Cancer Stem Cells: Devil or Savior—Looking behind the Scenes of Immunotherapy Failure

 ,
,  ,
, 

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
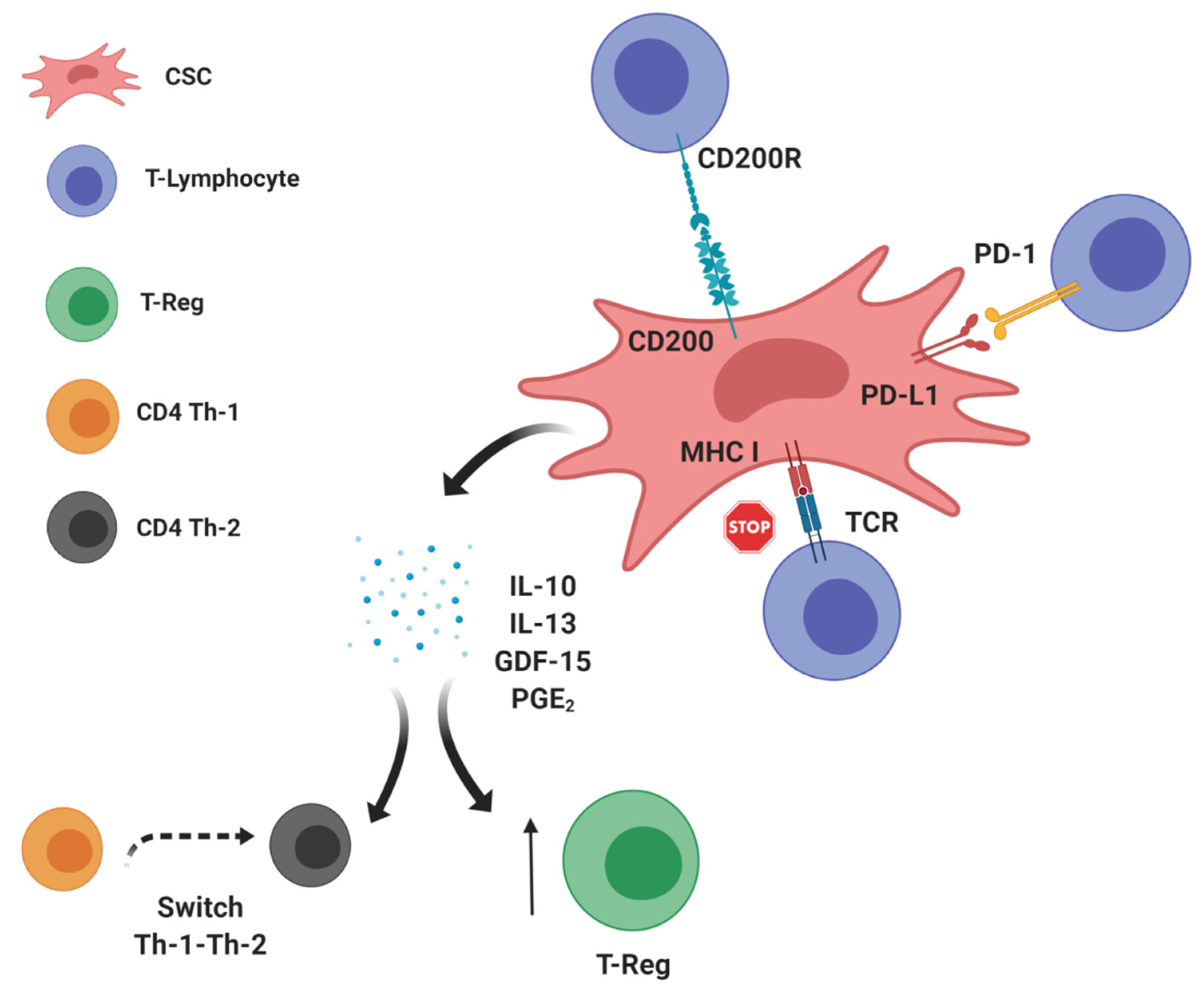
2. CSCs: Leading Actors in the Intratumor Immunosuppressive Scenario?
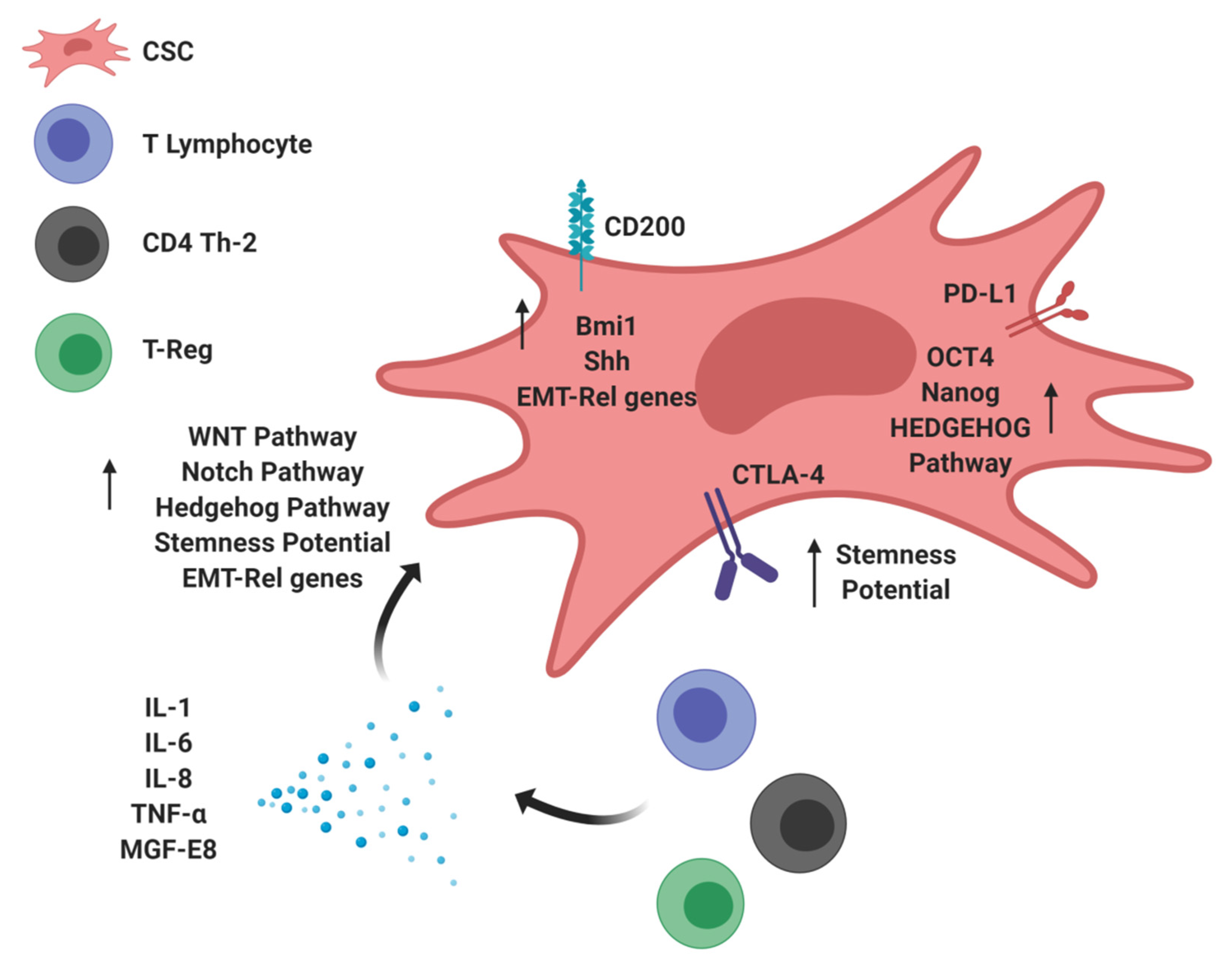
3. Immunomodulatory Molecules Stimulating Tumor Stemness: The Dark Side of CSCs
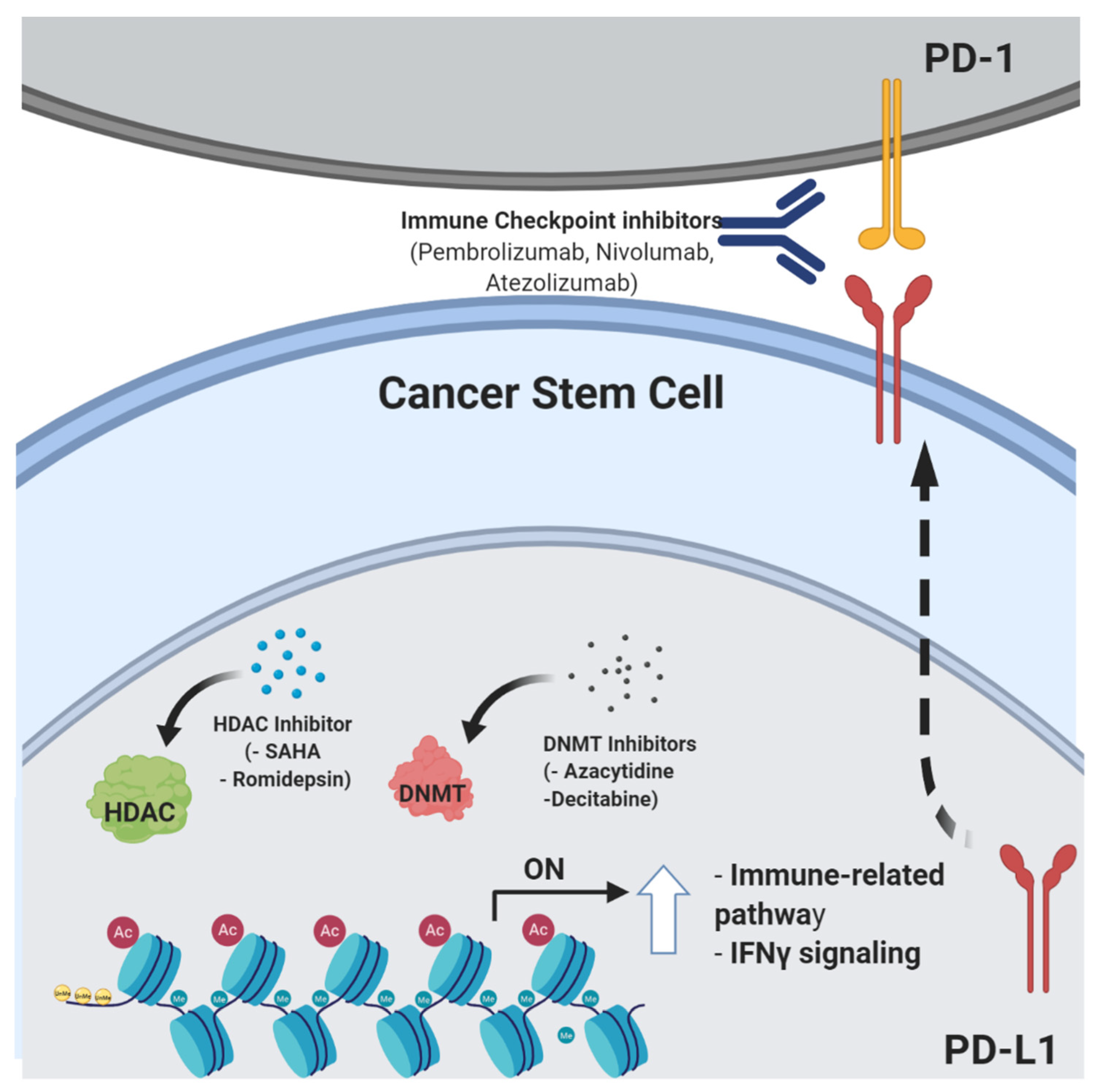
4. Epigenetic Control of Stemness Features and Immune Escape
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- O’Donnell, J.S.; Teng, M.W.L.; Smyth, M.J. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol. 2018, 16, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genome Res. 2018, 32, 1267–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gun, S.Y.; Lee, S.W.L.; Sieow, J.L.; Wong, S.C. Targeting immune cells for cancer therapy. Redox Boil. 2019, 25, 101174. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Favero, F.; De Bruin, E.C.; Birkbak, N.J.; Szallasi, Z.; Swanton, C. Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci. Transl. Med. 2015, 7, 283ra54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Jiang, E.; Yan, T.; Xu, Z.; Shang, Z. Tumor Microenvironment and Cell Fusion. BioMed Res. Int. 2019, 2019, 5013592-12. [Google Scholar] [CrossRef]
- Ciardiello, C.; Leone, A.; Budillon, A. The Crosstalk between Cancer Stem Cells and Microenvironment Is Critical for Solid Tumor Progression: The Significant Contribution of Extracellular Vesicles. Stem Cells Int. 2018, 2018, 1–11. [Google Scholar] [CrossRef]
- Greaves, M.; Mailey, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef]
- Kreso, A.; Dick, J.E. Evolution of the Cancer Stem Cell Model. Cell Stem Cell 2014, 14, 275–291. [Google Scholar] [CrossRef] [Green Version]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef]
- Shackleton, M.; Vaillant, F.; Simpson, K.J.; Stingl, J.; Smyth, G.K.; Asselin-Labat, M.-L.; Wu, L.; Lindeman, G.J.; Visvader, J.E. Generation of a functional mammary gland from a single stem cell. Nature 2006, 439, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Mose, E.S.; Montel, V.; Tarin, D. Dormant Cancer Cells Retrieved from Metastasis-Free Organs Regain Tumorigenic and Metastatic Potency. Am. J. Pathol. 2006, 169, 673–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phi, L.T.H.; Sari, I.N.; Yang, Y.-G.; Lee, S.-H.; Jun, N.; Kim, K.S.; Lee, Y.K.; Kwon, H.Y. Cancer Stem Cells (CSCs) in Drug Resistance and their Therapeutic Implications in Cancer Treatment. Stem Cells Int. 2018, 2018, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayob, A.Z.; Ramasamy, T.S. Cancer stem cells as key drivers of tumour progression. J. Biomed. Sci. 2018, 25, 20. [Google Scholar] [CrossRef]
- Maccalli, C.; Rasul, K.I.; Elawad, M.; Ferrone, S. The role of cancer stem cells in the modulation of anti-tumor immune responses. Semin. Cancer Boil. 2018, 53, 189–200. [Google Scholar] [CrossRef]
- Boyd, A.S.; Rodrigues, N.P. Stem Cells Cycle toward Immune Surveillance. Immuniy 2018, 48, 187–190. [Google Scholar] [CrossRef] [Green Version]
- Miranda, A.; Hamilton, P.T.; Zhang, A.W.; Pattnaik, S.; Becht, E.; Mezheyeuski, A.; Bruun, J.; Micke, P.; De Reynies, A.; Nelson, B.H. Cancer stemness, intratumoral heterogeneity, and immune response across cancers. Proc. Natl. Acad. Sci. USA 2019, 116, 9020–9029. [Google Scholar] [CrossRef] [Green Version]
