Helicobacter Pylori Targets the EPHA2 Receptor Tyrosine Kinase in Gastric Cells Modulating Key Cellular Functions

 , , ,
, , , 
Abstract
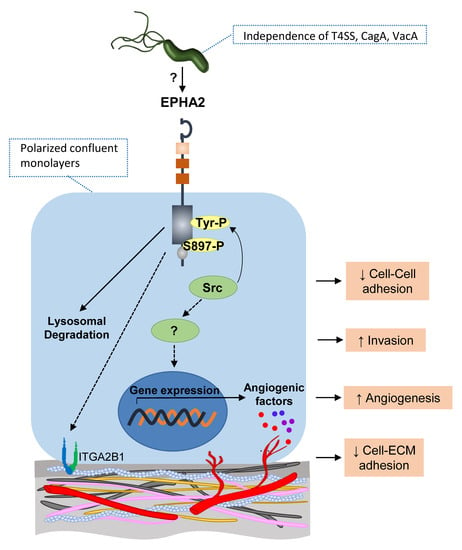
:
1. Introduction
2. Materials and Methods
2.1. Antibodies, Pharmacologic Inhibitors, and Chemicals
2.2. Cell Culture
2.3. Cell Viability/Proliferation Assay
2.4. H. Pylori Strains and Clinical Isolates
2.5. Infection of Gastric Cell Lines
2.6. Immunofluorescence
2.7. Immunoblotting
2.8. Detection of Tyrosine-Phosphorylated EPHA2 by Enzyme-Linked Immunosorbent Assay
2.9. RNA Extraction, cDNA Synthesis, and Quantitative RT-PCR (RT-qPCR)
2.10. Small Interfering RNA (siRNA) Transfections
2.11. Cell–Cell Adhesion Assay
2.12. Adhesion Assay to ECM Substrates
2.13. In Vitro Matrigel Invasion Assay
2.14. In Vitro Angiogenesis Assay—Endothelial Cell Capillary-Like Tube Formation Assay
2.15. In Vivo Angiogenesis Assay—Chicken Embryo Chorioallantoic Membrane (CAM)
2.16. Immunohistochemistry Analysis of CAM
2.17. Angiogenesis Array
2.18. Quantification of IL-8 Secretion by Enzyme-Linked Immunosorbent Assay
2.19. Statistical Analysis
3. Results
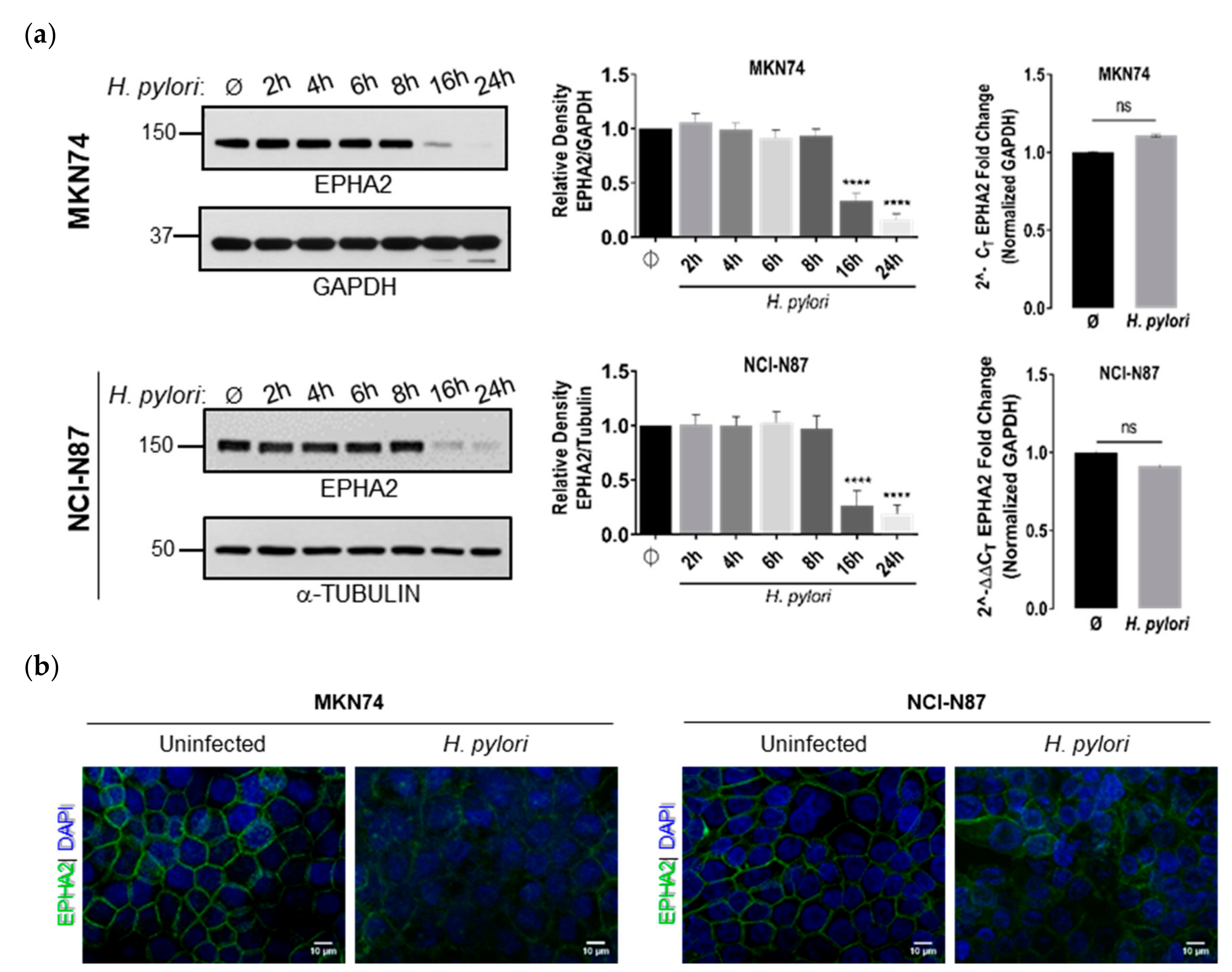
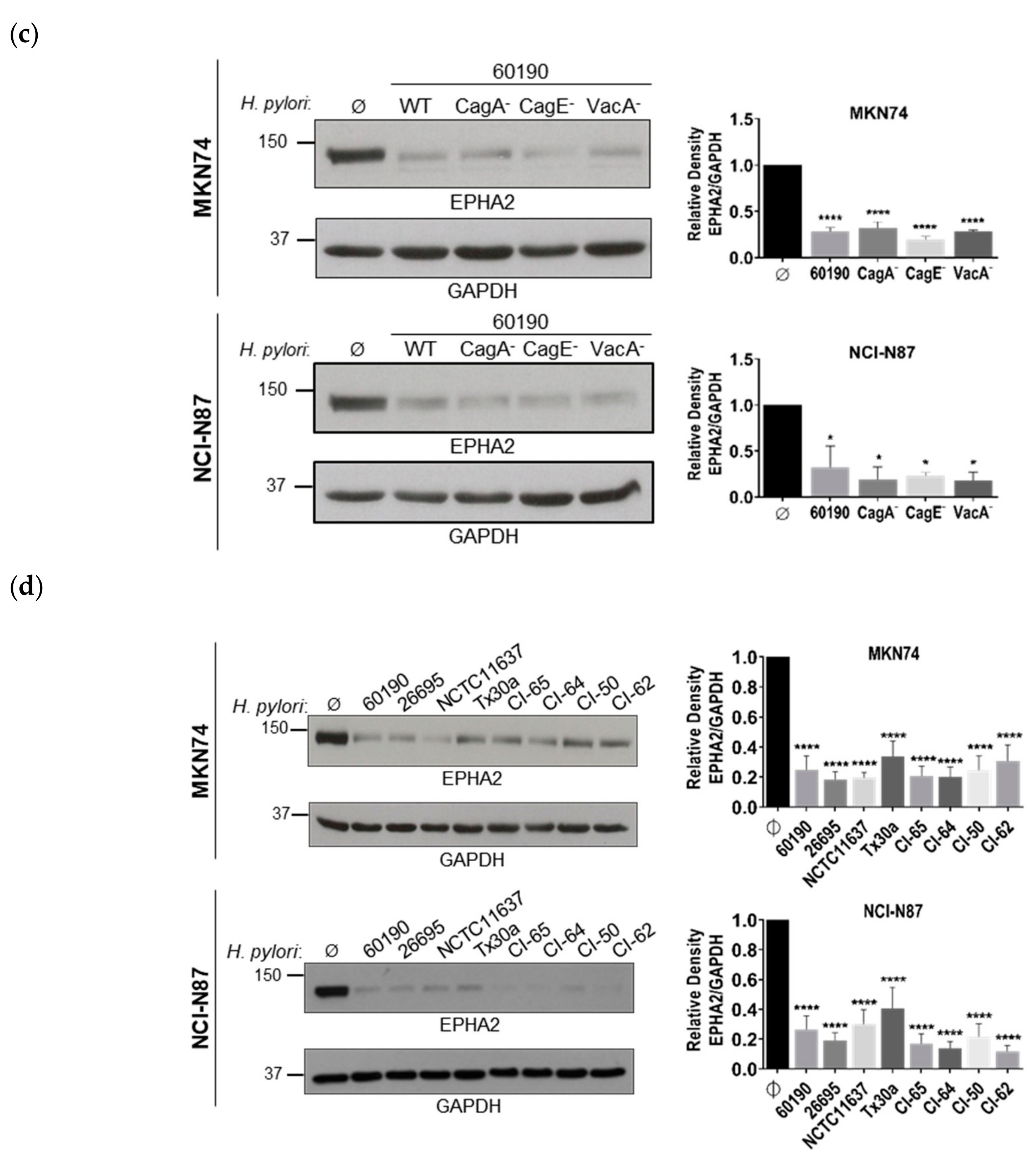
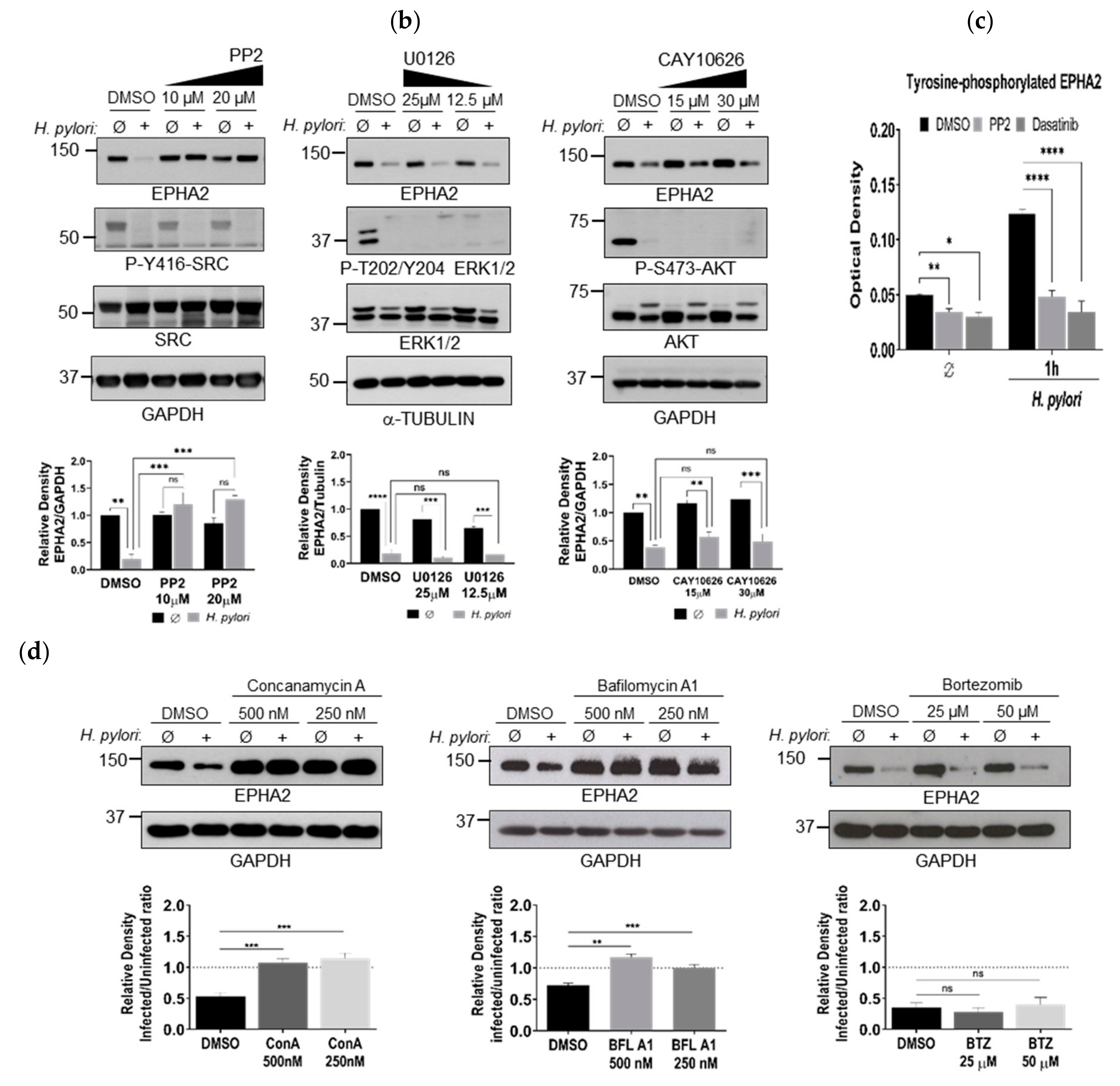
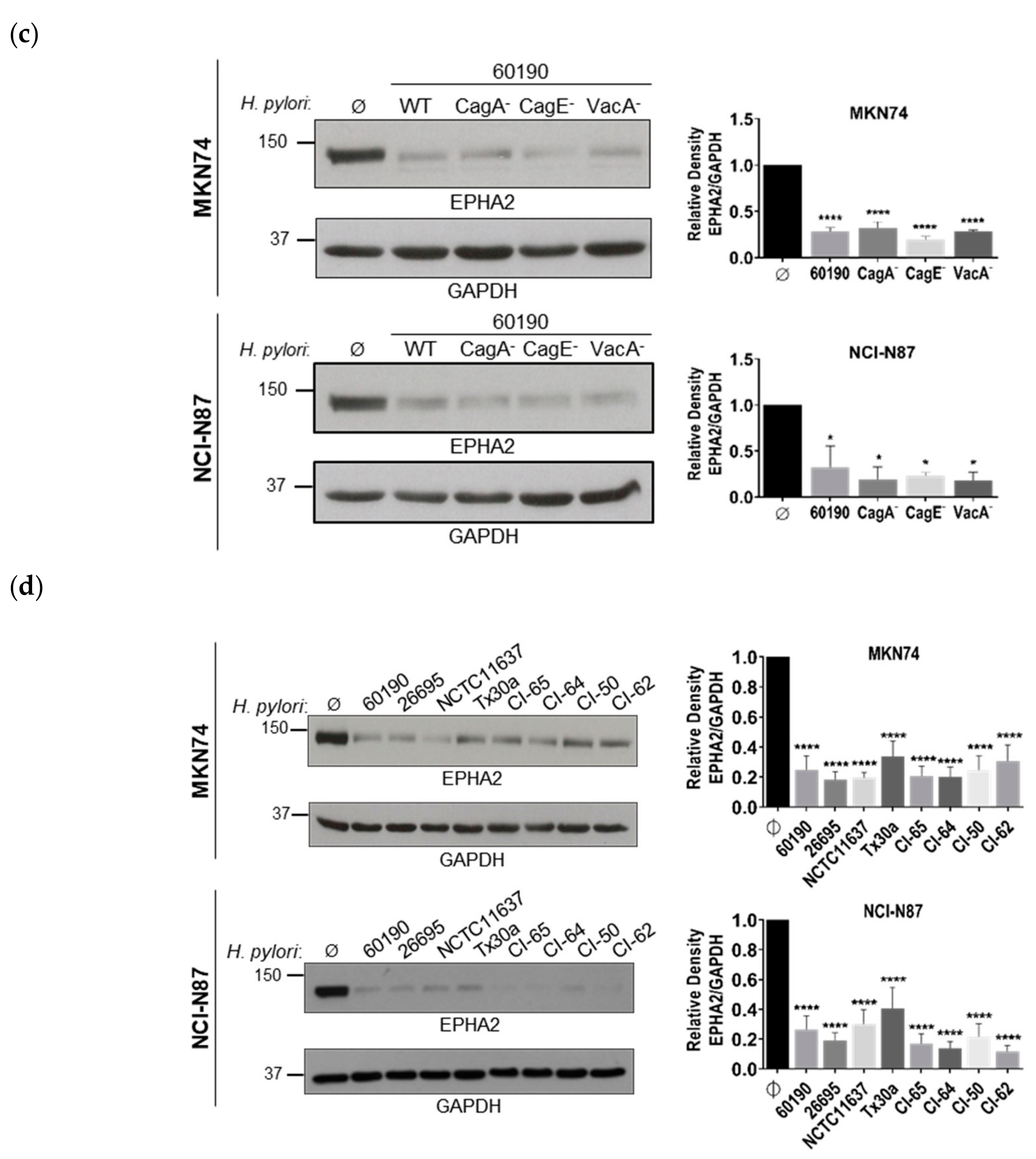
3.1. Infection of Polarized Gastric Epithelial Cells by H. pylori Induces Downregulation of EPHA2 Protein Independently of T4SS, CagA, and VacA Virulence Factors
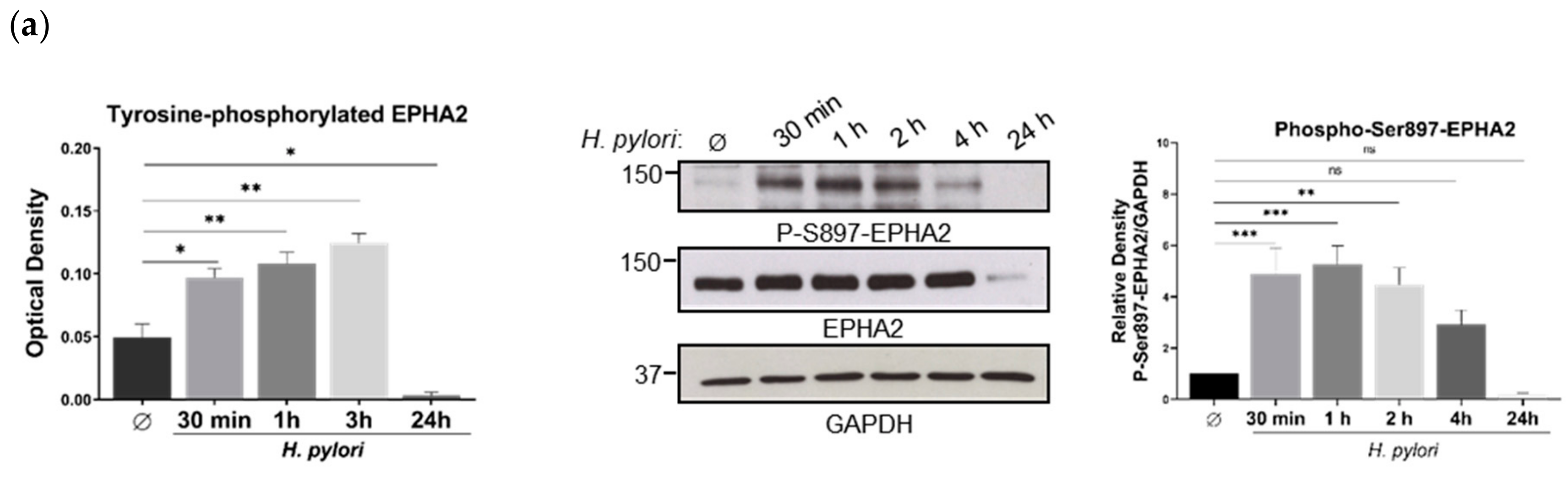
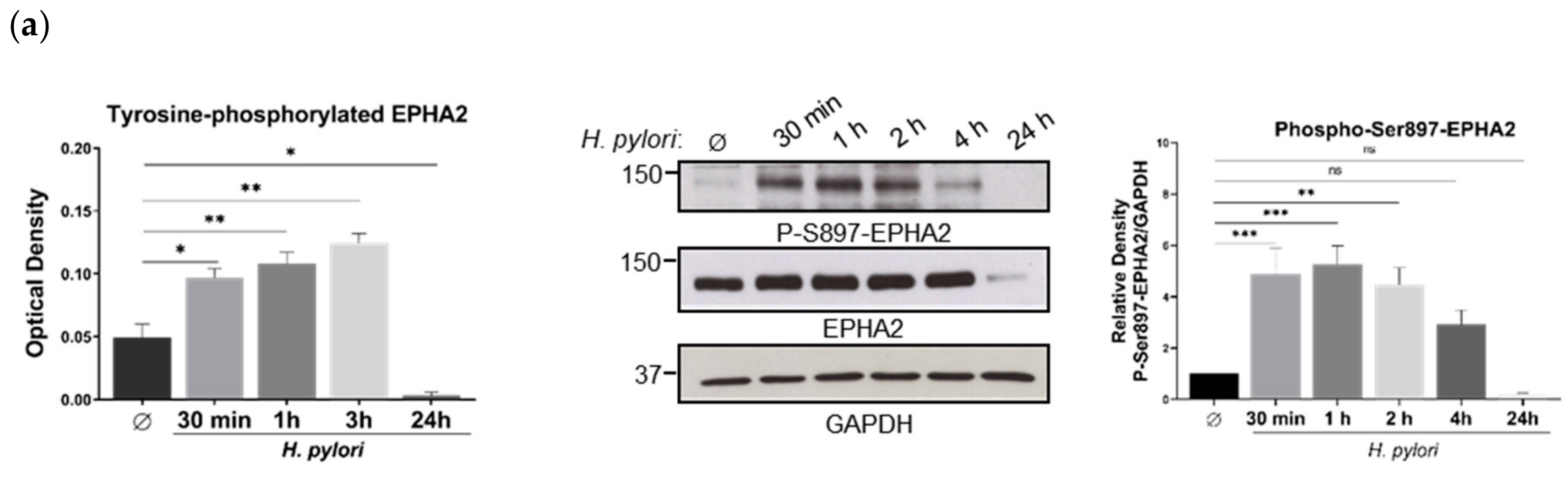
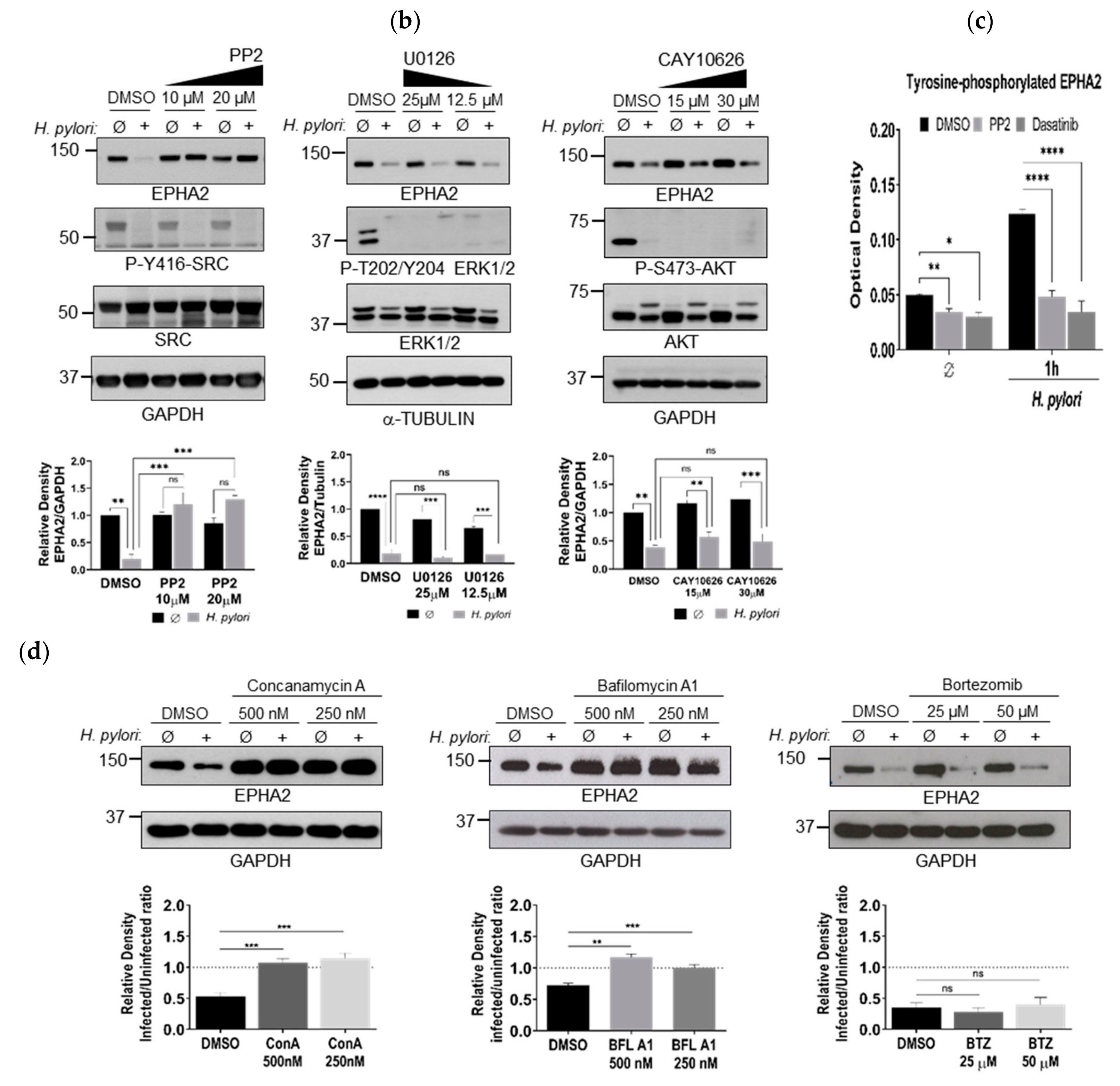
3.2. H. pylori-Mediated EPHA2 Downregulation is Preceded by Activation of the EPHA2 Receptor by Phosphorylation and Followed by Protein Degradation via the Lysosomal Pathway
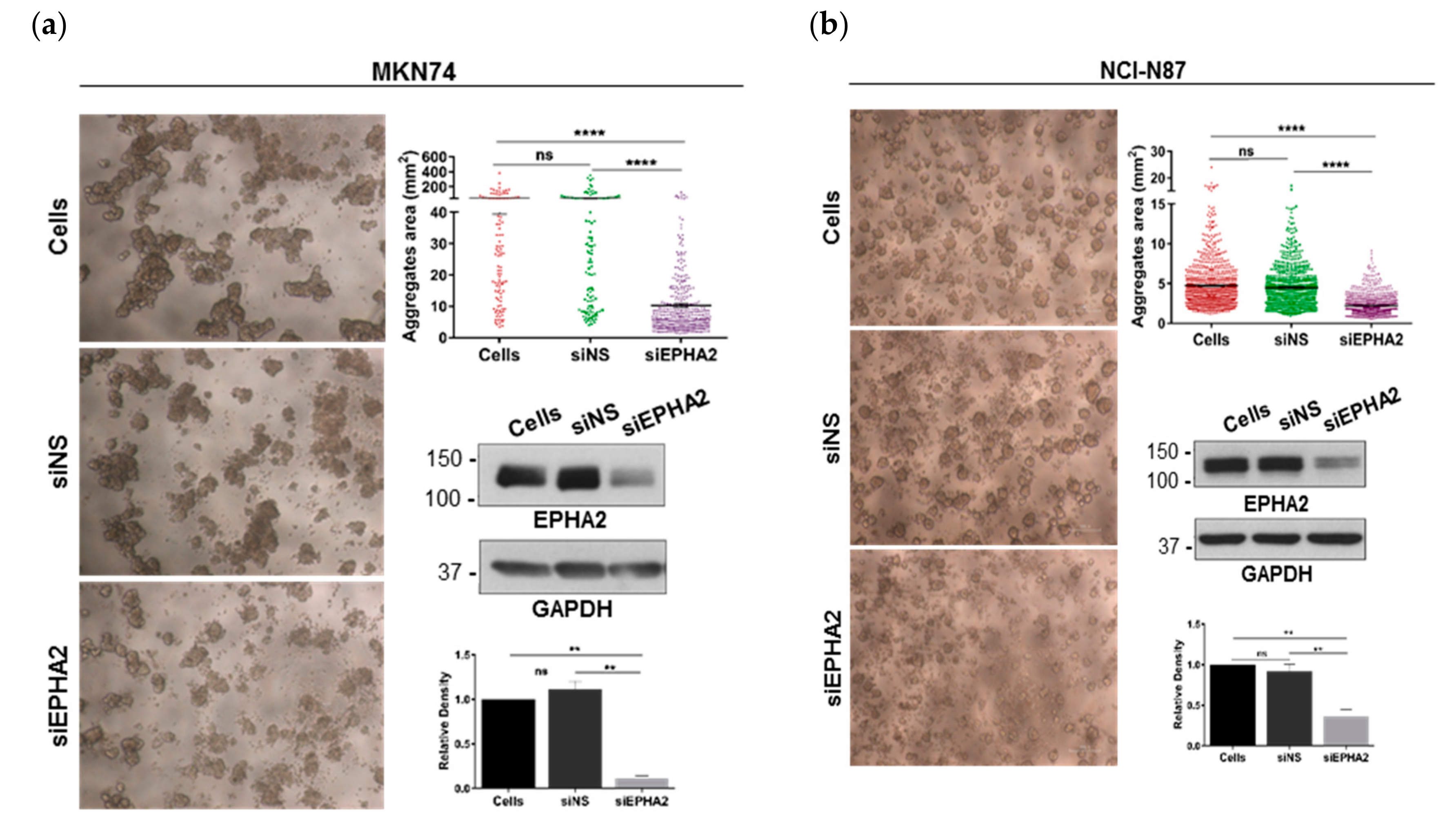
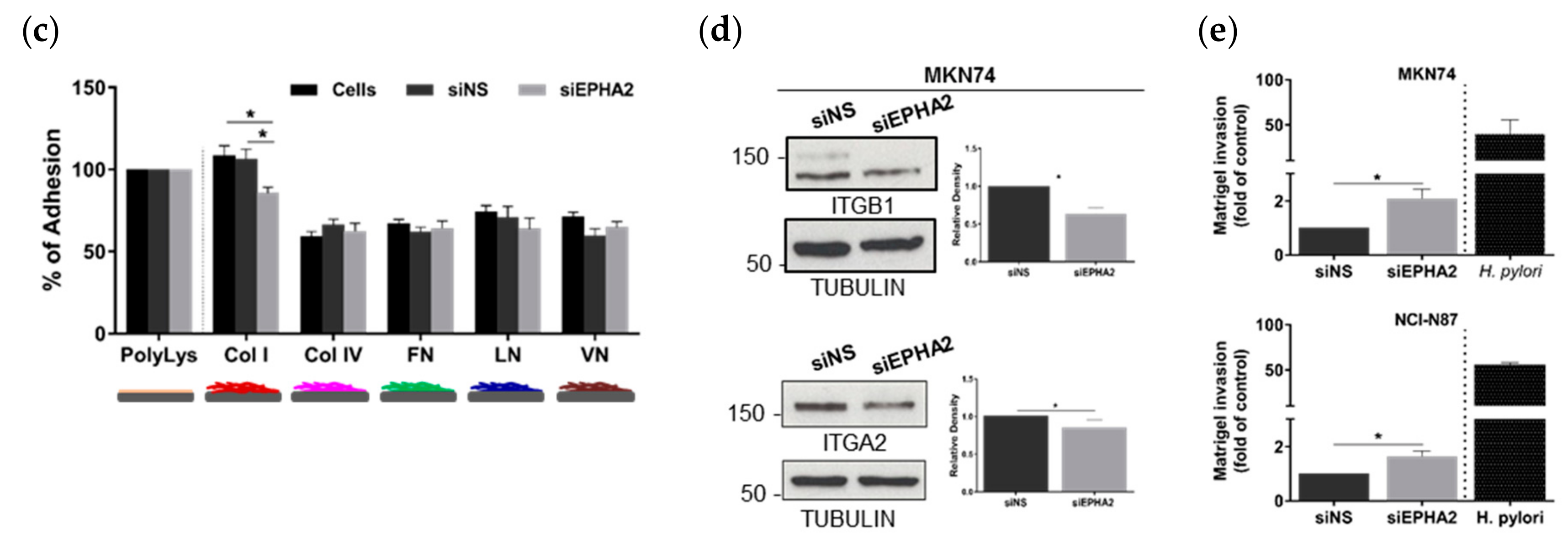
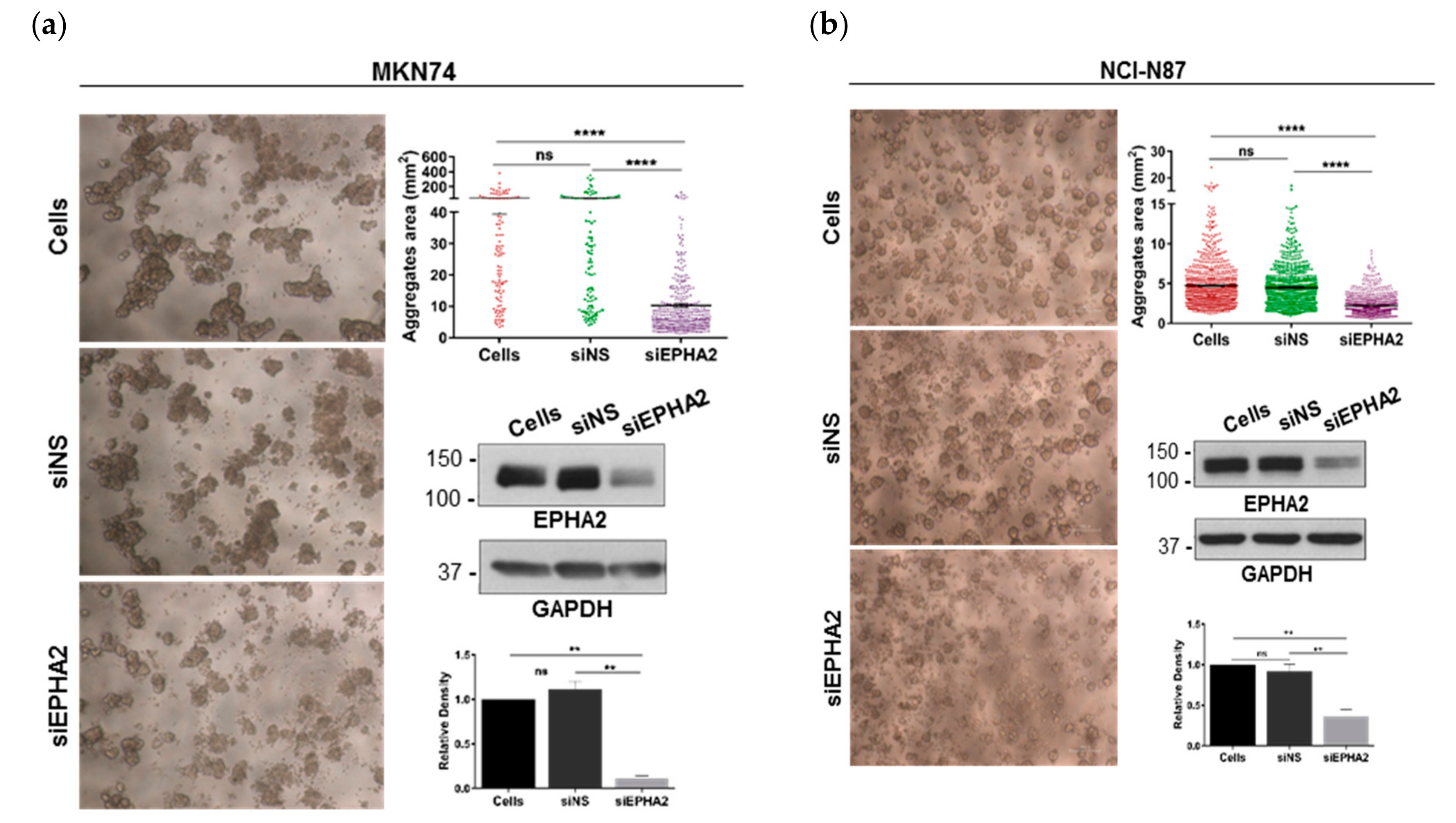
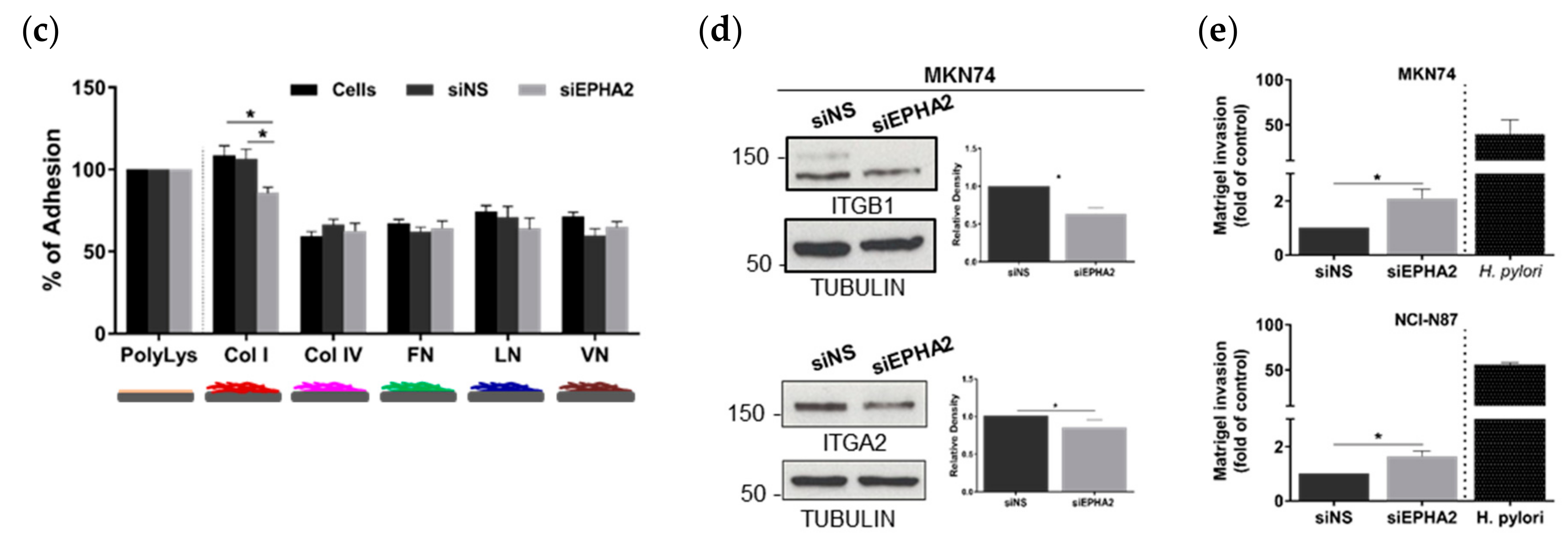
3.3. EPHA2-Silencing by siRNA on Gastric MKN74 and NCI-N87 Cell Lines Impairs Cell–Cell and Cell–Matrix Adhesion and Favours Cell Invasion
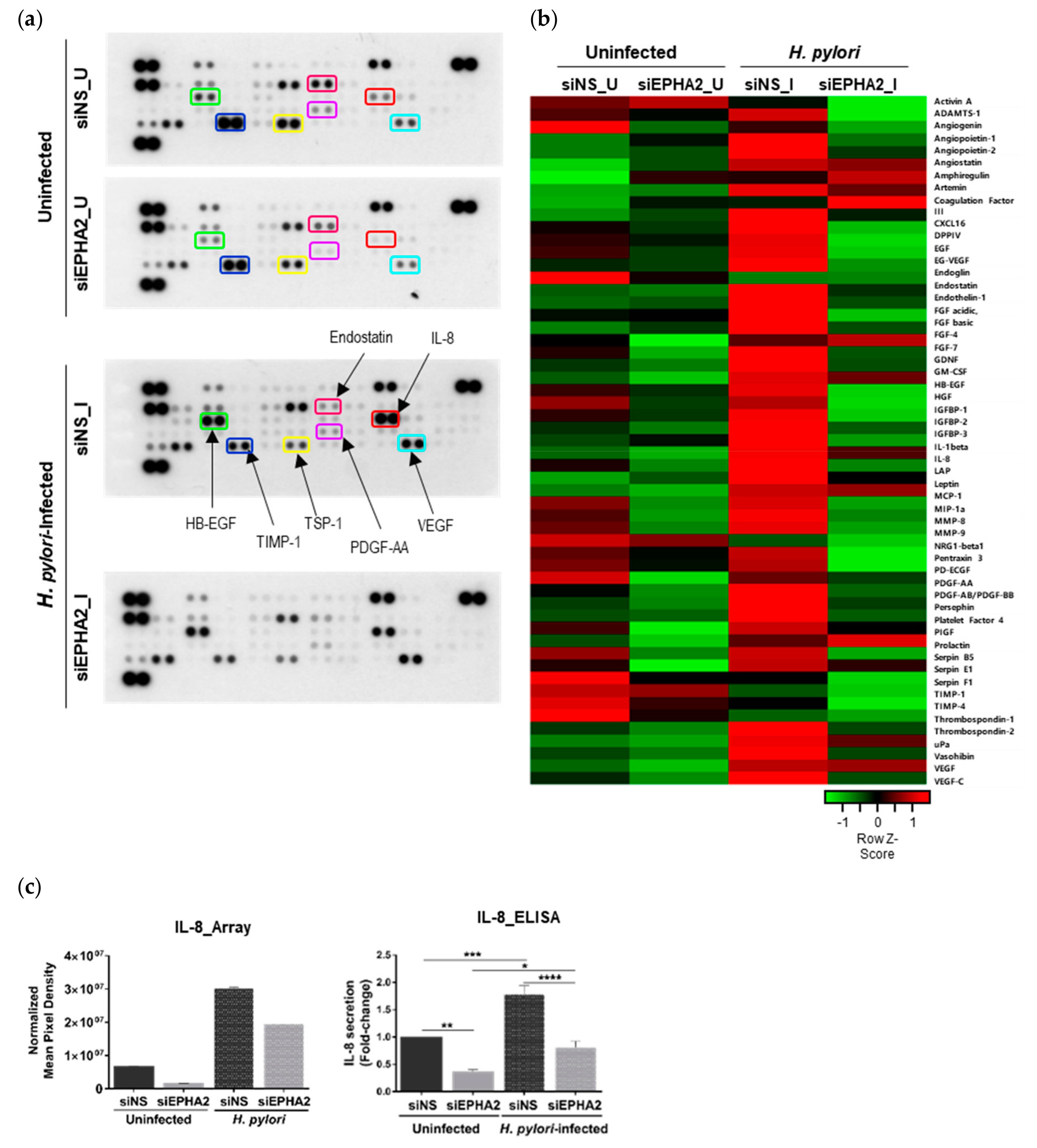
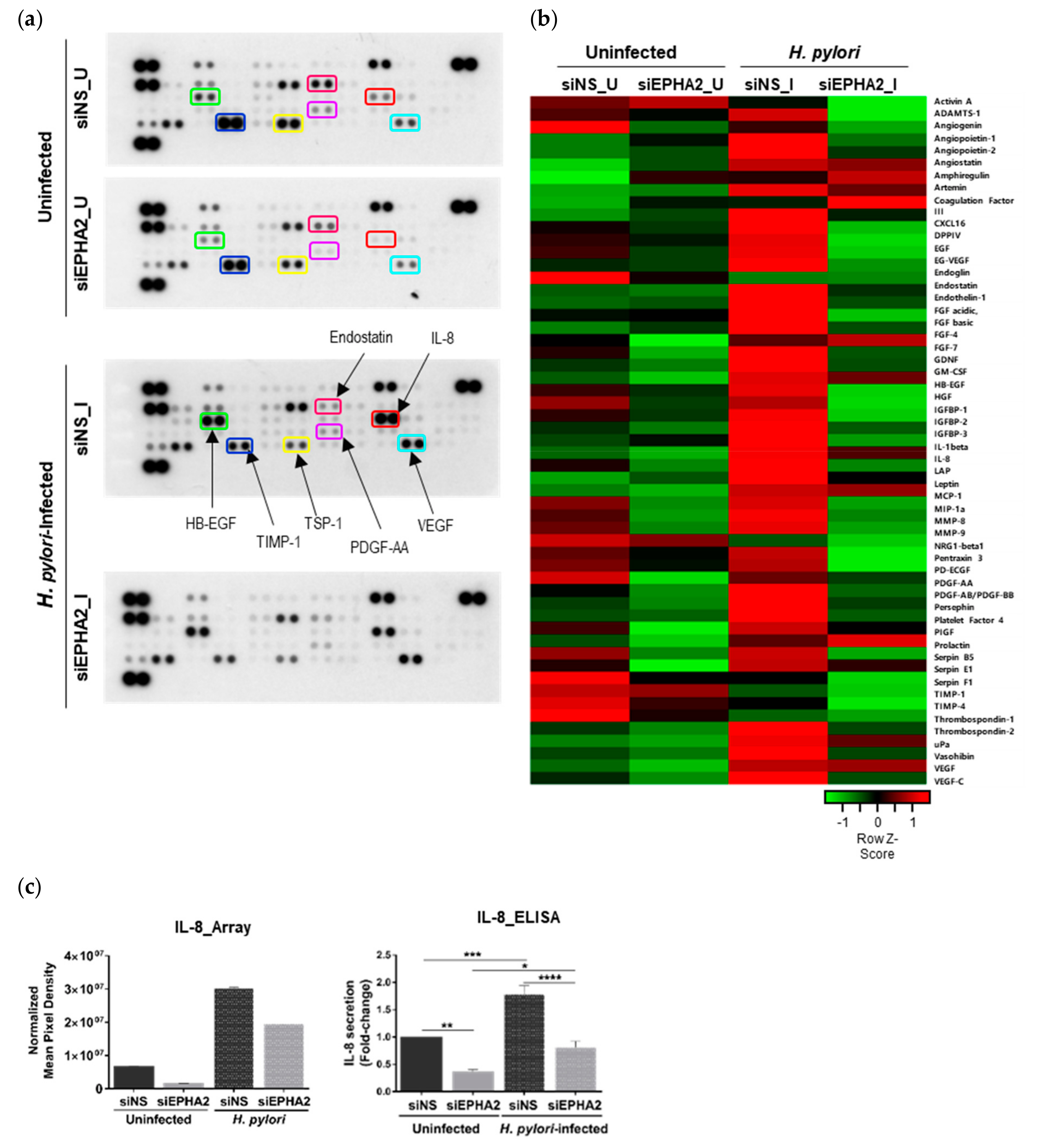
3.4. EPHA2 Receptor Influences the Angiogenic Expression Profile of Gastric Epithelial Cells
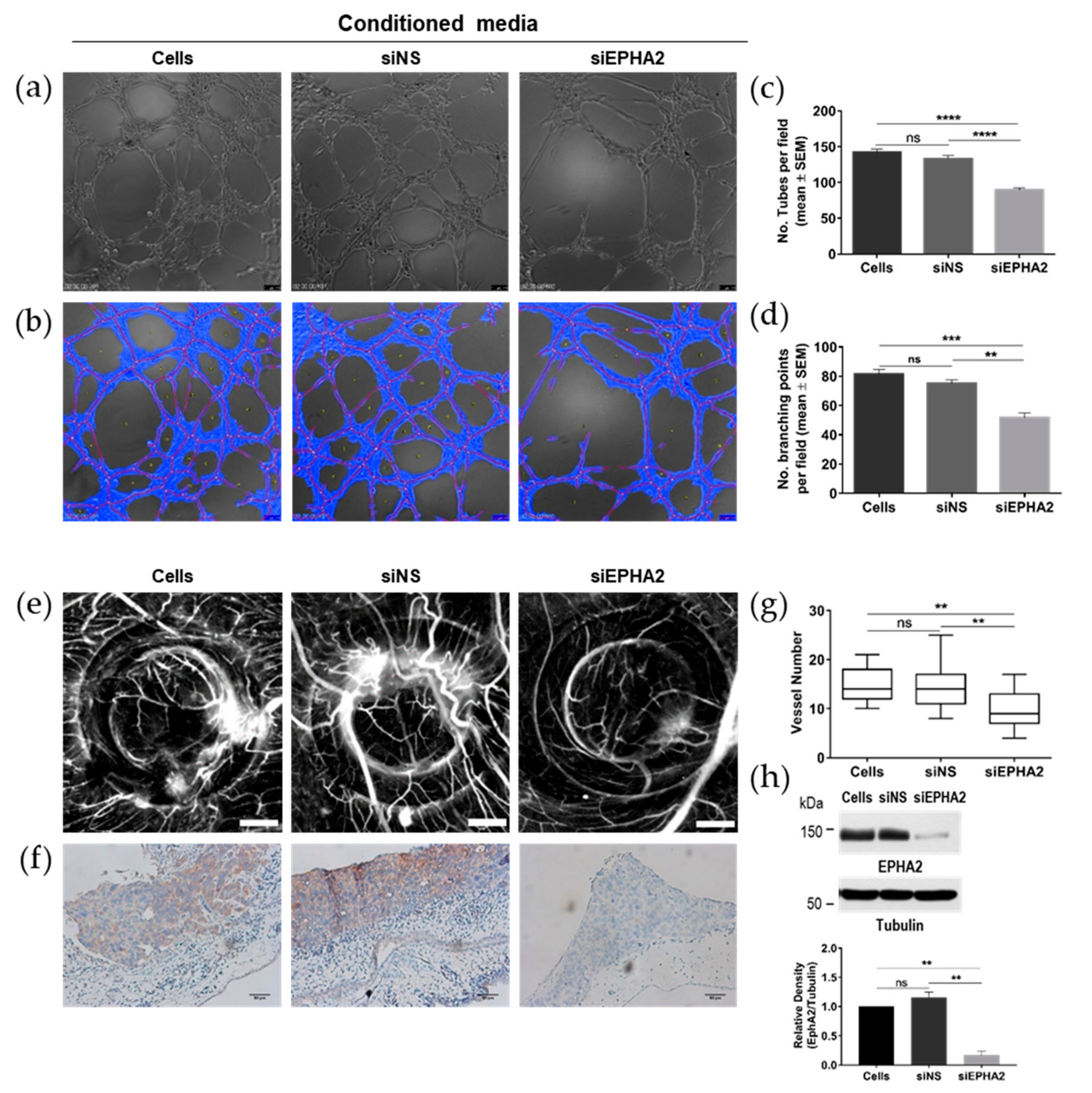
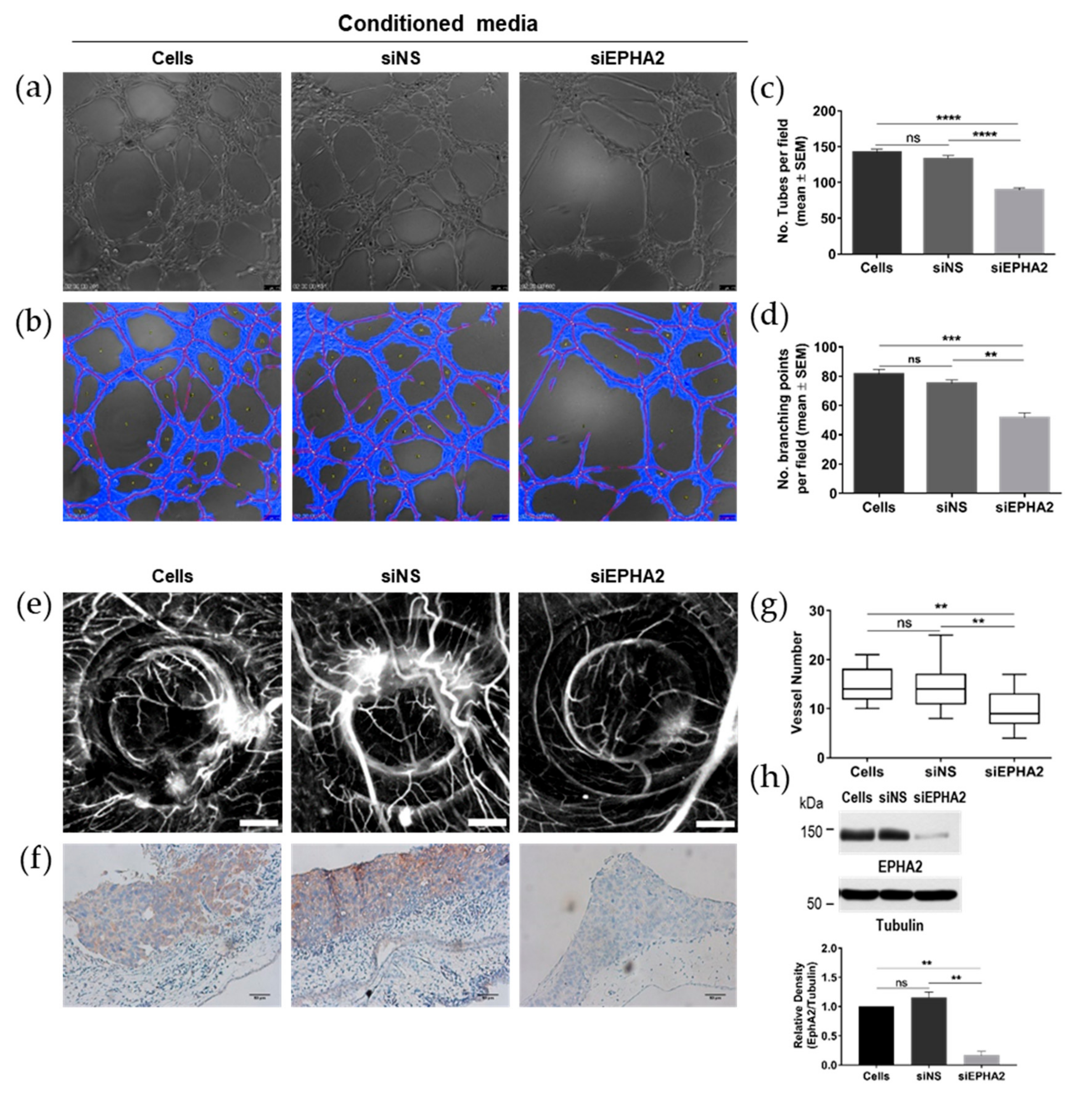
3.5. Knockdown of EPHA2 Receptor in Gastric Cells Impairs Angiogenesis In Vivo and In Vitro
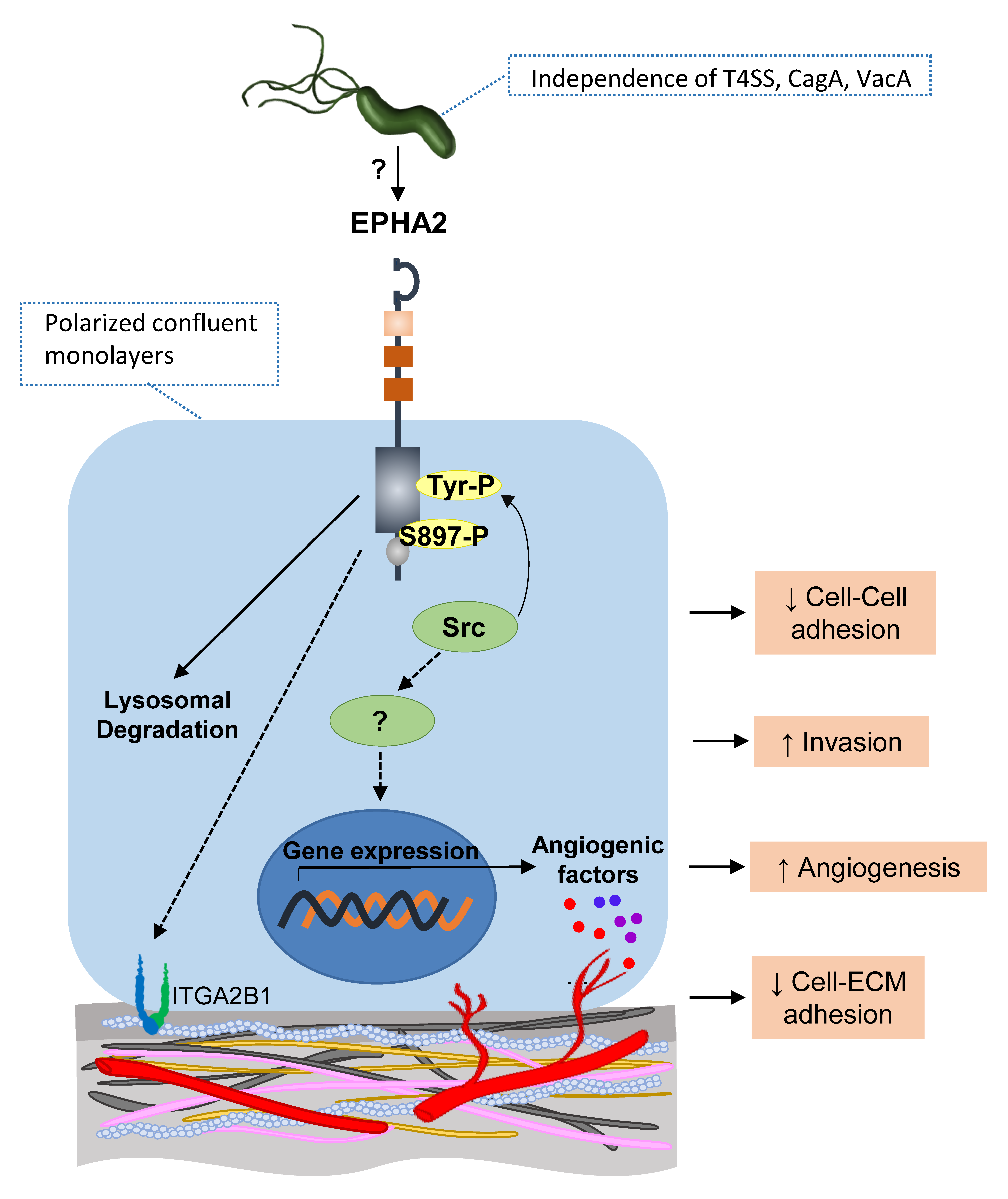
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Atherton, J.C. The pathogenesis ofhelicobacter pylori–induced gastro-duodenal diseases. Annu. Rev. Pathol. Mech. Dis. 2006, 1, 63–96. [Google Scholar] [CrossRef]
- Naumann, M.; Sokolova, O.; Tegtmeyer, N.; Backert, S. Helicobacter pylori: A Paradigm Pathogen for Subverting Host Cell Signal Transmission. Trends Microbiol. 2017, 25, 316–328. [Google Scholar] [CrossRef]
