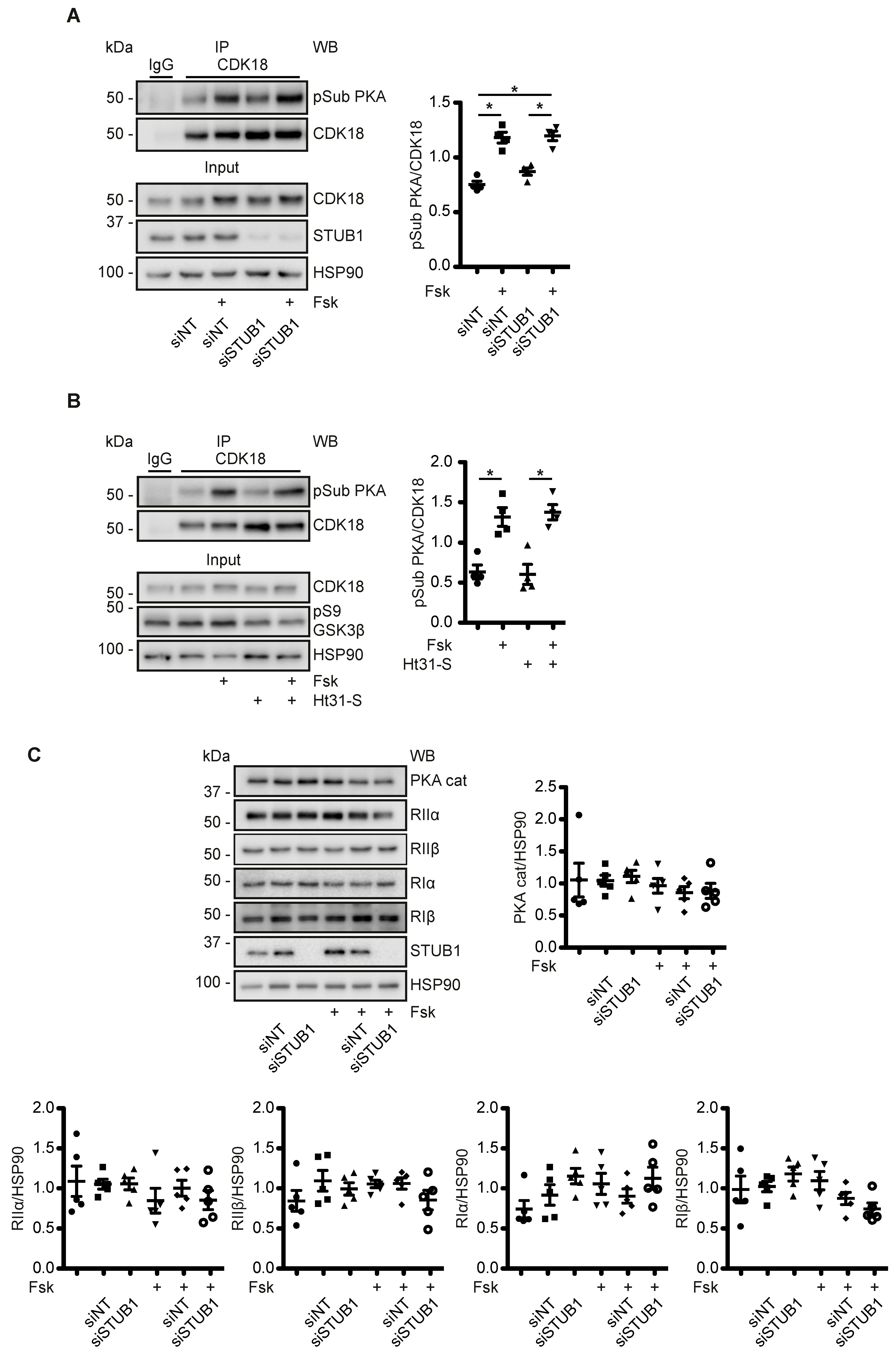
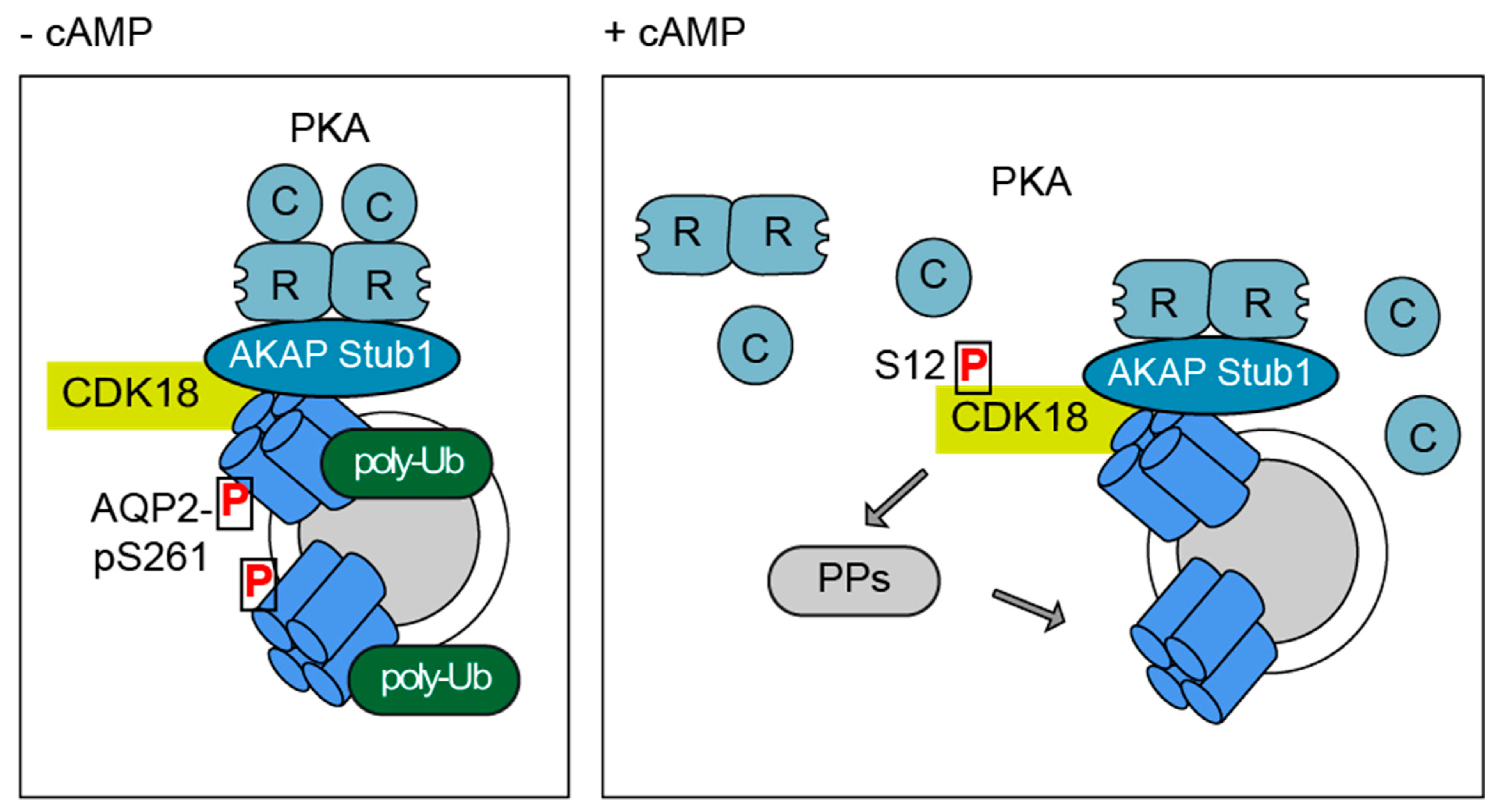
3.6. A CDK18-Containing Signalosome with AQP2 and PKA Is Organised by STUB1, Which Functions as an AKAP
3.7. A Cytosolic Pool of PKA Phosphorylates CDK18 - PKA within the AQP2/CDK18/STUB1/PKA Complex Does Not Coordinate PKA Phosphorylation of CDK18 or AQP2
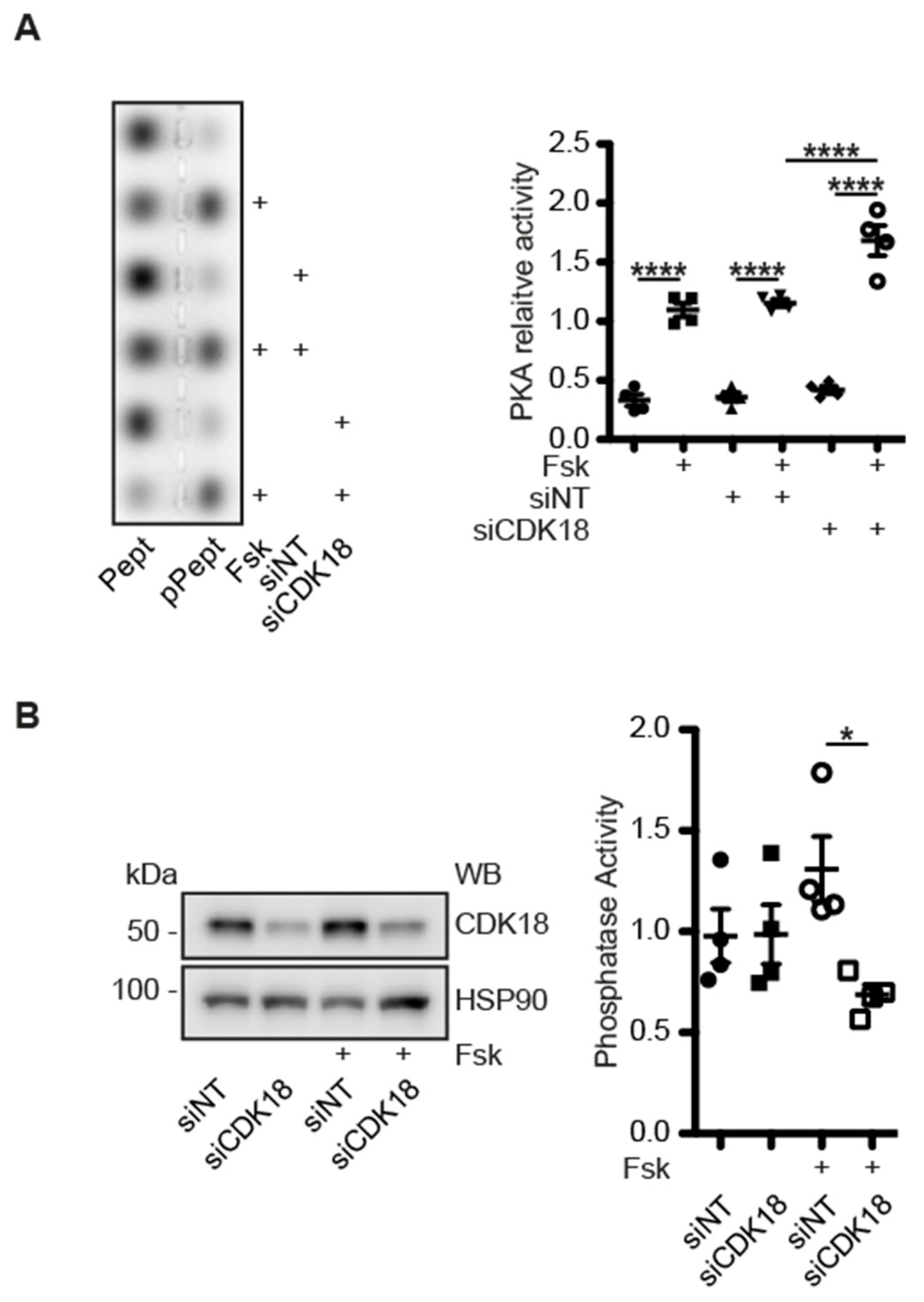
3.8. CDK18 Is Involved in Feedback Control of PKA and Phosphatase Activity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vukicevic, T.; Schulz, M.; Faust, D.; Klussmann, E. The Trafficking of the Water Channel Aquaporin-2 in Renal Principal Cells-a Potential Target for Pharmacological Intervention in Cardiovascular Diseases. Front. Pharmacol. 2016, 7, 23. [Google Scholar] [CrossRef] [PubMed]
- Ranieri, M.; Di Mise, A.; Tamma, G.; Valenti, G. Vasopressin-aquaporin-2 pathway: Recent advances in understanding water balance disorders. F1000Res. 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Baltzer, S.; Klussmann, E. Small molecules for modulating the localisation of the water channel aquaporin-2-disease relevance and perspectives for targeting local cAMP signalling. Naunyn. Schmiedebergs Arch. Pharmacol. 2019, 392, 1049–1064. [Google Scholar] [CrossRef] [PubMed]
- Verbalis, J.G. An Antifungal for Antidiuresis? J. Am. Soc. Nephrol. 2019, 30, 717–718. [Google Scholar] [CrossRef] [PubMed]
- Vukicevic, T.; Hinze, C.; Baltzer, S.; Himmerkus, N.; Quintanova, C.; Zuhlke, K.; Compton, F.; Ahlborn, R.; Dema, A.; Eichhorst, J.; et al. Fluconazole Increases Osmotic Water Transport in Renal Collecting Duct through Effects on Aquaporin-2 Trafficking. J. Am. Soc. Nephrol. 2019, 30, 795–810. [Google Scholar] [CrossRef]
- Stefan, E.; Wiesner, B.; Baillie, G.S.; Mollajew, R.; Henn, V.; Lorenz, D.; Furkert, J.; Santamaria, K.; Nedvetsky, P.; Hundsrucker, C.; et al. Compartmentalization of cAMP-dependent signaling by phosphodiesterase-4D is involved in the regulation of vasopressin-mediated water reabsorption in renal principal cells. J. Am. Soc. Nephrol. 2007, 18, 199–212. [Google Scholar] [CrossRef]
- Henn, V.; Edemir, B.; Stefan, E.; Wiesner, B.; Lorenz, D.; Theilig, F.; Schmitt, R.; Vossebein, L.; Tamma, G.; Beyermann, M.; et al. Identification of a novel A-kinase anchoring protein 18 isoform and evidence for its role in the vasopressin-induced aquaporin-2 shuttle in renal principal cells. J. Biol. Chem. 2004, 279, 26654–26665. [Google Scholar] [CrossRef]
- Okutsu, R.; Rai, T.; Kikuchi, A.; Ohno, M.; Uchida, K.; Sasaki, S.; Uchida, S. AKAP220 colocalizes with AQP2 in the inner medullary collecting ducts. Kidney Int. 2008, 74, 1429–1433. [Google Scholar] [CrossRef]
- Whiting, J.L.; Ogier, L.; Forbush, K.A.; Bucko, P.; Gopalan, J.; Seternes, O.M.; Langeberg, L.K.; Scott, J.D. AKAP220 manages apical actin networks that coordinate aquaporin-2 location and renal water reabsorption. Proc. Natl. Acad. Sci. USA. 2016, 113, E4328–E4337. [Google Scholar] [CrossRef]