- Korkaya, H.; Liu, S.; Wicha, M.S. Regulation of cancer stem cells by cytokine networks: Attacking cancer’s inflammatory roots. Clin. Cancer Res. 2011, 17, 6125–6129. [Google Scholar] [CrossRef] [Green Version]
- Sultan, M.; Coyle, K.; Vidovic, D.; Thomas, M.L.; Gujar, S.; Marcato, P. Hide-and-seek: The interplay between cancer stem cells and the immune system. Carcinogenesis 2016, 38, 107–118. [Google Scholar] [CrossRef]
- Lu, X.; Kang, Y. Cell Fusion Hypothesis of the Cancer Stem Cell. Adv. Exp. Med. Biol. 2011, 950, 129–140. [Google Scholar]
- Nagler, C.; Zänker, K.S.; Dittmar, T. Cell Fusion, Drug Resistance and Recurrence CSCs. Adv. Exp. Med. Biol. 2011, 950, 173–182. [Google Scholar]
- Pires, B.; E Souza, L.D.; Rodrigues, J.A.; De Amorim, Í.S.S.; Mencalha, A.L. Targeting Cellular Signaling Pathways in Breast Cancer Stem Cells and its Implication for Cancer Treatment. Anticancer. Res. 2016, 36, 5681–5692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seliger, B.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar]
- Ebben, J.D.; Treisman, D.M.; Zorniak, M.; Kutty, R.G.; Clark, P.A.; Kuo, J.S. The cancer stem cell paradigm: A new understanding of tumor development and treatment. Expert Opin. Ther. Targets 2010, 14, 621–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Paulete, A.R.; Teijeira, A.; Cueto, F.J.; Garasa, S.; Perez-Gracia, J.L.; Sanchez-Arraez, A.; Sancho, D.; Melero, I. Antigen cross-presentation and T-cell cross-priming in cancer immunology and immunotherapy. Ann. Oncol. 2017, 28, xii74. [Google Scholar] [CrossRef]
- Cresswell, P.; Bangia, N.; Dick, T.; Diedrich, G. The nature of the MHC class I peptide loading complex. Immunol. Rev. 1999, 172, 21–28. [Google Scholar] [CrossRef]
- Rasool, S.; Rutella, S.; Ferrone, S.; Maccalli, C. Cancer Stem Cells: The Players of Immune Evasion from Immunotherapy. In Cancer Stem Cell Resistance to Targeted Therapy; Maccalli, C., Todaro, M., Ferrone, S., Eds.; Springer Nature: Basel, Switzerland, 2019; pp. 223–249. [Google Scholar]
- Rodríguez, J.A. HLA-mediated tumor escape mechanisms that may impair immunotherapy clinical outcomes via T-cell activation. Oncol. Lett. 2017, 14, 4415–4427. [Google Scholar] [CrossRef] [Green Version]
- Chikamatsu, K.; Takahashi, G.; Sakakura, K.; Ferrone, S.; Masuyama, K. Immunoregulatory properties of CD44+ cancer stem-like cells in squamous cell carcinoma of the head and neck. Head Neck 2011, 33, 208–215. [Google Scholar] [CrossRef] [Green Version]
- Volonté, A.; Di Tomaso, T.; Spinelli, M.; Todaro, M.; Sanvito, F.; Albarello, L.; Bissolati, M.; Ghirardelli, L.; Orsenigo, E.; Ferrone, S.; et al. Cancer-initiating cells from colorectal cancer patients escape from t cell–mediated immunosurveillance in vitro through membrane-bound IL-4. J. Immunol. 2014, 192, 523–532. [Google Scholar]
- Schatton, T.; Schütte, U.; Frank, N.Y.; Zhan, Q.; Hoerning, A.; Robles, S.C.; Zhou, J.; Hodi, F.S.; Spagnoli, G.C.; Murphy, G.F.; et al. Modulation of T-cell activation by malignant melanoma initiating cells. Cancer Res. 2010, 70, 697–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Tomaso, T.; Mazzoleni, S.; Wang, E.; Sovena, G.; Clavenna, D.; Franzin, A.; Mortini, P.; Ferrone, S.; Doglioni, C.; Marincola, F.M.; et al. Immunobiological characterization of cancer stem cells isolated from glioblastoma patients. Clin. Cancer Res. 2010, 16, 800–813. [Google Scholar] [CrossRef] [Green Version]