- Keates, S.; Sougioultzis, S.; Keates, A.C.; Zhao, D.; Peek, R.M.; Shaw, L.M.; Kelly, C.P. cag+ Helicobacter pyloriInduce Transactivation of the Epidermal Growth Factor Receptor in AGS Gastric Epithelial Cells. J. Boil. Chem. 2001, 276, 48127–48134. [Google Scholar] [CrossRef] [Green Version]
- Churin, Y.; Al-Ghoul, L.; Kepp, O.; Meyer, T.F.; Birchmeier, W.; Naumann, M. Helicobacter pylori CagA protein targets the c-Met receptor and enhances the motogenic response. J. Cell Boil. 2003, 161, 249–255. [Google Scholar] [CrossRef] [Green Version]
- Keates, S.; Keates, A.C.; Nath, S.; Peek, R.M.; Kelly, C.P. Transactivation of the epidermal growth factor receptor by cag+ Helicobacter pylori induces upregulation of the early growth response gene Egr-1 in gastric epithelial cells. Gut 2005, 54, 1363–1369. [Google Scholar] [CrossRef] [PubMed]
- Ashktorab, H.; Daremipouran, M.; Wilson, M.; Siddiqi, S.; Lee, E.L.; Rakhshani, N.; Malekzadeh, R.; Johnson, A.C.; Hewitt, S.M.; Smoot, D.T. Transactivation of the EGFR by AP-1 Is Induced by Helicobacter pylori in Gastric Cancer. Am. J. Gastroenterol. 2007, 102, 2135–2146. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Danjo, K.; Robinson, P.A.; Crabtree, J.E. In-Cell Western analysis of Helicobacter pylori-induced phosphorylation of extracellular-signal related kinase via the transactivation of the epidermal growth factor receptor. Microbes Infect. 2007, 9, 838–846. [Google Scholar] [CrossRef] [PubMed]
- Yan, F.; Cao, H.; Chaturvedi, R.; Krishna, U.; Hobbs, S.S.; Dempsey, P.J.; Peek, R.M.; Cover, T.L.; Washington, M.K.; Wilson, K.; et al. Epidermal growth factor receptor activation protects gastric epithelial cells from Helicobacter pylori-induced apoptosis. Gastroenterology 2009, 136, 1297–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, M.J.; Costa, A.; Costa, A.; Ferreira, R.M.; Sampaio, P.; Machado, J.C.; Seruca, R.; Mareel, M.; Figueiredo, C. CagA Associates with c-Met, E-Cadherin, and p120-Catenin in a Multiproteic Complex That Suppresses Helicobacter pylori–Induced Cell-Invasive Phenotype. J. Infect. Dis. 2009, 200, 745–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schirrmeister, W.; Gnad, T.; Wex, T.; Higashiyama, S.; Wolke, C.; Naumann, M.; Lendeckel, U. Ectodomain shedding of E-cadherin and c-Met is induced by Helicobacter pylori infection. Exp. Cell Res. 2009, 315, 3500–3508. [Google Scholar] [CrossRef]
- Tegtmeyer, N.; Hartig, R.; Delahay, R.M.; Rohde, M.; Brandt, S.; Conradi, J.; Takahashi, S.; Smolka, A.J.; Sewald, N.; Backert, S. A Small Fibronectin-mimicking Protein from Bacteria Induces Cell Spreading and Focal Adhesion Formation. J. Boil. Chem. 2010, 285, 23515–23526. [Google Scholar] [CrossRef] [Green Version]