- Klussmann, E.; Maric, K.; Wiesner, B.; Beyermann, M.; Rosenthal, W. Protein kinase A anchoring proteins are required for vasopressin-mediated translocation of aquaporin-2 into cell membranes of renal principal cells. J. Biol. Chem. 1999, 274, 4934–4938. [Google Scholar] [CrossRef]
- Fenton, R.A.; Moeller, H.B.; Hoffert, J.D.; Yu, M.J.; Nielsen, S.; Knepper, M.A. Acute regulation of aquaporin-2 phosphorylation at Ser-264 by vasopressin. Proc. Natl. Acad. Sci. USA. 2008, 105, 3134–3139. [Google Scholar] [CrossRef] [PubMed]
- Hoffert, J.D.; Fenton, R.A.; Moeller, H.B.; Simons, B.; Tchapyjnikov, D.; McDill, B.W.; Yu, M.J.; Pisitkun, T.; Chen, F.; Knepper, M.A. Vasopressin-stimulated increase in phosphorylation at Ser269 potentiates plasma membrane retention of aquaporin-2. J. Biol. Chem. 2008, 283, 24617–24627. [Google Scholar] [CrossRef] [PubMed]
- Fushimi, K.; Sasaki, S.; Marumo, F. Phosphorylation of serine 256 is required for cAMP-dependent regulatory exocytosis of the aquaporin-2 water channel. J. Biol. Chem. 1997, 272, 14800–14804. [Google Scholar] [CrossRef] [PubMed]
- Olesen, E.T.; Fenton, R.A. Aquaporin-2 membrane targeting: Still a conundrum. Am. J. Physiol. Renal Physiol. 2017, 312, F744–F747. [Google Scholar] [CrossRef]
- Nishimoto, G.; Zelenina, M.; Li, D.; Yasui, M.; Aperia, A.; Nielsen, S.; Nairn, A.C. Arginine vasopressin stimulates phosphorylation of aquaporin-2 in rat renal tissue. Am. J. Physiol. 1999, 276, F254–F259. [Google Scholar] [CrossRef]
- Isobe, K.; Jung, H.J.; Yang, C.R.; Claxton, J.; Sandoval, P.; Burg, M.B.; Raghuram, V.; Knepper, M.A. Systems-level identification of PKA-dependent signaling in epithelial cells. Proc. Natl. Acad. Sci. USA. 2017, 114, E8875–E8884. [Google Scholar] [CrossRef]
- Bradford, D.; Raghuram, V.; Wilson, J.L.; Chou, C.L.; Hoffert, J.D.; Knepper, M.A.; Pisitkun, T. Use of LC-MS/MS and Bayes’ theorem to identify protein kinases that phosphorylate aquaporin-2 at Ser256. Am. J. Physiol. Cell Physiol. 2014, 307, C123–C139. [Google Scholar] [CrossRef]
- Bouley, R.; Breton, S.; Sun, T.; McLaughlin, M.; Nsumu, N.N.; Lin, H.Y.; Ausiello, D.A.; Brown, D. Nitric oxide and atrial natriuretic factor stimulate cGMP-dependent membrane insertion of aquaporin 2 in renal epithelial cells. J. Clin. Invest. 2000, 106, 1115–1126. [Google Scholar] [CrossRef]
- Lu, H.; Sun, T.X.; Bouley, R.; Blackburn, K.; McLaughlin, M.; Brown, D. Inhibition of endocytosis causes phosphorylation (S256)-independent plasma membrane accumulation of AQP2. Am. J. Physiol. Renal Physiol. 2004, 286, F233–F243. [Google Scholar] [CrossRef]
- Lu, H.A.; Sun, T.X.; Matsuzaki, T.; Yi, X.H.; Eswara, J.; Bouley, R.; McKee, M.; Brown, D. Heat shock protein 70 interacts with aquaporin-2 and regulates its trafficking. J. Biol. Chem. 2007, 282, 28721–28732. [Google Scholar] [CrossRef]