- Pietra, G.; Manzini, C.; Vitale, M.; Balsamo, M.; Ognio, E.; Boitano, M.; Queirolo, P.; Moretta, L.; Mingari, M.C. Natural killer cells kill human melanoma cells with characteristics of cancer stem cells. Int. Immunol. 2009, 21, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Castriconi, R.; Daga, A.; Dondero, A.; Zona, G.; Poliani, P.L.; Melotti, A.; Griffero, F.; Marubbi, D.; Spaziante, R.; Bellora, F.; et al. NK Cells Recognize and Kill Human Glioblastoma Cells with Stem Cell-Like Properties. J. Immunol. 2009, 182, 3530–3539. [Google Scholar] [CrossRef] [Green Version]
- Gorczynski, R.; Yu, K.; Clark, D. Receptor engagement on cells expressing a ligand for the tolerance-inducing molecule OX2 induces an immunoregulatory population that inhibits alloreactivity in vitro and in vivo. J. Immunol. 2000, 165, 4854–4860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorczynski, R.; Chen, Z.; Kai, Y.; Lee, L.; Wong, S.; Marsden, P.A. CD200 is a ligand for all members of the CD200R family of immunoregulatory molecules. J. Immunol. 2004, 172, 7744–7749. [Google Scholar] [CrossRef]
- Kretz-Rommel, A.; Qin, F.; Dakappagari, N.; Ravey, E.P.; McWhirter, J.; Oltean, D.; Frederickson, S.; Maruyama, T.; Wild, M.A.; Nolan, M.-J.; et al. CD200 expression on tumor cells suppresses antitumor immunity: New approaches to cancer immunotherapy. J. Immunol. 2007, 178, 5595–5605. [Google Scholar] [CrossRef] [Green Version]
- Jung, Y.-S.; Vermeer, P.D.; Vermeer, D.W.; Lee, S.-J.; Goh, A.R.; Ahn, H.-J.; Lee, J.H. CD200: Association with cancer stem cell features and response to chemoradiation in head and neck squamous cell carcinoma. Head Neck 2014, 37, 327–335. [Google Scholar] [CrossRef] [Green Version]
- Deonarain, M.P.; Kousparou, C.A.; Epenetos, A.A. Antibodies targeting cancer stem cells: A new paradigm in immunotherapy? MAbs 2009, 1, 12–25. [Google Scholar] [CrossRef] [Green Version]
- Gupta, H.B.; A Clark, C.; Yuan, B.; Sareddy, G.; Pandeswara, S.; Padron, A.S.; Hurez, V.; Conejo-Garcia, J.; Vadlamudi, R.; Li, R.; et al. Tumor cell-intrinsic PD-L1 promotes tumor-initiating cell generation and functions in melanoma and ovarian cancer. Signal Transduct. Target. Ther. 2016, 1, 16030. [Google Scholar] [CrossRef] [Green Version]
- Castagnoli, L.; Cancila, V.; Cordoba-Romero, S.L.; Faraci, S.; Talarico, G.; Belmonte, B.; Iorio, M.; Milani, M.; Volpari, T.; Chiodoni, C.; et al. WNT signaling modulates PD-L1 expression in the stem cell compartment of triple-negative breast cancer. Oncogene 2019, 38, 4047–4060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Chen, C.; Xu, Z.P.; Gu, W. Increased PD-L1 Expression in Breast and Colon Cancer Stem Cells. Clin. Exp. Pharmacol. Physiol. 2017, 44, 602–604. [Google Scholar] [CrossRef] [PubMed]
- Raniszewska, A.; Polubiec-Kownacka, M.; Rutkowska, E.; Domagala-Kulawik, J. PD-L1 Expression on Lung Cancer Stem Cells in Metastatic Lymph Nodes Aspirates. Stem Cell Rev. Rep. 2018, 15, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Amarnath, S.; Mangus, C.W.; Wang, J.C.M.; Wei, F.; He, A.; Kapoor, V.; Foley, J.E.; Massey, P.R.; Felizardo, T.C.; Riley, J.; et al. The PDL1-PD1 Axis Converts Human TH1 Cells into Regulatory T Cells. Sci. Transl. Med. 2011, 3, 111ra120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francisco, L.M.; Sage, P.T.; Sharpe, A.H. The PD-1 pathway in tolerance and autoimmunity. Immunol. Rev. 2010, 236, 219–242. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Ge, J.; Xiang, B.; Wu, X.; Ma, J.; Zhou, M.; Li, X.; et al. Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol. Cancer 2019, 18, 10. [Google Scholar] [CrossRef] [Green Version]