- Chaturvedi, R.; Asim, M.; Piazuelo, M.B.; Yan, F.; Barry, D.P.; Sierra, J.C.; Delgado, A.G.; Hill, S.; Casero, R.A.; Bravo, L.E.; et al. Activation of EGFR and ERBB2 by Helicobacter pylori results in survival of gastric epithelial cells with DNA damage. Gastroenterology 2014, 146, 1739–1751.e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaidi, S.F.; Refaat, A.; Zhou, Y.; Muhammad, J.S.; Shin, M.-S.; Saiki, I.; Sakurai, H.; Sugiyama, T. Helicobacter pyloriInduces Serine Phosphorylation of EGFR via Novel TAK1-p38 Activation Pathway in an HB-EGF-Independent Manner. Helicobacter 2015, 20, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Che, Y.; Geng, B.; Xu, Y.; Miao, X.; Chen, L.; Mu, X.; Pan, J.; Zhang, C.; Zhao, T.; Wang, C.; et al. Helicobacter pylori-induced exosomal MET educates tumour-associated macrophages to promote gastric cancer progression. J. Cell. Mol. Med. 2018, 22, 5708–5719. [Google Scholar] [CrossRef] [PubMed]
- Kullander, K.; Klein, R. Mechanisms and functions of eph and ephrin signalling. Nat. Rev. Mol. Cell Boil. 2002, 3, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Himanen, J.-P.; Nikolov, D.B. Eph signaling: A structural view. Trends Neurosci. 2003, 26, 46–51. [Google Scholar] [CrossRef]
- Pasquale, E.B. Eph receptor signalling casts a wide net on cell behaviour. Nat. Rev. Mol. Cell Boil. 2005, 6, 462–475. [Google Scholar] [CrossRef]
- Wybenga-Groot, L.E.; Baskin, B.; Ong, S.H.; Tong, J.; Pawson, T.; Sicheri, F. Structural Basis for Autoinhibition of the EphB2 Receptor Tyrosine Kinase by the Unphosphorylated Juxtamembrane Region. Cell 2001, 106, 745–757. [Google Scholar] [CrossRef] [Green Version]
- Klein, R. Eph/ephrin signalling during development. Development 2012, 139, 4105–4109. [Google Scholar] [CrossRef] [Green Version]
- Holmberg, J.; Frisén, J. Ephrins are not only unattractive. Trends Neurosci. 2002, 25, 239–243. [Google Scholar] [CrossRef]
- Cheng, N.; Brantley, D.M.; Chen, J. The ephrins and Eph receptors in angiogenesis. Cytokine Growth Factor Rev. 2002, 13, 75–85. [Google Scholar] [CrossRef]
- Brantley-Sieders, D.; Chen, J. Eph Receptor Tyrosine Kinases in Angiogenesis: From Development to Disease. Angiogenesis 2004, 7, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Wimmer-Kleikamp, S.; Lackmann, M. Eph-modulated Cell Morphology, Adhesion and Motility in Carcinogenesis. IUBMB Life 2005, 57, 421–431. [Google Scholar] [CrossRef] [PubMed]
- Pasquale, E.B. Eph-Ephrin Bidirectional Signaling in Physiology and Disease. Cell 2008, 133, 38–52. [Google Scholar] [CrossRef] [Green Version]
- Pasquale, E.B. Eph receptors and ephrins in cancer: bidirectional signalling and beyond. Nat. Rev. Cancer 2010, 10, 165–180. [Google Scholar] [CrossRef] [Green Version]
- Miao, H.; Wang, B. Eph/ephrin signaling in epithelial development and homeostasis. Int. J. Biochem. Cell Boil. 2008, 41, 762–770. [Google Scholar] [CrossRef] [Green Version]
- Campbell, T.N.; Robbins, S. The Eph receptor/ephrin system: an emerging player in the invasion game. Curr. Issues Mol. Boil. 2008, 10, 61–66. [Google Scholar]
- Park, J.E.; Son, A.I.; Zhou, R. Roles of EphA2 in Development and Disease. Genes 2013, 4, 334–357. [Google Scholar] [CrossRef] [Green Version]
