Pluripotent Stem Cell-Based Models: A Peephole into Virus Infections during Early Pregnancy



{kind=link}
{kind=link}
Abstract
1. New Perspectives for Congenital Virology
2. All Congenital Viral Infections Face an Efficient Barrier: The Placenta
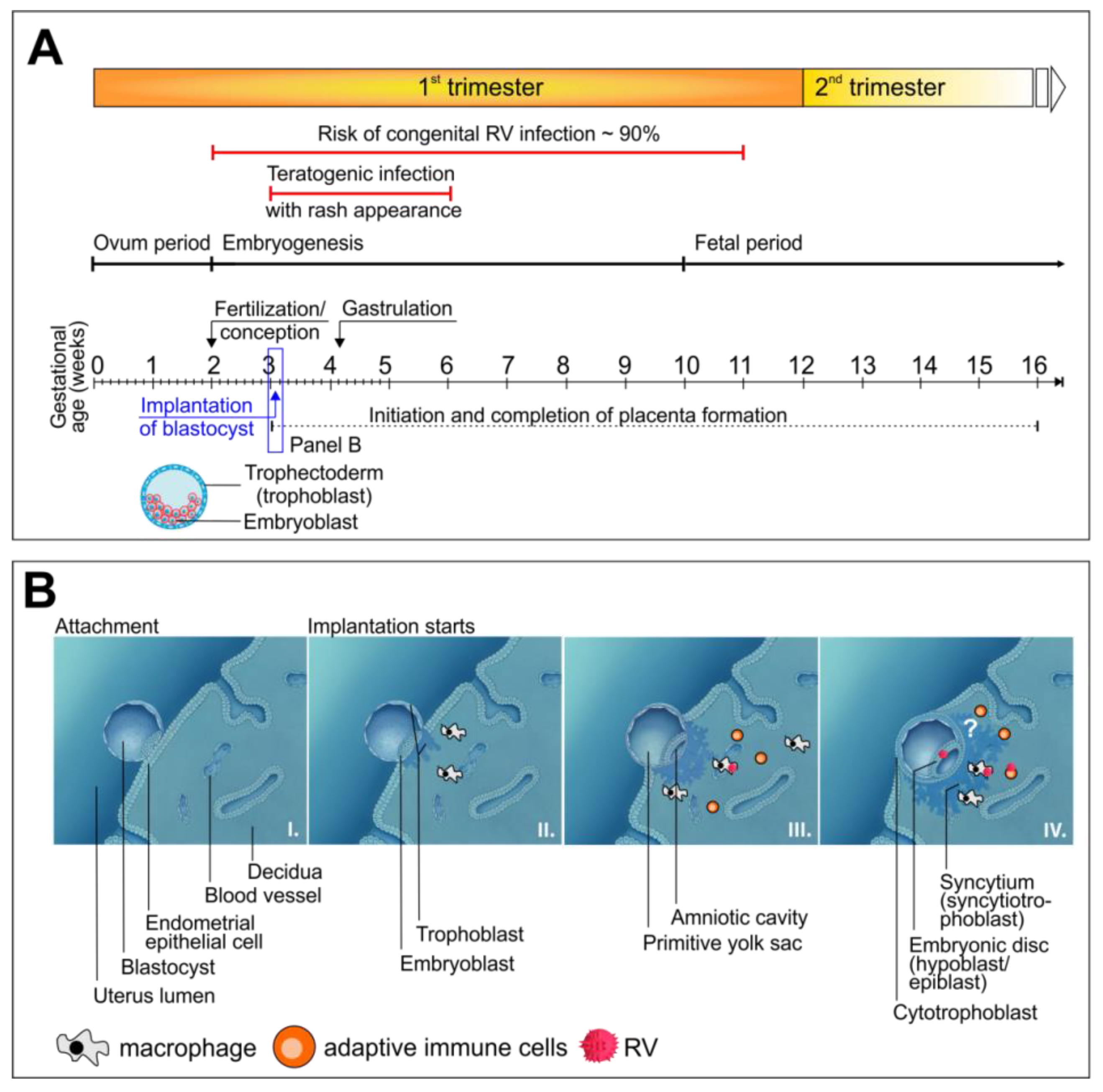
3. Characteristics of Congenital Virus Infections in Reference to RV as a Representative Teratogenic Virus
4. Implications of Genetic Variations of Teratogenic Viruses
5. Experimental Animal Models and the knowns and unknowns of Viral Teratogenicity in Humans
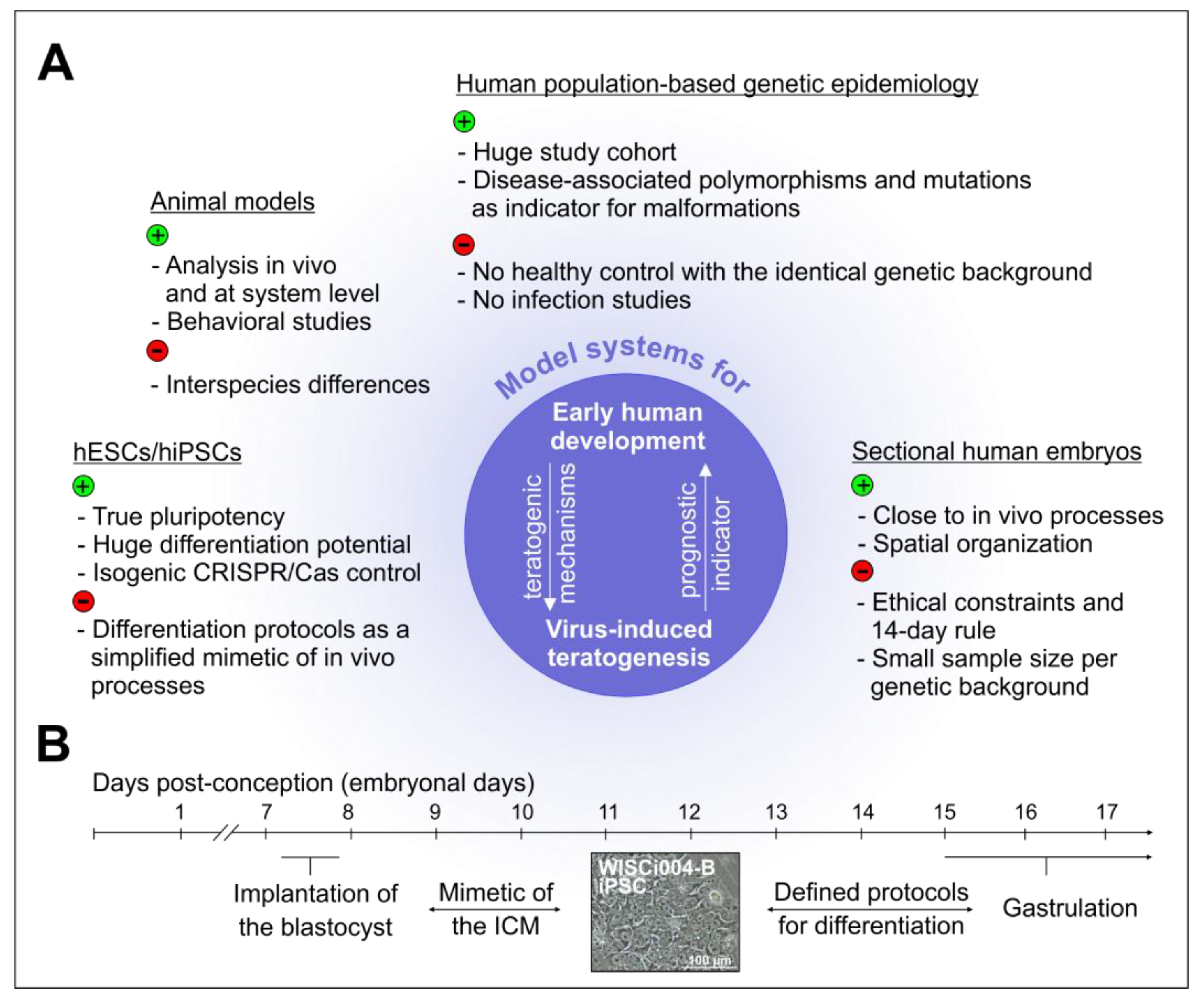
6. Novel Cell Culture Models to Address the unknown Mechanisms of Virus-Associated Alterations
6.1. The Advent of Induced Pluripotent Stem Cells and the Associated Implications for Developmental Biology
6.2. The infection of Pluripotent Stem Cell-Based Models of Human Embryogenesis with Teratogenic Viruses
6.3. Thoughts on the Limitations and Promises of the Continuously Advancing Array of Pluripotent Stem Cell-Based Models
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gregg, N.M. Congenital cataract following german measles in the mother. 1941. Problems Birth Defects 1991, 107, 170–180. [Google Scholar]
- Adam, M.P. The all-or-none phenomenon revisited. Birth Defects Res. Part. A Clin. Mol. Teratol. 2012, 94, 664–669. [Google Scholar] [CrossRef] [PubMed]
- Racicot, K.; Kwon, J.Y.; Aldo, P.; Silasi, M.; Mor, G. Understanding the complexity of the immune system during pregnancy. Am. J. Reprod. Immunol. 2014, 72, 107–116. [Google Scholar] [CrossRef]
- Chen, J.; Liang, Y.; Yi, P.; Xu, L.; Hawkins, H.K.; Rossi, S.L.; Soong, L.; Cai, J.; Menon, R.; Sun, J. Outcomes of congenital zika disease depend on timing of infection and maternal-fetal interferon action. Cell Rep. 2017, 21, 1588–1599. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Dao Thi, V.L.; Huang, Y.; Billerbeck, E.; Saha, D.; Hoffmann, H.H.; Wang, Y.; Silva, L.A.V.; Sarbanes, S.; Sun, T.; et al. Intrinsic immunity shapes viral resistance of stem cells. Cell 2018, 172, 423–438 e425. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef]
- Eggenberger, J.; Blanco-Melo, D.; Panis, M.; Brennand, K.J.; tenOever, B.R. Type i interferon response impairs differentiation potential of pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2019, 116, 1384–1393. [Google Scholar] [CrossRef]
- Dukhovny, A.; Lamkiewicz, K.; Chen, Q.; Fricke, M.; Jabrane-Ferrat, N.; Marz, M.; Jung, J.U.; Sklan, E.H. A crispr activation screen identifies genes that protect against zika virus infection. J. Virol. 2019. [Google Scholar] [CrossRef]
- Bayer, A.; Lennemann, N.J.; Ouyang, Y.S.; Bramley, J.C.; Morosky, S.; Marques, E.T.D.; Cherry, S.; Sadovsky, Y.; Coyne, C.B. Type iii interferons produced by human placental trophoblasts confer protection against zika virus infection. Cell Host Microbe 2016, 19, 705–712. [Google Scholar] [CrossRef]
- Corry, J.; Arora, N.; Good, C.A.; Sadovsky, Y.; Coyne, C.B. Organotypic models of type iii interferon-mediated protection from zika virus infections at the maternal-fetal interface. Proc. Natl. Acad. Sci. USA 2017, 114, 9433–9438. [Google Scholar] [CrossRef]
- Enders, G.; Nickerl-Pacher, U.; Miller, E.; Cradock-Watson, J.E. Outcome of confirmed periconceptional maternal rubella. Lancet 1988, 1, 1445–1447. [Google Scholar] [CrossRef]
- Bouthry, E.; Picone, O.; Hamdi, G.; Grangeot-Keros, L.; Ayoubi, J.M.; Vauloup-Fellous, C. Rubella and pregnancy: Diagnosis, management and outcomes. Prenat. Diagn. 2014, 34, 1246–1253. [Google Scholar] [CrossRef] [PubMed]
- Delorme-Axford, E.; Sadovsky, Y.; Coyne, C.B. The placenta as a barrier to viral infections. Annu. Rev. Virol. 2014, 1, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Arora, N.; Sadovsky, Y.; Dermody, T.S.; Coyne, C.B. Microbial vertical transmission during human pregnancy. Cell Host Microbe 2017, 21, 561–567. [Google Scholar] [CrossRef]