- Gianchecchi, E.; Fierabracci, A. Inhibitory Receptors and Pathways of Lymphocytes: The Role of PD-1 in Treg Development and Their Involvement in Autoimmunity Onset and Cancer Progression. Front. Immunol. 2018, 9, 2374. [Google Scholar] [CrossRef] [Green Version]
- Alsaab, H.O.; Sau, S.; Alzhrani, R.; Tatiparti, K.; Bhise, K.; Kashaw, S.K.; Iyer, A.K. PD-1 and PD-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome. Front. Pharmacol. 2017, 8, 8. [Google Scholar] [CrossRef]
- Hsu, J.-M.; Xia, W.; Hsu, Y.-H.; Chan, L.-C.; Yu, W.-H.; Cha, J.-H.; Chen, C.-T.; Liao, H.-W.; Kuo, C.-W.; Khoo, K.-H.; et al. STT3-dependent PD-L1 accumulation on cancer stem cells promotes immune evasion. Nat. Commun. 2018, 9, 1908. [Google Scholar] [CrossRef]
- Arce-Sillas, A.; Álvarez-Luquín, D.D.; Tamaya-Domínguez, B.; Gomez-Fuentes, S.; Trejo-García, A.; Melo-Salas, M.; Cárdenas, G.; Rodríguez-Ramírez, J.; Adalid-Peralta, L. Regulatory T Cells: Molecular Actions on Effector Cells in Immune Regulation. J. Immunol. Res. 2016, 2016, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Wynn, T.A. IL-13 effector functions. Annu. Rev. Immunol. 2003, 21, 425–456. [Google Scholar] [CrossRef] [PubMed]
- Maccalli, C.; Volontè, A.; Cimminiello, C.; Parmiani, G. Immunology of cancer stem cells in solid tumours. A review. Eur. J. Cancer 2014, 50, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, J.; Kong, J.; Tang, J.; Wu, Y.; Xu, E.; Zhang, H.; Lai, M. GDF15 promotes EMT and metastasis in colorectal cancer. Oncotarget 2015, 7, 860–872. [Google Scholar] [CrossRef] [PubMed]
- Roth, P.; Junker, M.; Tritschler, I.; Mittelbronn, M.; Dombrowski, Y.; Breit, S.N.; Tabatabai, G.; Wick, W.; Weller, M.; Wischhusen, J. GDF-15 Contributes to Proliferation and Immune Escape of Malignant Gliomas. Clin. Cancer Res. 2010, 16, 3851–3859. [Google Scholar] [CrossRef] [Green Version]
- Sasahara, A.; Tominaga, K.; Nishimura, T.; Yano, M.; Kiyokawa, E.; Noguchi, M.; Noguchi, M.; Kanauchi, H.; Ogawa, T.; Minato, H.; et al. An autocrine/paracrine circuit of growth differentiation factor (GDF) 15 has a role for maintenance of breast cancer stem-like cells. Oncotarget 2017, 8, 24869–24881. [Google Scholar] [CrossRef]
- Goessling, W.; North, T.E.; Loewer, S.; Lord, A.M.; Lee, S.; Stoick-Cooper, C.L.; Weidinger, G.; Puder, M.; Daley, G.Q.; Moon, R.; et al. Genetic Interaction of PGE2 and Wnt Signaling Regulates Developmental Specification of Stem Cells and Regeneration. Cell 2009, 136, 1136–1147. [Google Scholar] [CrossRef] [Green Version]
- Sakata, D.; Yao, C.; Narumiya, S. Emerging roles of prostanoids in T cell-mediated immunity. IUBMB Life 2010, 62, 591–596. [Google Scholar] [CrossRef]
- Wang, X.; Venugopal, C.; Manoranjan, B.; McFarlane, N.; O’Farrell, E.; Nolte, S.; Gunnarsson, T.; Hollenberg, R.; Kwiecien, J.; Northcott, P.; et al. Sonic hedgehog regulates Bmi1 in human medulloblastoma brain tumor-initiating cells. Oncogene 2011, 31, 187–199. [Google Scholar] [CrossRef] [Green Version]