- Arthur, J.; Huang, J.; Nomura, N.; Jin, W.W.; Li, W.; Cheng, X.; Brown, D.; Lu, H.J. Characterization of the putative phosphorylation sites of the AQP2 C terminus and their role in AQP2 trafficking in LLC-PK1 cells. Am. J. Physiol. Renal Physiol. 2015, 309, F673–F679. [Google Scholar] [CrossRef] [PubMed]
- Moeller, H.B.; Praetorius, J.; Rutzler, M.R.; Fenton, R.A. Phosphorylation of aquaporin-2 regulates its endocytosis and protein-protein interactions. Proc. Natl. Acad. Sci. USA. 2010, 107, 424–429. [Google Scholar] [CrossRef] [PubMed]
- Moeller, H.B.; Aroankins, T.S.; Slengerik-Hansen, J.; Pisitkun, T.; Fenton, R.A. Phosphorylation and ubiquitylation are opposing processes that regulate endocytosis of the water channel aquaporin-2. J. Cell Sci. 2014, 127, 3174–3183. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.J.; Lin, S.T.; Liu, S.H.; Kuo, K.T.; Hsu, C.H.; Knepper, M.A.; Yu, M.J. Vasopressin-induced serine 269 phosphorylation reduces Sipa1l1 (signal-induced proliferation-associated 1 like 1)-mediated aquaporin-2 endocytosis. J. Biol. Chem. 2017, 292, 7984–7993. [Google Scholar] [CrossRef] [PubMed]
- Nedvetsky, P.I.; Tabor, V.; Tamma, G.; Beulshausen, S.; Skroblin, P.; Kirschner, A.; Mutig, K.; Boltzen, M.; Petrucci, O.; Vossenkamper, A.; et al. Reciprocal regulation of aquaporin-2 abundance and degradation by protein kinase A and p38-MAP kinase. J. Am. Soc. Nephrol. 2010, 21, 1645–1656. [Google Scholar] [CrossRef] [PubMed]
- Bogum, J.; Faust, D.; Zuhlke, K.; Eichhorst, J.; Moutty, M.C.; Furkert, J.; Eldahshan, A.; Neuenschwander, M.; von Kries, J.P.; Wiesner, B.; et al. Small-molecule screening identifies modulators of aquaporin-2 trafficking. J. Am. Soc. Nephrol. 2013, 24, 744–758. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Iolascon, A.; Aglio, V.; Tamma, G.; D’Apolito, M.; Addabbo, F.; Procino, G.; Simonetti, M.C.; Montini, G.; Gesualdo, L.; Debler, E.W.; et al. Characterization of two novel missense mutations in the AQP2 gene causing nephrogenic diabetes insipidus. Nephron. Physiol. 2007, 105, p33–41. [Google Scholar] [CrossRef]
- Klussmann, E.; Tamma, G.; Lorenz, D.; Wiesner, B.; Maric, K.; Hofmann, F.; Aktories, K.; Valenti, G.; Rosenthal, W. An inhibitory role of Rho in the vasopressin-mediated translocation of aquaporin-2 into cell membranes of renal principal cells. J. Biol. Chem. 2001, 276, 20451–20457. [Google Scholar] [CrossRef]
- Atar, O.D.; Eisert, C.; Pokov, I.; Serebruany, V.L. Stability validation of paraformaldehyde-fixed samples for the assessment of the platelet PECAM-1, P-selectin, and PAR-1 thrombin receptor by flow cytometry. J. Thromb. Thrombolysis 2010, 30, 79–83. [Google Scholar] [CrossRef]
- Carpenter, A.E.; Jones, T.R.; Lamprecht, M.R.; Clarke, C.; Kang, I.H.; Friman, O.; Guertin, D.A.; Chang, J.H.; Lindquist, R.A.; Moffat, J.; et al. CellProfiler: Image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006, 7, R100. [Google Scholar] [CrossRef]