- Pereira, L. Congenital viral infection: Traversing the uterine-placental interface. Annu. Rev. Virol. 2018, 5, 273–299. [Google Scholar] [CrossRef]
- Weisblum, Y.; Panet, A.; Zakay-Rones, Z.; Haimov-Kochman, R.; Goldman-Wohl, D.; Ariel, I.; Falk, H.; Natanson-Yaron, S.; Goldberg, M.D.; Gilad, R.; et al. Modeling of human cytomegalovirus maternal-fetal transmission in a novel decidual organ culture. J. Virol. 2011, 85, 13204–13213. [Google Scholar] [CrossRef]
- Maidji, E.; McDonagh, S.; Genbacev, O.; Tabata, T.; Pereira, L. Maternal antibodies enhance or prevent cytomegalovirus infection in the placenta by neonatal fc receptor-mediated transcytosis. Am. J. Pathol. 2006, 168, 1210–1226. [Google Scholar] [CrossRef]
- Tan, L.; Lacko, L.A.; Zhou, T.; Tomoiaga, D.; Hurtado, R.; Zhang, T.; Sevilla, A.; Zhong, A.; Mason, C.E.; Noggle, S.; et al. Pre- and peri-implantation zika virus infection impairs fetal development by targeting trophectoderm cells. Nat. Commun. 2019, 10, 4155. [Google Scholar] [CrossRef]
- Zhang, S.; Lin, H.; Kong, S.; Wang, S.; Wang, H.; Wang, H.; Armant, D.R. Physiological and molecular determinants of embryo implantation. Mol. Aspects Med. 2013, 34, 939–980. [Google Scholar] [CrossRef]
- Massimiani, M.; Lacconi, V.; La Civita, F.; Ticconi, C.; Rago, R.; Campagnolo, L. Molecular signaling regulating endometrium-blastocyst crosstalk. Int. J. Mol. Sci. 2019, 21, 23. [Google Scholar] [CrossRef]
- Chantler, J.K.; Tingle, A.J. Replication and expression of rubella virus in human lymphocyte populations. J. Gen. Virol. 1980, 50, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Zobel, S.; Lorenz, M.; Frascaroli, G.; Bohnke, J.; Bilz, N.C.; Stanifer, M.L.; Boulant, S.; Bergs, S.; Liebert, U.G.; Claus, C. Rubella virus strain-associated differences in the induction of oxidative stress are independent of their interferon activation. Viruses 2018, 10, 540. [Google Scholar] [CrossRef] [PubMed]
- Trinh, Q.D.; Pham, N.T.K.; Takada, K.; Komine-Aizawa, S.; Hayakawa, S. Myelin oligodendrocyte glycoprotein-independent rubella infection of keratinocytes and resistance of first-trimester trophoblast cells to rubella virus in vitro. Viruses 2018, 10, 23. [Google Scholar] [CrossRef]
- Adamo, P.; Asis, L.; Silveyra, P.; Cuffini, C.; Pedranti, M.; Zapata, M. Rubella virus does not induce apoptosis in primary human embryo fibroblast cultures: A possible way of viral persistence in congenital infection. Viral Immunol. 2004, 17, 87–100. [Google Scholar] [CrossRef]
- Tondury, G.; Smith, D.W. Fetal rubella pathology. J. Pediatrics 1966, 68, 867–879. [Google Scholar] [CrossRef]
- Lazar, M.; Perelygina, L.; Martines, R.; Greer, P.; Paddock, C.D.; Peltecu, G.; Lupulescu, E.; Icenogle, J.; Zaki, S.R. Immunolocalization and distribution of rubella antigen in fatal congenital rubella syndrome. EBioMedicine 2016, 3, 86–92. [Google Scholar] [CrossRef]
- Wilson, J.G.; Beaudoin, A.R.; Free, H.J. Studies on the mechanism of teratogenic action of trypan blue. Anat. Rec. 1959, 133, 115–128. [Google Scholar] [CrossRef]
- Jena, M.K.; Nayak, N.; Chen, K.; Nayak, N.R. Role of macrophages in pregnancy and related complications. Arch. Immunol. Exp. 2019, 67, 295–309. [Google Scholar] [CrossRef]
- Medawar, P.B. Some immunological and endocrinological problems raised by the evolution of viviparity in vertebrates. Sym. Soc. Exp. Biol. 1953, 7, 320–338. [Google Scholar]
- Mor, G.; Aldo, P.; Alvero, A.B. The unique immunological and microbial aspects of pregnancy. Nat. Rev. Immunol. 2017, 17, 469–482. [Google Scholar] [CrossRef]
- Hanna, J.; Goldman-Wohl, D.; Hamani, Y.; Avraham, I.; Greenfield, C.; Natanson-Yaron, S.; Prus, D.; Cohen-Daniel, L.; Arnon, T.I.; Manaster, I.; et al. Decidual nk cells regulate key developmental processes at the human fetal-maternal interface. Nat. Med. 2006, 12, 1065–1074. [Google Scholar] [CrossRef] [PubMed]