- Carballo, G.B.; Honorato, J.R.; De Lopes, G.P.F.; Spohr, T.C.L.D.S.E. A highlight on Sonic hedgehog pathway. Cell Commun. Signal. 2018, 16, 11. [Google Scholar] [CrossRef]
- Shin, S.P.; Goh, A.R.; Kang, H.G.; Kim, S.J.; Kim, J.K.; Kim, K.T.; Lee, J.H.; Bae, Y.S.; Jung, Y.S.; Lee, S.J. CD200 Induces Epithelial-to-Mesenchymal Transition in Head and Neck Squamous Cell Carcinoma via β-Catenin-Mediated Nuclear Translocation. Cancers 2019, 11, 1583. [Google Scholar] [CrossRef] [Green Version]
- Stumpfova, M.; Ratner, D.; Desciak, E.B.; Eliezri, Y.D.; Owens, D.M. The immunosuppressive surface ligand CD200 augments the metastatic capacity of squamous cell carcinoma. Cancer Res. 2010, 70, 2962–2972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almozyan, S.; Colak, D.; Mansour, F.; Alaiya, A.; Al-Harazi, O.; Qattan, A.; Al-Mohanna, F.; Al-Alwan, M.; Ghebeh, H. PD-L1 promotes OCT4 and Nanog expression in breast cancer stem cells by sustaining PI3K/AKT pathway activation. Int. J. Cancer 2017, 141, 1402–1412. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.; Zhang, T.; Deng, S.-C.; Wei, J.-C.; Yang, P.; Wang, Q.; Chen, Z.-P.; Li, W.-L.; Chen, H.-C.; Hu, H.; et al. PD-L1 promotes colorectal cancer stem cell expansion by activating HMGA1-dependent signaling pathways. Cancer Lett. 2019, 450, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; Gladney, W.L. Immune escape mechanisms as a guide for cancer immunotherapy. Clin. Cancer Res. 2014, 21, 687–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Dang, J.; Ba, D.; Wang, C.; Han, J.; Zheng, F. Potential function of CTLA-4 in the tumourigenic capacity of melanoma stem cells. Oncol. Lett. 2018, 16, 6163–6170. [Google Scholar] [CrossRef]
- Mantovani, A.; Dinarello, C.A.; Molgora, M.; Garlanda, C. Interleukin-1 and Related Cytokines in the Regulation of Inflammation and Immunity. Immunity 2019, 50, 778–795. [Google Scholar] [CrossRef] [Green Version]
- Caucheteux, S.M.; Hu-Li, J.; Guo, L.; Bhattacharyya, N.; Crank, M.; Collins, M.T.; Paul, W.E. IL-1b enhances inflammatory TH2 differentiation. J. Allergy Clin. Immunol. 2016, 138, 898–901. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wang, L.; Pappan, L.; Galliher-Beckley, A.; Shi, J. IL-1b promotes stemness and invasiveness of colon cancer cells through Zeb1 activation. Mol. Cancer 2012, 11, 87. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui, I.; Erreni, M.; Kamal, M.A.; Porta, C.; Marchesi, F.; Pesce, S.; Pasqualini, F.; Schiarea, S.; Chiabrando, C.; Mantovani, A.; et al. Differential role of Interleukin-1 and Interleukin-6 in K-Ras-driven pancreatic carcinoma undergoing mesenchymal transition. OncoImmunology 2017, 7, e1388485. [Google Scholar] [CrossRef] [Green Version]
- Fisher, D.T.; Appenheimer, M.M.; Evans, S.S. The two faces of IL-6 in the tumor microenvironment. Semin. Immunol. 2014, 26, 38–47. [Google Scholar] [CrossRef] [Green Version]
- Wei, H. Interleukin 6 signaling maintains the stem-like properties of bladder cancer stem cells. Transl. Cancer Res. 2019, 8, 557–566. [Google Scholar] [CrossRef]