- Manning, T.; Sleator, R.D.; Walsh, P. Biologically inspired intelligent decision making: A commentary on the use of artificial neural networks in bioinformatics. Bioengineered 2013, 5, 80–95. [Google Scholar] [CrossRef] [PubMed]
- Boland, M.V.; Murphy, R.F. A neural network classifier capable of recognizing the patterns of all major subcellular structures in fluorescence microscope images of HeLa cells. Bioinformatics 2001, 17, 1213–1223. [Google Scholar] [CrossRef] [PubMed]
- Faust, D.; Geelhaar, A.; Eisermann, B.; Eichhorst, J.; Wiesner, B.; Rosenthal, W.; Klussmann, E. Culturing primary rat inner medullary collecting duct cells. J. Vis. Exp. 2013, 21, e50366. [Google Scholar] [CrossRef] [PubMed]
- Dema, A.; Schroeter, M.F.; Perets, E.; Skroblin, P.; Moutty, M.C.; Deak, V.A.; Birchmeier, W.; Klussmann, E. The AKAP GSKIP regulates beta-catenin through both its interactions with PKA and GSK3beta. J. Biol. Chem. 2016, 2, jbc-M116. [Google Scholar]
- Maric, K.; Oksche, A.; Rosenthal, W. Aquaporin-2 expression in primary cultured rat inner medullary collecting duct cells. Am. J. Physiol. 1998, 275, F796–F801. [Google Scholar] [CrossRef] [PubMed]
- Hundsrucker, C.; Skroblin, P.; Christian, F.; Zenn, H.M.; Popara, V.; Joshi, M.; Eichhorst, J.; Wiesner, B.; Herberg, F.W.; Reif, B.; et al. Glycogen synthase kinase 3beta interaction protein functions as an A-kinase anchoring protein. J. Biol. Chem. 2010, 285, 5507–5521. [Google Scholar] [CrossRef]
- Schäfer, G.; Milic, J.; Eldahshan, A.; Götz, F.; Zuhlke, K.; Schillinger, C.; Kreuchwig, A.; Elkins, J.M.; Abdul Azeez, K.R.; Oder, A.; et al. Highly functionalized terpyridines as competitive inhibitors of AKAP-PKA interactions. Angew. Chem. Int. Ed. Engl. 2013, 52, 12187–12191. [Google Scholar] [CrossRef]
- McAvoy, T.; Nairn, A.C. Serine/threonine protein phosphatase assays. Curr. Protoc. Mol. Biol. 2010, 92, 18.18.1–18.18.11. [Google Scholar] [CrossRef]
- Maass, P.G.; Aydin, A.; Luft, F.C.; Schachterle, C.; Weise, A.; Stricker, S.; Lindschau, C.; Vaegler, M.; Qadri, F.; Toka, H.R.; et al. PDE3A mutations cause autosomal dominant hypertension with brachydactyly. Nat. Genet. 2015, 47, 647–653. [Google Scholar] [CrossRef]
- Klussmann, E. Protein-protein interactions of PDE4 family members - Functions, interactions and therapeutic value. Cell. Signalling 2016, 28, 713–718. [Google Scholar] [CrossRef]
- Shevchenko, A.; Tomas, H.; Havlis, J.; Olsen, J.V.; Mann, M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 2006, 1, 2856–2860. [Google Scholar] [CrossRef] [PubMed]
- Rappsilber, J.; Mann, M.; Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2007, 2, 1896–1906. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef]