- Care, A.S.; Diener, K.R.; Jasper, M.J.; Brown, H.M.; Ingman, W.V.; Robertson, S.A. Macrophages regulate corpus luteum development during embryo implantation in mice. J. Clin. Investig. 2013, 123, 3472–3487. [Google Scholar] [CrossRef] [PubMed]
- Hübschen, J.M.; Muller, C.P. Rubella and Congenital Rubella; Elsevier: Philadelphia, PA, USA, 2018; pp. 608–610. [Google Scholar]
- Lee, J.Y.; Bowden, D.S. Rubella virus replication and links to teratogenicity. Clin. Microbiol. Rev. 2000, 13, 571–587. [Google Scholar] [CrossRef] [PubMed]
- Dudgeon, J.A. Viral infections. J. Clin. Pathol. Suppl. 1976, 10, 99–106. [Google Scholar] [CrossRef]
- Dietrich, M.L.; Schieffelin, J.S. Congenital cytomegalovirus infection. Ochsner J. 2019, 19, 123–130. [Google Scholar] [CrossRef]
- Grosse, S.D.; Ross, D.S.; Dollard, S.C. Congenital cytomegalovirus (cmv) infection as a cause of permanent bilateral hearing loss: A quantitative assessment. J. Clin. Virol. 2008, 41, 57–62. [Google Scholar] [CrossRef]
- Robinson, N.; Mayorquin Galvan, E.E.; Zavala Trujillo, I.G.; Zavala-Cerna, M.G. Congenital zika syndrome: Pitfalls in the placental barrier. Rev. Med. Virol. 2018, 28, e1985. [Google Scholar] [CrossRef]
- Kenneson, A.; Cannon, M.J. Review and meta-analysis of the epidemiology of congenital cytomegalovirus (cmv) infection. Rev. Med. Virol. 2007, 17, 253–276. [Google Scholar] [CrossRef]
- Boppana, S.B.; Britt, W.J.; Fowler, K.; Hutto, S.C.; James, S.H.; Kimberlin, D.W.; Poole, C.; Ross, S.A.; Whitley, R.J. Pathogenesis of non-zika congenital viral infections. J. Infect. Dis. 2017, 216, S912–S918. [Google Scholar] [CrossRef]
- Enders, G.; Daiminger, A.; Bader, U.; Exler, S.; Enders, M. Intrauterine transmission and clinical outcome of 248 pregnancies with primary cytomegalovirus infection in relation to gestational age. J. Clin. Virol. 2011, 52, 244–246. [Google Scholar] [CrossRef]
- Cao-Lormeau, V.M.; Roche, C.; Teissier, A.; Robin, E.; Berry, A.L.; Mallet, H.P.; Sall, A.A.; Musso, D. Zika virus, french polynesia, south pacific, 2013. Emerg. Infect. Dis. 2014, 20, 1085–1086. [Google Scholar] [CrossRef] [PubMed]
- Franca, G.V.; Schuler-Faccini, L.; Oliveira, W.K.; Henriques, C.M.; Carmo, E.H.; Pedi, V.D.; Nunes, M.L.; Castro, M.C.; Serruya, S.; Silveira, M.F.; et al. Congenital zika virus syndrome in brazil: A case series of the first 1501 livebirths with complete investigation. Lancet 2016, 388, 891–897. [Google Scholar] [CrossRef]
- Besnard, M.; Lastere, S.; Teissier, A.; Cao-Lormeau, V.M.; Musso, D. Evidence of perinatal transmission of zika virus, french polynesia, december 2013 and february 2014. Eurosurveillance 2014, 19, 13–16. [Google Scholar] [CrossRef]
- Rasmussen, S.A.; Jamieson, D.J.; Honein, M.A.; Petersen, L.R. Zika virus and birth defects--reviewing the evidence for causality. N. Engl. J. Med. 2016, 374, 1981–1987. [Google Scholar] [CrossRef]
- Pomar, L.; Malinger, G.; Benoist, G.; Carles, G.; Ville, Y.; Rousset, D.; Hcini, N.; Pomar, C.; Jolivet, A.; Lambert, V. Association between zika virus and fetopathy: A prospective cohort study in french guiana. Ultrasound Obstet. Gynecol.: Off. J. Int. Soc. Ultrasound Obstet. Gynecol. 2017, 49, 729–736. [Google Scholar]
- Tang, H.; Hammack, C.; Ogden, S.C.; Wen, Z.; Qian, X.; Li, Y.; Yao, B.; Shin, J.; Zhang, F.; Lee, E.M.; et al. Zika virus infects human cortical neural progenitors and attenuates their growth. Cell Stem Cell 2016, 18, 587–590. [Google Scholar]
- Xu, M.; Lee, E.M.; Wen, Z.; Cheng, Y.; Huang, W.K.; Qian, X.; Tcw, J.; Kouznetsova, J.; Ogden, S.C.; Hammack, C.; et al. Identification of small-molecule inhibitors of zika virus infection and induced neural cell death via a drug repurposing screen. Nat. Med. 2016, 22, 1101–1107. [Google Scholar] [CrossRef]