- Wan, S.; Zhao, E.; Kryczek, I.; Vatan, L.; Sadovskaya, A.; Ludema, G.; Simeone, D.M.; Zou, W.; Welling, T.H. Tumor-associated macrophages produce interleukin 6 and signal via STAT3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology 2014, 147, 1393–1404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishibashi, K.; Koguchi, T.; Matsuoka, K.; Onagi, A.; Tanji, R.; Takinami-Honda, R.; Hoshi, S.; Onoda, M.; Kurimura, Y.; Hata, J.; et al. Interleukin-6 induces drug resistance in renal cell carcinoma. Fukushima J. Med Sci. 2018, 64, 103–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zong, X.; Mitra, S.; Mitra, A.K.; Matei, D.; Nephew, K.P. IL-6 mediates platinum-induced enrichment of ovarian cancer stem cells. JCI Insight 2018, 3, 122360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, J.M.; Dominguez, C.; Hamilton, D.H.; Palena, C. The IL-8/IL-8R axis: A double agent in tumor immune resistance. Vaccines 2016, 4, 22. [Google Scholar] [CrossRef] [Green Version]
- Jin, F.; Miao, Y.; Xu, P.; Qiu, X. IL-8 regulates the stemness properties of cancer stem cells in the small-cell lung cancer cell line H446. OncoTargets Ther. 2018, 11, 5723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Fan, J.; Chen, H.; Meng, Z.; Chen, Z.; Wang, P.; Liu, L. The IL-8/CXCR1 axis is associated with cancer stem cell-like properties and correlates with clinical prognosis in human pancreatic cancer cases. Sci. Rep. 2014, 4, 5911. [Google Scholar] [CrossRef]
- Singh, J.K.; Simões, B.M.; Howell, S.J.; Farnie, G.; Clarke, R. Recent advances reveal IL-8 signaling as a potential key to targeting breast cancer stem cells. Breast Cancer Res. 2013, 15, 210. [Google Scholar] [CrossRef] [Green Version]
- Salomon, B.L.; Leclerc, M.; Tosello, J.; Ronin, E.; Piaggio, E.; Cohen, J.L. Tumor Necrosis Factor +¦ and Regulatory T Cells in Oncoimmunology. Front. Immunol. 2018, 9, 444. [Google Scholar] [CrossRef]
- Wang, H.; Wang, H.S.; Zhou, B.H.; Li, C.L.; Zhang, F.; Wang, X.F.; Zhang, G.; Bu, X.Z.; Cai, S.H.; Du, J. Epithelial-mesenchymal transition (EMT) induced by TNF-+¦ requires AKT/GSK-3+¦-mediated stabilization of snail in colorectal cancer. PLoS ONE 2013, 8, e56664. [Google Scholar] [CrossRef]
- Jinushi, M.; Sato, M.; Kanamoto, A.; Itoh, A.; Nagai, S.; Koyasu, S.; Dranoff, G.; Tahara, H. Milk fat globule epidermal growth factor–8 blockade triggers tumor destruction through coordinated cell-autonomous and immune-mediated mechanisms. J. Exp. Med. 2009, 206, 1317–1326. [Google Scholar] [CrossRef] [PubMed]
- Jinushi, M.; Chiba, S.; Yoshiyama, H.; Masutomi, K.; Kinoshita, I.; Dosaka-Akita, H.; Yagita, H.; Takaoka, A.; Tahara, H. Tumor-associated macrophages regulate tumorigenicity and anticancer drug responses of cancer stem/initiating cells. Proc. Natl. Acad. Sci. USA 2011, 108, 12425–12430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolffe, A.P.; Matzke, M.A. Epigenetics: Regulation Through Repression. Science 1999, 286, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Luger, K.; Mäder, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef] [Green Version]
- McCulloch, E.A.; Siminovitch, L.; Till, J.; Locke, M. Spleen-Colony Formation in Anemic Mice of Genotype WW. Science 1964, 144, 844–846. [Google Scholar] [CrossRef] [Green Version]
- Seita, J.; Weissman, I.L. Hematopoietic stem cell: Self-renewal versus differentiation. Wiley Interdiscip. Rev. Syst. Boil. Med. 2010, 2, 640–653. [Google Scholar] [CrossRef] [Green Version]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.-Y.; Jones, D.T.W.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.A.K.; Tönjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef]
- Wu, C.; Allis, C.D. Nucleosomes, histones & chromatin part B. Preface. Methods Enzymol. 2012, 513, xv–xvi. [Google Scholar]