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell Proteomics 2014, 13, 2513–2526. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic. Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Ashby, C.D.; Walsh, D.A. Characterization of the interaction of a protein inhibitor with adenosine 3’,5’-monophosphate-dependent protein kinases. I. Interaction with the catalytic subunit of the protein kinase. J. Biol. Chem. 1972, 247, 6637–6642. [Google Scholar]
- Fantozzi, D.A.; Harootunian, A.T.; Wen, W.; Taylor, S.S.; Feramisco, J.R.; Tsien, R.Y.; Meinkoth, J.L. Thermostable inhibitor of cAMP-dependent protein kinase enhances the rate of export of the kinase catalytic subunit from the nucleus. J. Biol. Chem. 1994, 269, 2676–2686. [Google Scholar]
- Korsse, S.E.; Peppelenbosch, M.P.; van Veelen, W. Targeting LKB1 signaling in cancer. Biochim. Biophys. Acta 2013, 1835, 194–210. [Google Scholar] [CrossRef]
- Herskovits, A.Z.; Davies, P. The regulation of tau phosphorylation by PCTAIRE 3: Implications for the pathogenesis of Alzheimer’s disease. Neurobiol. Dis. 2006, 23, 398–408. [Google Scholar] [CrossRef]
- Chaput, D.; Kirouac, L.; Stevens, S.M., Jr.; Padmanabhan, J. Potential role of PCTAIRE-2, PCTAIRE-3 and P-Histone H4 in amyloid precursor protein-dependent Alzheimer pathology. Oncotarget 2016, 7, 8481–8497. [Google Scholar] [CrossRef] [PubMed]
- Barone, G.; Staples, C.J.; Ganesh, A.; Patterson, K.W.; Bryne, D.P.; Myers, K.N.; Patil, A.A.; Eyers, C.E.; Maslen, S.; Skehel, J.M.; et al. Human CDK18 promotes replication stress signaling and genome stability. Nucleic. Acids Res. 2016, 44, 8772–8785. [Google Scholar] [CrossRef] [PubMed]
- Ning, J.F.; Stanciu, M.; Humphrey, M.R.; Gorham, J.; Wakimoto, H.; Nishihara, R.; Lees, J.; Zou, L.; Martuza, R.L.; Wakimoto, H.; et al. Myc targeted CDK18 promotes ATR and homologous recombination to mediate PARP inhibitor resistance in glioblastoma. Nat. Commun. 2019, 10, 2910. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, S.; Kominato, K.; Koide-Yoshida, S.; Miyamoto, K.; Isshiki, K.; Tsuji, A.; Yuasa, K. PCTAIRE kinase 3/cyclin-dependent kinase 18 is activated through association with cyclin A and/or phosphorylation by protein kinase A. J. Biol. Chem. 2014, 289, 18387–18400. [Google Scholar] [CrossRef]
- Edkins, A.L. CHIP: A co-chaperone for degradation by the proteasome. Subcell Biochem. 2015, 78, 219–242. [Google Scholar]
- Wu, Q.; Moeller, H.B.; Stevens, D.A.; Sanchez-Hodge, R.; Childers, G.; Kortenoeven, M.L.A.; Cheng, L.; Rosenbaek, L.L.; Rubel, C.; Patterson, C.; et al. CHIP Regulates Aquaporin-2 Quality Control and Body Water Homeostasis. J. Am. Soc. Nephrol. 2018, 29, 936–948. [Google Scholar] [CrossRef]
- Centrone, M.; Ranieri, M.; Di Mise, A.; Berlingerio, S.P.; Russo, A.; Deen, P.M.T.; Staub, O.; Valenti, G.; Tamma, G. AQP2 Abundance is Regulated by the E3-Ligase CHIP Via HSP70. Cell Physiol. Biochem. 2017, 44, 515–531. [Google Scholar] [CrossRef]
- Walker-Gray, R.; Stengel, F.; Gold, M.G. Mechanisms for restraining cAMP-dependent protein kinase revealed by subunit quantitation and cross-linking approaches. Proc. Natl. Acad. Sci. USA. 2017, 114, 10414–10419. [Google Scholar] [CrossRef]