- Chowdhury, R.; Allan, M.F.; Maranas, C.D. Optmaven-2.0: De novo design of variable antibody regions against targeted antigen epitopes. Antibodies 2018, 7, 23. [Google Scholar] [CrossRef]
- Wilkinson, G.W.; Davison, A.J.; Tomasec, P.; Fielding, C.A.; Aicheler, R.; Murrell, I.; Seirafian, S.; Wang, E.C.; Weekes, M.; Lehner, P.J.; et al. Human cytomegalovirus: Taking the strain. Med. Microbiol. Immunol. 2015, 204, 273–284. [Google Scholar]
- Suarez, N.M.; Wilkie, G.S.; Hage, E.; Camiolo, S.; Holton, M.; Hughes, J.; Maabar, M.; Vattipally, S.B.; Dhingra, A.; Gompels, U.A.; et al. Human cytomegalovirus genomes sequenced directly from clinical material: Variation, multiple-strain infection, recombination, and gene loss. J. Infect. Dis. 2019, 220, 781–791. [Google Scholar] [CrossRef]
- Dobbins, G.C.; Patki, A.; Chen, D.; Tiwari, H.K.; Hendrickson, C.; Britt, W.J.; Fowler, K.; Chen, J.Y.; Boppana, S.B.; Ross, S.A. Association of cmv genomic mutations with symptomatic infection and hearing loss in congenital cmv infection. BMC Infect. Dis. 2019, 19, 1046. [Google Scholar] [CrossRef] [PubMed]
- Udenze, D.; Trus, I.; Berube, N.; Gerdts, V.; Karniychuk, U. The african strain of zika virus causes more severe in utero infection than asian strain in a porcine fetal transmission model. Emerg Microbes Infec. 2019, 8, 1098–1107. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Huang, X.Y.; Liu, Z.Y.; Zhang, F.; Zhu, X.L.; Yu, J.Y.; Ji, X.; Xu, Y.P.; Li, G.; Li, C.; et al. A single mutation in the prm protein of zika virus contributes to fetal microcephaly. Science 2017, 358, 933–936. [Google Scholar] [CrossRef] [PubMed]
- Esser-Nobis, K.; Aarreberg, L.D.; Roby, J.A.; Fairgrieve, M.R.; Green, R.; Gale, M., Jr. Comparative analysis of african and asian lineage-derived zika virus strains reveals differences in activation of and sensitivity to antiviral innate immunity. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Bilz, N.C.; Jahn, K.; Lorenz, M.; Lüdtke, A.; Hübschen, J.M.; Geyer, H.; Mankertz, A.; Hübner, D.; Liebert, U.G.; Claus, C. Rubella viruses shift cellular bioenergetics to a more oxidative and glycolytic phenotype with a strain-specific requirement for glutamine. J. Virol. 2018, 92, e00934-18. [Google Scholar] [CrossRef]
- WHO. Rubella virus nomenclature update: 2013. Wkly. Epidemiol. Rec. 2013, 88, 337–343. [Google Scholar]
- Brown, K.E.; Rota, P.A.; Goodson, J.L.; Williams, D.; Abernathy, E.; Takeda, M.; Mulders, M.N. Genetic characterization of measles and rubella viruses detected through global measles and rubella elimination surveillance, 2016-2018. Mmwr. Morb. Mortal. Wkly. Rep. 2019, 68, 587–591. [Google Scholar] [CrossRef]
- Zhu, Z.; Abernathy, E.; Cui, A.; Zhang, Y.; Zhou, S.; Zhang, Z.; Wang, C.; Wang, T.; Ling, H.; Zhao, C.; et al. Rubella virus genotypes in the people’s republic of china between 1979 and 2007: A shift in endemic viruses during the 2001 rubella epidemic. J. Clin. Microbiol. 2010, 48, 1775–1781. [Google Scholar] [CrossRef]
- Vauloup-Fellous, C.; Hubschen, J.M.; Abernathy, E.S.; Icenogle, J.; Gaidot, N.; Dubreuil, P.; Parent-du-Chatelet, I.; Grangeot-Keros, L.; Muller, C.P. Phylogenetic analysis of rubella viruses involved in congenital rubella infections in france between 1995 and 2009. J. Clin. Microbiol. 2010, 48, 2530–2535. [Google Scholar] [CrossRef]
- Martinez-Torres, A.O.; Mosquera, M.M.; De Ory, F.; Gonzalez-Praetorius, A.; Echevarria, J.E. Genetic characterization of rubella virus strains detected in spain, 1998-2014. PLoS ONE 2016, 11, e0162403. [Google Scholar] [CrossRef]
- Wang, C.; Zhu, Z.; Xu, Q.; Fang, X.; Liu, X.; Xiong, P.; Song, L.; Xu, W.; Xu, A. Progress towards rubella elimination after implementation of rubella immunization for over 20 years in shandong province, china. Sci. Rep. 2017, 7, 17982. [Google Scholar] [CrossRef] [PubMed]