- Butler, N.; Schmidt, N.W. Erythropoietin-producing hepatocellular receptor B2 receptor tyrosine kinase: A novel regulator of infection- and inflammation-induced liver fibrosis. Hepatology 2015, 62, 680–683. [Google Scholar] [CrossRef] [Green Version]
- Wykosky, J.; Debinski, W. The EphA2 receptor and ephrinA1 ligand in solid tumors: function and therapeutic targeting. Mol. Cancer Res. 2008, 6, 1795–1806. [Google Scholar] [CrossRef] [Green Version]
- Hafner, C.; Schmitz, G.; Meyer, S.; Bataille, F.; Hau, P.; Langmann, T.; Dietmaier, W.; Landthaler, M.; Vogt, T. Differential Gene Expression of Eph Receptors and Ephrins in Benign Human Tissues and Cancers. Clin. Chem. 2004, 50, 490–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, R.; Kataoka, H.; Sato, N.; Kanamori, M.; Ihara, M.; Igarashi, H.; Ravshanov, S.; Wang, Y.J.; Li, Z.Y.; Shimamura, T.; et al. Epha2/efna1 expression in human gastric cancer. Cancer Sci. 2005, 96, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.; Chen, Z.; Wu, S.; Ge, J.; Chang, S.; Wang, X.; Chen, J.; Chen, Z. Expression of EphA2 and E-cadherin in Gastric Cancer: Correlated with Tumor Progression and Lymphogenous Metastasis. Pathol. Oncol. Res. 2008, 15, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Hou, F.; Yuan, W.; Huang, J.; Qian, L.; Chen, Z.; Ge, J.; Wu, S.; Chen, J.; Wang, J.; Chen, Z. Overexpression of EphA2 correlates with epithelial–mesenchymal transition-related proteins in gastric cancer and their prognostic importance for postoperative patients. Med Oncol. 2011, 29, 2691–2700. [Google Scholar] [CrossRef]
- Miyazaki, K.; Inokuchi, M.; Takagi, Y.; Kato, K.; Kojima, K.; Sugihara, K. EphA4 is a prognostic factor in gastric cancer. BMC Clin. Pathol. 2013, 13, 19. [Google Scholar] [CrossRef] [Green Version]
- Inokuchi, M.; Nakagawa, M.; Baogok, N.; Takagi, Y.; Tanioka, T.; Gokita, K.; Okuno, K.; Kojima, K. Prognostic Significance of High EphA1-4 Expression Levels in Locally Advanced Gastric Cancer. Anticancer Res. 2018, 38, 1685–1693. [Google Scholar]
- Peng, Q.; Chen, L.; Wu, W.; Wang, J.; Zheng, X.; Chen, Z.; Jiang, Q.; Han, J.; Wei, L.; Wang, L.; et al. Eph receptor a2 governs a feedback loop that activates wnt/beta-catenin signaling in gastric cancer. Cell Death Dis. 2018, 9, 1146. [Google Scholar] [CrossRef] [Green Version]
- Oki, M.; Yamamoto, H.; Taniguchi, H.; Adachi, Y.; Imai, K.; Shinomura, Y. Overexpression of the receptor tyrosine kinase EphA4 in human gastric cancers. World J. Gastroenterol. 2008, 14, 5650–5656. [Google Scholar] [CrossRef]
- Chakraborty, S.; Veettil, M.V.; Bottero, V.; Chandran, B. Kaposi’s sarcoma-associated herpesvirus interacts with EphrinA2 receptor to amplify signaling essential for productive infection. Proc. Natl. Acad. Sci. USA 2012, 109, E1163–E1172. [Google Scholar] [CrossRef] [Green Version]