- Dema, A.; Perets, E.; Schulz, M.S.; Deak, V.A.; Klussmann, E. Pharmacological targeting of AKAP-directed compartmentalized cAMP signalling. Cell Signal. 2015, 27, 2474–2487. [Google Scholar] [CrossRef]
- Skroblin, P.; Grossmann, S.; Schäfer, G.; Rosenthal, W.; Klussmann, E. Mechanisms of protein kinase a anchoring. Int. Rev. Cell Mol. Biol. 2010, 283, 235–330. [Google Scholar]
- Scott, J.D.; Dessauer, C.W.; Tasken, K. Creating order from chaos: Cellular regulation by kinase anchoring. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 187–210. [Google Scholar] [CrossRef] [PubMed]
- Götz, F.; Roske, Y.; Schulz, M.S.; Autenrieth, K.; Bertinetti, D.; Faelber, K.; Zuhlke, K.; Kreuchwig, A.; Kennedy, E.J.; Krause, G.; et al. AKAP18:PKA-RIIalpha structure reveals crucial anchor points for recognition of regulatory subunits of PKA. Biochem. J. 2016, 473, 1881–1894. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.D.; Stofko, R.E.; McDonald, J.R.; Comer, J.D.; Vitalis, E.A.; Mangili, J.A. Type II regulatory subunit dimerization determines the subcellular localization of the cAMP-dependent protein kinase. J. Biol. Chem. 1990, 265, 21561–21566. [Google Scholar]
- Hundsrucker, C.; Krause, G.; Beyermann, M.; Prinz, A.; Zimmermann, B.; Diekmann, O.; Lorenz, D.; Stefan, E.; Nedvetsky, P.; Dathe, M.; et al. High-affinity AKAP7delta-protein kinase A interaction yields novel protein kinase A-anchoring disruptor peptides. Biochem. J. 2006, 396, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Hundsrucker, C.; Rosenthal, W.; Klussmann, E. Peptides for disruption of PKA anchoring. Biochem. Soc. Trans. 2006, 34, 472–473. [Google Scholar] [CrossRef] [PubMed]
- Eccles, R.L.; Czajkowski, M.T.; Barth, C.; Muller, P.M.; McShane, E.; Grunwald, S.; Beaudette, P.; Mecklenburg, N.; Volkmer, R.; Zuhlke, K.; et al. Bimodal antagonism of PKA signalling by ARHGAP36. Nat. Commun. 2016, 7, 12963. [Google Scholar] [CrossRef]
- Bachmann, V.A.; Mayrhofer, J.E.; Ilouz, R.; Tschaikner, P.; Raffeiner, P.; Rock, R.; Courcelles, M.; Apelt, F.; Lu, T.W.; Baillie, G.S.; et al. Gpr161 anchoring of PKA consolidates GPCR and cAMP signaling. Proc. Natl. Acad. Sci. USA. 2016, 113, 7786–7791. [Google Scholar] [CrossRef]
- Rinaldi, L.; Delle Donne, R.; Catalanotti, B.; Torres-Quesada, O.; Enzler, F.; Moraca, F.; Nistico, R.; Chiuso, F.; Piccinin, S.; Bachmann, V.; et al. Feedback inhibition of cAMP effector signaling by a chaperone-assisted ubiquitin system. Nat. Commun. 2019, 10, 2572. [Google Scholar] [CrossRef]
- Medvar, B.; Raghuram, V.; Pisitkun, T.; Sarkar, A.; Knepper, M.A. Comprehensive database of human E3 ubiquitin ligases: Application to aquaporin-2 regulation. Physiol. Genomics 2016, 48, 502–512. [Google Scholar] [CrossRef]
- Cheung, P.W.; Ueberdiek, L.; Day, J.; Bouley, R.; Brown, D. Protein phosphatase 2C is responsible for VP-induced dephosphorylation of AQP2 serine 261. Am. J. Physiol. Renal Physiol. 2017, 313, F404–F413. [Google Scholar] [CrossRef]