- Funato, K.; Major, T.; Lewis, P.; Allis, C.D.; Tabar, V. Use of human embryonic stem cells to model pediatric gliomas with H3.3K27M histone mutation. Science 2014, 346, 1529–1533. [Google Scholar] [CrossRef] [Green Version]
- Band, V.; Zhao, X.; Malhotra, G.K.; Band, H. Shared signaling pathways in normal and breast cancer stem cells. J. Carcinog. 2011, 10, 38. [Google Scholar] [CrossRef]
- Handle, F.; Erb, H.H.H.; Luef, B.; Hoefer, J.; Dietrich, D.; Parson, W.; Kristiansen, G.; Santer, F.; Culig, Z. SOCS3 Modulates the Response to Enzalutamide and is Regulated by AR Signaling and CpG Methylation in Prostate Cancer Cells. Mol. Cancer Res. 2016, 14, 574–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersson, E.R.; Sandberg, R.; Lendahl, U. Notch signaling: Simplicity in design, versatility in function. Development 2011, 138, 3593–3612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghoshal, P.; Nganga, A.J.; Moran-Giuati, J.; Szafranek, A.; Johnson, T.R.; Bigelow, A.J.; Houde, C.M.; Avet-Loiseau, H.; Smiraglia, D.J.; Ersing, N.; et al. Loss of the SMRT/NCoR2 Corepressor Correlates with JAG2 Overexpression in Multiple Myeloma. Cancer Res. 2009, 69, 4380–4387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregorieff, A.; Clevers, H. Wnt signaling in the intestinal epithelium: From endoderm to cancer. Genes Dev. 2005, 19, 877–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, P.A.; Baylin, S.B. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 2002, 3, 415–428. [Google Scholar] [CrossRef]
- Ying, J.; Li, H.; Yu, J.; Ng, K.M.; Poon, F.F.; Wong, S.C.; Chan, A.T.; Sung, J.J.; Tao, Q. WNT5A exhibits tumor-suppressive activity through antagonizing the Wnt/beta-catenin signaling, and is frequently methylated in colorectal cancer. Clin. Cancer Res. 2008, 14, 55–61. [Google Scholar] [CrossRef] [Green Version]
- Laird, P.W. Cancer epigenetics. Hum. Mol. Genet. 2005, 14, R65–R76. [Google Scholar] [CrossRef] [Green Version]
- Ansel, K.M.; Lee, D.U.; Rao, A. An epigenetic view of helper T cell differentiation. Nat. Immunol. 2003, 4, 616–623. [Google Scholar] [CrossRef]
- Bergman, Y.; Cedar, H. A stepwise epigenetic process controls immunoglobulin allelic exclusion. Nat. Rev. Immunol. 2004, 4, 753–761. [Google Scholar] [CrossRef]
- Smale, S.T.; Fisher, A.G. Chromatin structure and gene regulation in the immune system. Annu Rev Immunol. 2002, 20, 427–462. [Google Scholar] [CrossRef] [PubMed]
- Sigalotti, L.; Fratta, E.; Coral, S.; Maio, M. Epigenetic drugs as immunomodulators for combination therapies in solid tumors. Pharmacol. Ther. 2014, 142, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Héninger, E.; Krueger, T.E.; Lang, J.M. Augmenting Antitumor Immune Responses with Epigenetic Modifying Agents. Front. Immunol. 2015, 6, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terranova-Barberio, M.; Thomas, S.; Munster, P.N. Epigenetic modifiers in immunotherapy: A focus on checkpoint inhibitors. Immunotherapy 2016, 8, 705–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.C.; Campoli, M.; Restifo, N.P.; Wang, X.; Ferrone, S. Immune selection of hot-spot beta 2-microglobulin gene mutations, HLA-A2 allospecificity loss, and antigen-processing machinery component down-regulation in melanoma cells derived from recurrent metastases following immunotherapy. J. Immunol. 