- Hahn, A.S.; Kaufmann, J.K.; Wies, E.; Naschberger, E.; Panteleev-Ivlev, J.; Schmidt, K.; Holzer, A.; Schmidt, M.; Chen, J.; König, S.; et al. The ephrin receptor tyrosine kinase A2 is a cellular receptor for Kaposi’s sarcoma–associated herpesvirus. Nat. Med. 2012, 18, 961–966. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Sathiyamoorthy, K.; Zhang, X.; Schaller, S.; Perez-White, B.; Jardetzky, T.S.; Longnecker, R. Ephrin receptor A2 is a functional entry receptor for Epstein-Barr virus. Nat. Microbiol. 2018, 3, 172–180. [Google Scholar] [CrossRef]
- Zhang, H.; Li, Y.; Wang, H.-B.; Zhang, A.; Chen, M.-L.; Fang, Z.-X.; Dong, X.-D.; Li, S.-B.; Du, Y.; Xiong, D.; et al. Ephrin receptor A2 is an epithelial cell receptor for Epstein–Barr virus entry. Nat. Microbiol. 2018, 3, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Subbarayal, P.; Karunakaran, K.; Winkler, A.-C.; Rother, M.; Gonzalez, E.; Meyer, T.F.; Rudel, T. EphrinA2 Receptor (EphA2) Is an Invasion and Intracellular Signaling Receptor for Chlamydia trachomatis. PLoS Pathog. 2015, 11, e1004846. [Google Scholar] [CrossRef] [PubMed]
- Swidergall, M.; Solis, N.V.; Lionakis, M.S.; Filler, S.G. Epha2 is an epithelial cell pattern recognition receptor for fungal beta-glucans. Nat. Microbiol. 2017, 3, 53–61. [Google Scholar] [CrossRef] [Green Version]
- Glowinski, F.; Holland, C.; Thiede, B.; Jungblut, P.R.; Meyer, T.F. Analysis of T4SS-induced signaling by H. pylori using quantitative phosphoproteomics. Front. Microbiol. 2014, 5, 356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative pcr and the 2(-delta delta c(t)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Busk, M.; Pytela, R.; Sheppard, D. Characterization of the integrin alpha v beta 6 as a fibronectin-binding protein. J. Boil. Chem. 1992, 267, 5790–5796. [Google Scholar]
- Ribatti, D.; Nico, B.; Vacca, A.; Roncali, L.; Burri, P.H.; Djonov, V. Chorioallantoic membrane capillary bed: a useful target for studying angiogenesis and anti-angiogenesis in vivo. Anat. Rec. Adv. Integr. Anat. Evol. Boil. 2001, 264, 317–324. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- Walker-Daniels, J.; Riese, D.J.; Kinch, M.S. c-Cbl-dependent EphA2 protein degradation is induced by ligand binding. Mol. Cancer Res. 2002, 1, 79–87. [Google Scholar]
- Zhuang, G.; Hunter, S.; Hwang, Y.; Chen, J. Regulation of EphA2 Receptor Endocytosis by SHIP2 Lipid Phosphatase via Phosphatidylinositol 3-Kinase-dependent Rac1 Activation. J. Boil. Chem. 2006, 282, 2683–2694. [Google Scholar] [CrossRef] [Green Version]
- Goh, L.K.; Sorkin, A. Endocytosis of Receptor Tyrosine Kinases. Cold Spring Harb. Perspect. Boil. 2013, 5, a017459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bin Fang, W.; Brantley-Sieders, D.M.; Hwang, Y.; Ham, A.-J.L.; Chen, J. Identification and functional analysis of phosphorylated tyrosine residues within EphA2 receptor tyrosine kinase. J. Boil. Chem. 2008, 283, 16017–16026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, H.; Li, D.