- Tamma, G.; Robben, J.H.; Trimpert, C.; Boone, M.; Deen, P.M. Regulation of AQP2 localization by S256 and S261 phosphorylation and ubiquitination. Am. J. Physiol. Cell Physiol. 2011, 300, C636–C646. [Google Scholar] [CrossRef] [PubMed]
- Kamsteeg, E.J.; Hendriks, G.; Boone, M.; Konings, I.B.; Oorschot, V.; van der Sluijs, P.; Klumperman, J.; Deen, P.M. Short-chain ubiquitination mediates the regulated endocytosis of the aquaporin-2 water channel. Proc. Natl. Acad. Sci. USA. 2006, 103, 18344–18349. [Google Scholar] [CrossRef] [PubMed]
- Gharbi-Ayachi, A.; Labbe, J.C.; Burgess, A.; Vigneron, S.; Strub, J.M.; Brioudes, E.; Van-Dorsselaer, A.; Castro, A.; Lorca, T. The substrate of Greatwall kinase, Arpp19, controls mitosis by inhibiting protein phosphatase 2A. Science 2010, 330, 1673–1677. [Google Scholar] [CrossRef] [PubMed]
- Jo, I.; Ward, D.T.; Baum, M.A.; Scott, J.D.; Coghlan, V.M.; Hammond, T.G.; Harris, H.W. AQP2 is a substrate for endogenous PP2B activity within an inner medullary AKAP-signaling complex. Am. J. Physiol. Renal Physiol. 2001, 281, F958–F965. [Google Scholar] [CrossRef] [PubMed]
- Valenti, G.; Procino, G.; Carmosino, M.; Frigeri, A.; Mannucci, R.; Nicoletti, I.; Svelto, M. The phosphatase inhibitor okadaic acid induces AQP2 translocation independently from AQP2 phosphorylation in renal collecting duct cells. J. Cell Sci. 2000, 113, 1985–1992. [Google Scholar]
- McSorley, T.; Stefan, E.; Henn, V.; Wiesner, B.; Baillie, G.S.; Houslay, M.D.; Rosenthal, W.; Klussmann, E. Spatial organisation of AKAP18 and PDE4 isoforms in renal collecting duct principal cells. Eur. J. Cell Biol. 2006, 85, 673–678. [Google Scholar] [CrossRef]
- Szaszak, M.; Christian, F.; Rosenthal, W.; Klussmann, E. Compartmentalized cAMP signalling in regulated exocytic processes in non-neuronal cells. Cell Signal. 2008, 20, 590–601. [Google Scholar] [CrossRef]
- Klussmann, E.; Rosenthal, W. Role and identification of protein kinase A anchoring proteins in vasopressin-mediated aquaporin-2 translocation. Kidney Int. 2001, 60, 446–449. [Google Scholar] [CrossRef][Green Version]
- Nedvetsky, P.I.; Stefan, E.; Frische, S.; Santamaria, K.; Wiesner, B.; Valenti, G.; Hammer, J.A., 3rd; Nielsen, S.; Goldenring, J.R.; Rosenthal, W.; et al. A Role of myosin Vb and Rab11-FIP2 in the aquaporin-2 shuttle. Traffic 2007, 8, 110–123. [Google Scholar] [CrossRef]
- Matsuda, S.; Kawamoto, K.; Miyamoto, K.; Tsuji, A.; Yuasa, K. PCTK3/CDK18 regulates cell migration and adhesion by negatively modulating FAK activity. Sci. Rep. 2017, 7, 45545. [Google Scholar] [CrossRef]
- Varjosalo, M.; Keskitalo, S.; Van Drogen, A.; Nurkkala, H.; Vichalkovski, A.; Aebersold, R.; Gstaiger, M. The protein interaction landscape of the human CMGC kinase group. Cell Rep. 2013, 3, 1306–1320. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Viability (%) | Protein Symbol | Gene Name | Accession | |