- Cekinovic, D.; Golemac, M.; Pugel, E.P.; Tomac, J.; Cicin-Sain, L.; Slavuljica, I.; Bradford, R.; Misch, S.; Winkler, T.H.; Mach, M.; et al. Passive immunization reduces murine cytomegalovirus-induced brain pathology in newborn mice. J. Virol. 2008, 82, 12172–12180. [Google Scholar] [CrossRef] [PubMed]
- Schleiss, M.R. Comparison of vaccine strategies against congenital cmv infection in the guinea pig model. J. Clin. Virol. : Off. Publ. Pan Am. Soc. Clin. Virol. 2008, 41, 224–230. [Google Scholar] [CrossRef]
- Bradford, R.D.; Yoo, Y.G.; Golemac, M.; Pugel, E.P.; Jonjic, S.; Britt, W.J. Murine cmv-induced hearing loss is associated with inner ear inflammation and loss of spiral ganglia neurons. PLoS Pathog. 2015, 11, e1004774. [Google Scholar] [CrossRef]
- Fowler, K.B.; McCollister, F.P.; Dahle, A.J.; Boppana, S.; Britt, W.J.; Pass, R.F. Progressive and fluctuating sensorineural hearing loss in children with asymptomatic congenital cytomegalovirus infection. J. Pediatrics 1997, 130, 624–630. [Google Scholar] [CrossRef]
- Fowler, K.B.; Boppana, S.B. Congenital cytomegalovirus (cmv) infection and hearing deficit. J. Clin. Virol. : Off. Publ. Pan Am. Soc. Clin. Virol. 2006, 35, 226–231. [Google Scholar] [CrossRef]
- Lazarini, F.; Katsimpardi, L.; Levivien, S.; Wagner, S.; Gressens, P.; Teissier, N.; Lledo, P.M. Congenital cytomegalovirus infection alters olfaction before hearing deterioration in mice. J. Neurosci. : Off. J. Soc. Neurosci. 2018, 38, 10424–10437. [Google Scholar] [CrossRef]
- Marin-Lopez, A.; Calvo-Pinilla, E.; Moreno, S.; Utrilla-Trigo, S.; Nogales, A.; Brun, A.; Fikrig, E.; Ortego, J. Modeling arboviral infection in mice lacking the interferon alpha/beta receptor. Viruses 2019, 11, 35. [Google Scholar] [CrossRef]
- Rossi, S.L.; Tesh, R.B.; Azar, S.R.; Muruato, A.E.; Hanley, K.A.; Auguste, A.J.; Langsjoen, R.M.; Paessler, S.; Vasilakis, N.; Weaver, S.C. Characterization of a novel murine model to study zika virus. Am. J. Trop. Med. Hyg. 2016, 94, 1362–1369. [Google Scholar] [CrossRef]
- Grant, A.; Ponia, S.S.; Tripathi, S.; Balasubramaniam, V.; Miorin, L.; Sourisseau, M.; Schwarz, M.C.; Sanchez-Seco, M.P.; Evans, M.J.; Best, S.M.; et al. Zika virus targets human stat2 to inhibit type i interferon signaling. Cell Host Microbe 2016, 19, 882–890. [Google Scholar] [CrossRef]
- Gorman, M.J.; Caine, E.A.; Zaitsev, K.; Begley, M.C.; Weger-Lucarelli, J.; Uccellini, M.B.; Tripathi, S.; Morrison, J.; Yount, B.L.; Dinnon, K.H.; et al. An immunocompetent mouse model of zika virus infection. Cell Host Microbe 2018, 23, 672–685. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.J.; Blackman, M.A.; Lin, J.S. Pre-clinical pregnancy models for evaluating zika vaccines. Trop. Med. Infect. Dis. 2019, 4, 58. [Google Scholar] [CrossRef] [PubMed]
- Caine, E.A.; Jagger, B.W.; Diamond, M.S. Animal models of zika virus infection during pregnancy. Viruses 2018, 10, 598. [Google Scholar] [CrossRef] [PubMed]
- Parkman, P.D.; Phillips, P.E.; Meyer, H.M., Jr. Experimental rubella virus infection in pregnant monkeys. Am. J. Dis. Child. 1965, 110, 390–394. [Google Scholar] [CrossRef]
- Delahunt, C.S.; Rieser, N. Rubella-induced embryopathies in monkeys. Am. J. Obstet. Gynecol. 1967, 99, 580–588. [Google Scholar] [CrossRef]
- Zhu, Z.; Huangfu, D. Human pluripotent stem cells: An emerging model in developmental biology. Development 2013, 140, 705–717. [Google Scholar] [CrossRef]
- Yue, F.; Cheng, Y.; Breschi, A.; Vierstra, J.; Wu, W.; Ryba, T.; Sandstrom, R.; Ma, Z.; Davis, C.; Pope, B.D.; et al. A comparative encyclopedia of DNA elements in the mouse genome. Nature 2014, 515, 355–364. [Google Scholar] [CrossRef]