-Q.; Mukherjee, A.; Guo, H.; Petty, A.; Cutter, J.; Basilion, J.P.; Sedor, J.; Wu, J.; Danielpour, D.; et al. EphA2 Mediates Ligand-Dependent Inhibition and Ligand-Independent Promotion of Cell Migration and Invasion via a Reciprocal Regulatory Loop with Akt. Cancer Cell 2009, 16, 9–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jokinen, J.; Ohtaki, A.; Mizuno, M.; Tonozuka, T.; Sakano, Y.; Kamitori, S.; Dadu, E.; Nykvist, P.; Käpylä, J.; White, D.J.; et al. Integrin-mediated Cell Adhesion to Type I Collagen Fibrils. J. Boil. Chem. 2004, 279, 31956–31963. [Google Scholar] [CrossRef] [Green Version]
- Fagiani, E.; Christofori, G. Angiopoietins in angiogenesis. Cancer Lett. 2013, 328, 18–26. [Google Scholar] [CrossRef]
- Eftang, L.L.; Esbensen, Y.; Tannæs, T.; Bukholm, I.R.K.; Bukholm, G. Interleukin-8 is the single most up-regulated gene in whole genome profiling of H. pylori exposed gastric epithelial cells. BMC Microbiol. 2012, 12, 9. [Google Scholar] [CrossRef] [Green Version]
- Krachler, A.M.; Woolery, A.R.; Orth, K. Manipulation of kinase signaling by bacterial pathogens. J. Cell Boil. 2011, 195, 1083–1092. [Google Scholar] [CrossRef] [Green Version]
- Tegtmeyer, N.; Neddermann, M.; Asche, C.I.; Backert, S. Subversion of host kinases: A key network in cellular signaling hijacked byHelicobacter pyloriCagA. Mol. Microbiol. 2017, 105, 358–372. [Google Scholar] [CrossRef] [Green Version]
- McPherson, P.S.; Kay, B.K.; Hussain, N.K. Signaling on the endocytic pathway. Traffic 2001, 2, 375–384. [Google Scholar] [CrossRef]
- Sabet, O.; Stockert, R.; Xouri, G.; Brüggemann, Y.; Stanoev, A.; Bastiaens, P.I.H. Ubiquitination switches EphA2 vesicular traffic from a continuous safeguard to a finite signalling mode. Nat. Commun. 2015, 6, 8047. [Google Scholar] [CrossRef] [Green Version]
- Boissier, P.; Chen, J.; Huynh-Do, U. EphA2 signaling following endocytosis: Role of Tiam1. Traffic 2013, 14, 1255–1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker-Daniels, J.; Hess, A.R.; Hendrix, M.J.; Kinch, M. Differential Regulation of EphA2 in Normal and Malignant Cells. Am. J. Pathol. 2003, 162, 1037–1042. [Google Scholar] [CrossRef] [Green Version]
- Brantley-Sieders, D.M.; Caughron, J.; Hicks, D.; Pozzi, A.; Ruiz, J.C.; Chen, J. EphA2 receptor tyrosine kinase regulates endothelial cell migration and vascular assembly through phosphoinositide 3-kinase-mediated Rac1 GTPase activation. J. Cell Sci. 2004, 117, 2037–2049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lisabeth, E.M.; Falivelli, G.; Pasquale, E.B. Eph Receptor Signaling and Ephrins. Cold Spring Harb. Perspect. Boil. 2013, 5, a009159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, L.-Y.; Patel, O.; Janes, P.W.; Murphy, J.M.; Lucet, I. Eph receptor signalling: from catalytic to non-catalytic functions. Oncogene 2019, 38, 6567–6584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, S.; Gale, N.; Aldrich, T.; Maisonpierre, P.; Lhotak, V.; Pawson, T.; Goldfarb, M.; Yancopoulos, G. Ligands for EPH-related receptor tyrosine kinases that require membrane attachment or clustering for activity. Science 1994, 266, 816–819. [Google Scholar] [CrossRef]