|---|---|---|---|---|
| 1 | 97 | Pkia | protein kinase inhibitor alpha | NM_008862.3 |
| 2 | 87 | Stk11 | serine/threonine kinase 11 | NM_011492.3 |
| 3 | 79 | Fgfr1 | fibroblast growth factor receptor 1 | NM_010206.2 |
| 4 | 78 | Ephb3 | Eph receptor B3 | NM_010143.1 |
| 5 | 75 | Bmpr1b | bone morphogenetic protein receptor type 1B | NM_007560.3 |
| 6 | 70 | Vrk3 | vaccinia related kinase 3 | NM_133945.1 |
| 7 | 70 | Trim27 | tripartite motif-containing 27 | NM_009054.3 |
| 8 | 70 | Cdk18 | cyclin-dependent kinase 18 | NM_008795.2 |
| 9 | 69 | Ak3 | adenylate kinase 3 | NM_021299.1 |
| 10 | 69 | Syk | spleen tyrosine kinase | NM_011518.2 |
| 11 | 66 | Tk2 | thymidine kinase 2 mitochondrial | NM_021028.3 |
| 12 | 65 | Dusp2 | dual specificity phosphatase 2 | NM_010090.2 |
| 13 | 64 | PFKP | Phosphofructokinase 1 | NM_019703 |
| 14 | 52 | Trpm6 | transient receptor potential cation channel subfamily M member 6 | NM_153417.1 |
| 15 | 52 | Aurka | aurora kinase A | NM_011497.3 |
| 16 | 50 | Evi5l | ecotropic viral integration site 5 like | NM_001039578.3 |
| 17 | 33 | Aurkb | aurora kinase B | NM_011496.1 |
| 18 | 25 | Copb2 | coatomer protein complex subunit beta 2 (beta prime) | NM_015827.2 |
| 19 | 18 | Wee1 | WEE 1 homolog 1 (S. pombe) | NM_009516.3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dema, A.; Faust, D.; Lazarow, K.; Wippich, M.; Neuenschwander, M.; Zühlke, K.; Geelhaar, A.; Pallien, T.; Hallscheidt, E.; Eichhorst, J.; et al. Cyclin-Dependent Kinase 18 Controls Trafficking of Aquaporin-2 and Its Abundance through Ubiquitin Ligase STUB1, Which Functions as an AKAP. Cells 2020, 9, 673. https://doi.org/10.3390/cells9030673
Dema A, Faust D, Lazarow K, Wippich M, Neuenschwander M, Zühlke K, Geelhaar A, Pallien T, Hallscheidt E, Eichhorst J, et al. Cyclin-Dependent Kinase 18 Controls Trafficking of Aquaporin-2 and Its Abundance through Ubiquitin Ligase STUB1, Which Functions as an AKAP. Cells. 2020; 9(3):673. https://doi.org/10.3390/cells9030673
Chicago/Turabian StyleDema, Alessandro, Dörte Faust, Katina Lazarow, Marc Wippich, Martin Neuenschwander, Kerstin Zühlke, Andrea Geelhaar, Tamara Pallien, Eileen Hallscheidt, Jenny Eichhorst, and et al. 2020. "Cyclin-Dependent Kinase 18 Controls Trafficking of Aquaporin-2 and Its Abundance through Ubiquitin Ligase STUB1, Which Functions as an AKAP" Cells 9, no. 3: 673. https://doi.org/10.3390/cells9030673
APA StyleDema, A., Faust, D., Lazarow, K., Wippich, M., Neuenschwander, M., Zühlke, K., Geelhaar, A., Pallien, T., Hallscheidt, E., Eichhorst, J., Wiesner, B., Černecká, H., Popp, O., Mertins, P., Dittmar, G., von Kries, J. P., & Klussmann, E. (2020). Cyclin-Dependent Kinase 18 Controls Trafficking of Aquaporin-2 and Its Abundance through Ubiquitin Ligase STUB1, Which Functions as an AKAP. Cells, 9(3), 673. https://doi.org/10.3390/cells9030673