- Gerrelli, D.; Lisgo, S.; Copp, A.J.; Lindsay, S. Enabling research with human embryonic and fetal tissue resources. Development 2015, 142, 3073–3076. [Google Scholar] [CrossRef]
- Fougerousse, F.; Bullen, P.; Herasse, M.; Lindsay, S.; Richard, I.; Wilson, D.; Suel, L.; Durand, M.; Robson, S.; Abitbol, M.; et al. Human-mouse differences in the embryonic expression patterns of developmental control genes and disease genes. Hum. Mol. Genet. 2000, 9, 165–173. [Google Scholar] [CrossRef]
- Fogarty, N.M.E.; McCarthy, A.; Snijders, K.E.; Powell, B.E.; Kubikova, N.; Blakeley, P.; Lea, R.; Elder, K.; Wamaitha, S.E.; Kim, D.; et al. Genome editing reveals a role for oct4 in human embryogenesis. Nature 2017, 550, 67–73. [Google Scholar] [CrossRef]
- Shahbazi, M.N.; Jedrusik, A.; Vuoristo, S.; Recher, G.; Hupalowska, A.; Bolton, V.; Fogarty, N.N.M.; Campbell, A.; Devito, L.; Ilic, D.; et al. Self-organization of the human embryo in the absence of maternal tissues. Nat. Cell Biol. 2016, 18, 700–708. [Google Scholar] [CrossRef] [PubMed]
- Deglincerti, A.; Croft, G.F.; Pietila, L.N.; Zernicka-Goetz, M.; Siggia, E.D.; Brivanlou, A.H. Self-organization of the in vitro attached human embryo. Nature 2016, 533, 251–254. [Google Scholar] [CrossRef] [PubMed]
- Wujcicka, W.; Paradowska, E.; Studzinska, M.; Wilczynski, J.; Nowakowska, D. Tlr2 2258 g>a single nucleotide polymorphism and the risk of congenital infection with human cytomegalovirus. Virol. J. 2017, 14, 12. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.D.; Faucz, F.R.; Melo, A.; Pezzuto, P.; de Azevedo, G.S.; Schamber-Reis, B.L.F.; Tavares, J.S.; Mattapallil, J.J.; Tanuri, A.; Aguiar, R.S.; et al. Variations in maternal adenylate cyclase genes are associated with congenital zika syndrome in a cohort from northeast, brazil. J. Intern. Med. 2019, 285, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.N.O.; Ribeiro, D.R.; Alves, J.C.; Cazzaniga, R.A.; Magalhaes, L.S.; de Souza, M.S.F.; Fonseca, A.B.L.; Bispo, A.J.B.; Porto, R.L.S.; dos Santos, C.A.; et al. Association between zika virus microcephaly in newborns with the rs3775291 variant in toll-like receptor 3 and rs1799964 variant at tumor necrosis factor-alpha gene. J. Infect. Dis. 2019, 220, 1797–1801. [Google Scholar] [CrossRef]
- Caires, L.C.; Goulart, E.; Melo, U.S.; Araujo, B.H.S.; Alvizi, L.; Soares-Schanoski, A.; de Oliveira, D.F.; Kobayashi, G.S.; Griesi-Oliveira, K.; Musso, C.M.; et al. Discordant congenital zika syndrome twins show differential in vitro viral susceptibility of neural progenitor cells (vol 9, 2018). Nat. Commun. 2018, 9, 475. [Google Scholar] [CrossRef]
- Dvash, T.; Benvenisty, N. Human embryonic stem cells as a model for early human development. Best Pract. Res. Clin. Obstet. Gynaecol. 2004, 18, 929–940. [Google Scholar] [CrossRef]
- Achberger, K.; Haderspeck, J.C.; Kleger, A.; Liebau, S. Stem cell-based retina models. Adv. Drug Deliv. Rev. 2019, 140, 33–50. [Google Scholar] [CrossRef]
- Heemskerk, I.; Warmflash, A. Pluripotent stem cells as a model for embryonic patterning: From signaling dynamics to spatial organization in a dish. Dev. Dynam. 2016, 245, 976–990. [Google Scholar] [CrossRef]
- Zheng, Y.; Xue, X.; Shao, Y.; Wang, S.; Esfahani, S.N.; Li, Z.; Muncie, J.M.; Lakins, J.N.; Weaver, V.M.; Gumucio, D.L.; et al. Controlled modelling of human epiblast and amnion development using stem cells. Nature 2019, 573, 421–425. [Google Scholar] [CrossRef]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.H.; Chen, X.; Li, D.S.; Li, R.; Addicks, G.C.; Glennon, C.; Zwaka, T.P.; Thomson, J.A. Bmp4 initiates human embryonic stem cell differentiation to trophoblast. Nat. Biotechnol. 2002, 20, 1261–1264. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.C.; Moretto-Zita, M.; Soncin, F.; Wakeland, A.; Wolfe, L.; Leon-Garcia, S.; Pandian, R.; Pizzo, D.; Cui, L.; Nazor, K.; et al. Bmp4-directed trophoblast differentiation of human embryonic stem cells is mediated through delta np63(+) cytotrophoblast stem cell state. Development 2013, 140, 3965–3976. [Google Scholar] [CrossRef]