- Stein, E.; Lane, A.A.; Cerretti, D.P.; Schoecklmann, H.O.; Schroff, A.D.; Van Etten, R.L.; Daniel, T.O. Eph receptors discriminate specific ligand oligomers to determine alternative signaling complexes, attachment, and assembly responses. Genome Res. 1998, 12, 667–678. [Google Scholar] [CrossRef] [Green Version]
- Himanen, J.-P.; Rajashankar, K.R.; Lackmann, M.; Cowan, C.A.; Henkemeyer, M.; Nikolov, D.B. Crystal structure of an Eph receptor–ephrin complex. Nature 2001, 414, 933–938. [Google Scholar] [CrossRef]
- Vearing, C.J.; Lackmann, M. Eph receptor signalling; dimerisation just isn’t enough. Growth Factors 2005, 23, 67–76. [Google Scholar] [CrossRef]
- Egea, J.; Nissen, U.V.; Dufour, A.; Sahin, M.; Greer, P.; Kullander, K.; Mrsic-Flogel, T.; Greenberg, M.E.; Kiehn, O.; Vanderhaeghen, P.; et al. Regulation of EphA4 Kinase Activity Is Required for a Subset of Axon Guidance Decisions Suggesting a Key Role for Receptor Clustering in Eph Function. Neuron 2005, 47, 515–528. [Google Scholar] [CrossRef] [Green Version]
- Xu, Q.; Lin, W.-C.; Petit, R.S.; Groves, J.T. EphA2 Receptor Activation by Monomeric Ephrin-A1 on Supported Membranes. Biophys. J. 2011, 101, 2731–2739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, D.R.; Kanvinde, P.; King, C.; Pasquale, E.B.; Hristova, K. The EphA2 receptor is activated through induction of distinct, ligand-dependent oligomeric structures. Commun. Boil. 2018, 1, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murai, K.K.; Pasquale, E.B. `Eph’ective signaling: forward, reverse and crosstalk. J. Cell Sci. 2003, 116, 2823–2832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arvanitis, D.; Davy, A. Eph/ephrin signaling: networks. Genome Res. 2008, 22, 416–429. [Google Scholar] [CrossRef] [Green Version]
- Lemmon, M.; Schlessinger, J. Cell Signaling by Receptor Tyrosine Kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [Green Version]
- Zou, J.X.; Wang, B.; Kalo, M.S.; Zisch, A.H.; Pasquale, E.B.; Ruoslahti, E. An Eph receptor regulates integrin activity through R-Ras. Proc. Natl. Acad. Sci. USA 1999, 96, 13813–13818. [Google Scholar] [CrossRef] [Green Version]
- Miao, H.; Burnett, E.; Kinch, M.; Simon, E.; Wang, B. Activation of EphA2 kinase suppresses integrin function and causes focal-adhesion-kinase dephosphorylation. Nat. Cell Biol. 1999, 2, 62–69. [Google Scholar] [CrossRef]
- Hu, M.; Carles-Kinch, K.L.; Zelinski, D.P.; Kinch, M.S. EphA2 induction of fibronectin creates a permissive microenvironment for malignant cells. Mol. Cancer Res. 2004, 2, 533–540. [Google Scholar]
- Chen, Z.; Oh, N.; Biswas, K.H.; Yu, C.-H.; Luxenburg, C.; Groves, J.T. Spatially modulated ephrinA1:EphA2 signaling increases local contractility and global focal adhesion dynamics to promote cell motility. Proc. Natl. Acad. Sci. USA 2018, 115, E5696–E5705. [Google Scholar] [CrossRef] [Green Version]