2005, 174, 1462–1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fratta, E.; Coral, S.; Covre, A.; Parisi, G.; Colizzi, F.; Danielli, R.; Nicolay, H.; Sigalotti, L.; Maio, M. The biology of cancer testis antigens: Putative function, regulation and therapeutic potential. Mol. Oncol. 2011, 5, 164–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wrangle, J.; Wang, W.; Koch, A.; Easwaran, H.; Mohammad, H.P.; Vendetti, F.; VanCriekinge, W.; Demeyer, T.; Du, Z.; Parsana, P.; et al. Alterations of immune response of non-small cell lung cancer with Azacytidine. Oncotarget 2013, 4, 2067–2079. [Google Scholar] [CrossRef] [Green Version]
- Booth, L.; Roberts, J.L.; Sander, C.; Lee, J.; Kirkwood, J.M.; Poklepovic, A.; Dent, P. The HDAC inhibitor AR42 interacts with pazopanib to kill trametinib/dabrafenib-resistant melanoma cells in vitro and in vivo. Oncotarget 2017, 8, 16367–16386. [Google Scholar] [CrossRef] [Green Version]
- Beg, A.A.; E Gray, J. HDAC inhibitors with PD-1 blockade: A promising strategy for treatment of multiple cancer types? Epigenomics 2016, 8, 1015–1017. [Google Scholar] [CrossRef]
- Yang, H.; Lan, P.; Hou, Z.; Guan, Y.; Zhang, J.; Xu, W.; Tian, Z.; Zhang, C. Histone deacetylase inhibitor SAHA epigenetically regulates miR-17-92 cluster and MCM7 to upregulate MICA expression in hepatoma. Br. J. Cancer 2014, 112, 112–121. [Google Scholar] [CrossRef] [Green Version]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome — Biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, N.; Easwaran, H.; Baylin, S.B. Harnessing the potential of epigenetic therapy to target solid tumors. J. Clin. Investig. 2014, 124, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Chiappinelli, K.B.; Strissel, P.L.; Desrichard, A.; Li, H.; Henke, C.; Akman, B.; Hein, A.; Rote, N.S.; Cope, L.M.; Snyder, A.; et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 2015, 162, 974–986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krummel, M.F.; Allison, J.P. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J. Exp. Med. 1995, 182, 459–465. [Google Scholar] [CrossRef] [Green Version]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. New Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Collins, D.C.; Sundar, R.; Lim, J.S.; Yap, T.A. Towards Precision Medicine in the Clinic: From Biomarker Discovery to Novel Therapeutics. Trends Pharmacol. Sci. 2017, 38, 25–40. [Google Scholar] [CrossRef] [PubMed]
- Hasin, Y.; Seldin, M.; Lusis, A.J. Multi-omics approaches to disease. Genome Boil. 2017, 18, 83. [Google Scholar] [CrossRef] [PubMed]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castagnoli, L.; De Santis, F.; Volpari, T.; Vernieri, C.; Tagliabue, E.; Di Nicola, M.; Pupa, S.M. Cancer Stem Cells: Devil or Savior—Looking behind the Scenes of Immunotherapy Failure. Cells 2020, 9, 555. https://doi.org/10.3390/cells9030555
Castagnoli L, De Santis F, Volpari T, Vernieri C, Tagliabue E, Di Nicola M, Pupa SM. Cancer Stem Cells: Devil or Savior—Looking behind the Scenes of Immunotherapy Failure. Cells. 2020; 9(3):555. https://doi.org/10.3390/cells9030555
Chicago/Turabian StyleCastagnoli, Lorenzo, Francesca De Santis, Tatiana Volpari, Claudio Vernieri, Elda Tagliabue, Massimo Di Nicola, and Serenella M. Pupa. 2020. "Cancer Stem Cells: Devil or Savior—Looking behind the Scenes of Immunotherapy Failure" Cells 9, no. 3: 555. https://doi.org/10.3390/cells9030555