- Gonczol, E.; Andrews, P.W.; Plotkin, S.A. Cytomegalovirus replicates in differentiated but not in undifferentiated human embryonal carcinoma cells. Science 1984, 224, 159–161. [Google Scholar] [CrossRef]
- Hong, S.; Hwang, D.Y.; Yoon, S.; Isacson, O.; Ramezani, A.; Hawley, R.G.; Kim, K.S. Functional analysis of various promoters in lentiviral vectors at different stages of in vitro differentiation of mouse embryonic stem cells. Mol. Ther. : J. Am. Soc. Gene Ther. 2007, 15, 1630–1639. [Google Scholar] [CrossRef]
- Kim, S.; Kim, G.J.; Miyoshi, H.; Moon, S.H.; Ahn, S.E.; Lee, J.H.; Lee, H.J.; Cha, K.Y.; Chung, H.M. Efficiency of the elongation factor-1alpha promoter in mammalian embryonic stem cells using lentiviral gene delivery systems. Stem Cells Dev. 2007, 16, 537–545. [Google Scholar] [CrossRef]
- Xia, X.; Zhang, Y.; Zieth, C.R.; Zhang, S.C. Transgenes delivered by lentiviral vector are suppressed in human embryonic stem cells in a promoter-dependent manner. Stem Cells Dev. 2007, 16, 167–176. [Google Scholar] [CrossRef]
- Wang, R.; Liang, J.; Jiang, H.; Qin, L.J.; Yang, H.T. Promoter-dependent egfp expression during embryonic stem cell propagation and differentiation. Stem Cells Dev. 2008, 17, 279–289. [Google Scholar] [CrossRef]
- Matsukage, S.; Kosugi, I.; Kawasaski, H.; Miura, K.; Kitani, H.; Tsutsui, Y. Mouse embryonic stem cells are not susceptible to cytomegalovirus but acquire susceptibility during differentiation. Birth Defects Res. Part. A Clin. Mol. Teratol. 2006, 76, 115–125. [Google Scholar] [CrossRef]
- Belzile, J.P.; Stark, T.J.; Yeo, G.W.; Spector, D.H. Human cytomegalovirus infection of human embryonic stem cell-derived primitive neural stem cells is restricted at several steps but leads to the persistence of viral DNA. J. Virol. 2014, 88, 4021–4039. [Google Scholar] [CrossRef]
- D’Aiuto, L.; Di Maio, R.; Heath, B.; Raimondi, G.; Milosevic, J.; Watson, A.M.; Bamne, M.; Parks, W.T.; Yang, L.; Lin, B.; et al. Human induced pluripotent stem cell-derived models to investigate human cytomegalovirus infection in neural cells. PLoS ONE 2012, 7, e49700. [Google Scholar] [CrossRef] [PubMed]
- Berger, A.A.; Gil, Y.; Panet, A.; Weisblum, Y.; Oiknine-Djian, E.; Gropp, M.; Steiner, D.; Reubinoff, B.E.; Wolf, D.G. Transition toward human cytomegalovirus susceptibility in early human embryonic stem cell-derived neural precursors. J. Virol. 2015, 89, 11159–11164. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.M.; Rana, P.; Jaeger, H.K.; O’Dowd, J.M.; Balemba, O.B.; Fortunato, E.A. Human cytomegalovirus compromises development of cerebral organoids. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed]
- Teissier, N.; Fallet-Bianco, C.; Delezoide, A.L.; Laquerriere, A.; Marcorelles, P.; Khung-Savatovsky, S.; Nardelli, J.; Cipriani, S.; Csaba, Z.; Picone, O.; et al. Cytomegalovirus-induced brain malformations in fetuses. J. Neuropathol. Exp. Neurol. 2014, 73, 143–158. [Google Scholar] [CrossRef]
- Bilz, N.C.; Willscher, E.; Binder, H.; Bohnke, J.; Stanifer, M.L.; Hubner, D.; Boulant, S.; Liebert, U.G.; Claus, C. Teratogenic rubella virus alters the endodermal differentiation capacity of human induced pluripotent stem cells. Cells 2019, 8, 870. [Google Scholar] [CrossRef]
- Liu, L.; Chen, Z.; Zhang, X.; Li, S.; Hui, Y.; Feng, H.; Du, Y.; Jin, G.; Zhou, X.; Zhang, X. Protection of zikv infection-induced neuropathy by abrogation of acute antiviral response in human neural progenitors. Cell Death Differ. 2019, 26, 2607–2621. [Google Scholar] [CrossRef]
- Dang, J.; Tiwari, S.K.; Lichinchi, G.; Qin, Y.; Patil, V.S.; Eroshkin, A.M.; Rana, T.M. Zika virus depletes neural progenitors in human cerebral organoids through activation of the innate immune receptor tlr3. Cell Stem Cell 2016, 19, 258–265. [Google Scholar] [CrossRef]
- Salinas, S.; Erkilic, N.; Damodar, K.; Moles, J.P.; Fournier-Wirth, C.; Van de Perre, P.; Kalatzis, V.; Simonin, Y. Zika virus efficiently replicates in human retinal epithelium and disturbs its permeability. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Simonin, Y.; Erkilic, N.; Damodar, K.; Cle, M.; Desmetz, C.; Bollore, K.; Taleb, M.; Torriano, S.; Barthelemy, J.; Dubois, G.; et al. Zika virus induces strong inflammatory responses and impairs homeostasis and function of the human retinal pigment epithelium. EBioMedicine 2019, 39, 315–331. [Google Scholar] [CrossRef]
- Zhu, Z.; Mesci, P.; Bernatchez, J.A.; Gimple, R.C.; Wang, X.; Schafer, S.T.; Wettersten, H.I.; Beck, S.; Clark, A.E.; Wu, Q.; et al. Zika virus targets glioblastoma stem cells through a sox2-integrin alphavbeta5 axis. Cell Stem Cell 2020, 26, 187–204 e110. [Google Scholar] [CrossRef]
- Lough, J.; Sugi, Y. Endoderm and heart development. Dev. Dyn. : Off. Publ. Am. Assoc. Anat. 2000, 217, 327–342. [Google Scholar] [CrossRef]
- Rohani, L.; Johnson, A.A.; Naghsh, P.; Rancourt, D.E.; Ulrich, H.; Holland, H. Concise review: Molecular cytogenetics and quality control: Clinical guardians for pluripotent stem cells. Stem Cells Transl. Med. 2018, 7, 867–875. [Google Scholar] [CrossRef] [PubMed]
- Berg, J.; Hiller, T.; Kissner, M.S.; Qazi, T.H.; Duda, G.N.; Hocke, A.C.; Hippenstiel, S.; Elomaa, L.; Weinhart, M.; Fahrenson, C.; et al. Optimization of cell-laden bioinks for 3d bioprinting and efficient infection with influenza a virus. Sci. Rep. 2018, 8, 13877. [Google Scholar] [CrossRef] [PubMed]
- Shen, H. The labs growing human embryos for longer than ever before. Nature 2018, 559, 19–22. [Google Scholar] [CrossRef]
- Blake, L.E.; Thomas, S.M.; Blischak, J.D.; Hsiao, C.J.; Chavarria, C.; Myrthil, M.; Gilad, Y.; Pavlovic, B.J. A comparative study of endoderm differentiation in humans and chimpanzees. Genome Biol. 2018, 19, 162. [Google Scholar] [CrossRef]
- Krendl, C.; Shaposhnikov, D.; Rishko, V.; Ori, C.; Ziegenhain, C.; Sass, S.; Simon, L.; Muller, N.S.; Straub, T.; Brooks, K.E.; et al. Gata2/3-tfap2a/c transcription factor network couples human pluripotent stem cell differentiation to trophectoderm with repression of pluripotency. Proc. Natl. Acad. Sci. USA 2017, 114, E9579–E9588. [Google Scholar] [CrossRef]
- Chen, H.I.; Song, H.; Ming, G.L. Applications of human brain organoids to clinical problems. Dev. Dyn. : Off. Publ. Am. Assoc. Anat. 2019, 248, 53–64. [Google Scholar] [CrossRef]
- Watanabe, M.; Buth, J.E.; Vishlaghi, N.; de la Torre-Ubieta, L.; Taxidis, J.; Khakh, B.S.; Coppola, G.; Pearson, C.A.; Yamauchi, K.; Gong, D.; et al. Self-organized cerebral organoids with human-specific features predict effective drugs to combat zika virus infection. Cell Rep. 2017, 21, 517–532. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Claus, C.; Jung, M.; Hübschen, J.M. Pluripotent Stem Cell-Based Models: A Peephole into Virus Infections during Early Pregnancy. Cells 2020, 9, 542. https://doi.org/10.3390/cells9030542
Claus C, Jung M, Hübschen JM. Pluripotent Stem Cell-Based Models: A Peephole into Virus Infections during Early Pregnancy. Cells. 2020; 9(3):542. https://doi.org/10.3390/cells9030542
Chicago/Turabian StyleClaus, Claudia, Matthias Jung, and Judith M. Hübschen. 2020. "Pluripotent Stem Cell-Based Models: A Peephole into Virus Infections during Early Pregnancy" Cells 9, no. 3: 542. https://doi.org/10.3390/cells9030542
APA StyleClaus, C., Jung, M., & Hübschen, J. M. (2020). Pluripotent Stem Cell-Based Models: A Peephole into Virus Infections during Early Pregnancy. Cells, 9(3), 542. https://doi.org/10.3390/cells9030542