- Ieguchi, K.; Maru, Y. Roles of epha1/a2 and ephrin-a1 in cancer. Cancer Sci. 2019, 110, 841–848. [Google Scholar] [CrossRef]
- Ogawa, K.; Pasqualini, R.; A Lindberg, R.; Kain, R.; Freeman, A.L.; Pasquale, E.B. The ephrin-A1 ligand and its receptor, EphA2, are expressed during tumor neovascularization. Oncogene 2000, 19, 6043–6052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brantley, D.M.; Cheng, N.; Thompson, E.J.; Lin, Q.; Brekken, R.A.; Thorpe, P.E.; Muraoka, R.S.; Cerretti, D.P.; Pozzi, A.; Jackson, W.; et al. Soluble Eph A receptors inhibit tumor angiogenesis and progression in vivo. Oncogene 2002, 21, 7011–7026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brantley-Sieders, D.; Bin Fang, W.; Hicks, D.J.; Zhuang, G.; Shyr, Y.; Chen, J. Impaired tumor microenvironment in EphA2-deficient mice inhibits tumor angiogenesis and metastatic progression. FASEB J. 2005, 19, 1884–1886. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.G.; Han, L.Y.; Kamat, A.A.; Merritt, W.M.; Landen, C.N.; Deavers, M.T.; Fletcher, M.S.; Urbauer, D.L.; Kinch, M.; Sood, A.K. EphA2 overexpression is associated with angiogenesis in ovarian cancer. Cancer 2007, 109, 332–340. [Google Scholar] [CrossRef]
- Arienti, C.; Pignatta, S.; Tesei, A. Epidermal Growth Factor Receptor Family and its Role in Gastric Cancer. Front. Oncol. 2019, 9, 1308. [Google Scholar] [CrossRef]
- Inokuchi, M.; Otsuki, S.; Fujimori, Y.; Sato, Y.; Nakagawa, M.; Kojima, K. Clinical significance of MET in gastric cancer. World J. Gastrointest. Oncol. 2015, 7, 317–327. [Google Scholar] [CrossRef]
- Chiurillo, M.A. Role of the wnt/beta-catenin pathway in gastric cancer: An in-depth literature review. World J. Exp. Med. 2015, 5, 84–102. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pro-Angiogenic Factors | Anti-Angiogenic Factors |
|---|---|
| ANGPT1 (Angiopoietin-1) | PF4 (platelet factor 4) |
| ANGPT2 (Angiopoietin-2) * | PLG (angiostatin/plasminogen) |
| ARTN (Artemin) | THBS2 (thrombospondin 2) |
| CCL2 (C-C motif chemokine ligand 2, MCP-1) | VASH (vasohibin) |
| CSF2 (granulocyte-macrophage colony stimulating factor) | |
| CXCL8 (C-X-C motif chemokine ligand 8, IL-8) | |
| CXCL16 (C-X-C motif chemokine ligand 16) | |
| EGF (epidermal growth factor) | |
| EDN1 (endothelin-1) | |
| FGF1 (acidic fibroblast growth factor 1) | |
| FGF2 (basic FGF fibroblast growth factor 2) | |
| FGF-4 (fibroblast growth factor 4) | |
| GDNF (glial cell-derived neurotrophic factor) | |
| HBEGF (heparin binding EGF-like growth factor) | |
| IGFBP3 (insulin-like growth factor binding protein 3) | |
| IL1B (Interleukin 1 beta) | |
| PDGF-AB/-BB (platelet-derived growth factor subunit AB/BB) | |
| PLAU (plasminogen activator urokinase, uPa) | |
| PSPN (Persephin) | |
| VEGFA (vascular endothelial growth factor A) | |
| VEGFC (vascular endothelial growth factor C) |
| FC 1 ≤ 0.5 | FC 1 ≥ 1.5 |
|---|---|
| Pro-Angiogenic | Pro-Angiogenic |
| EGF | CCL2/MCP-1 |
| HGF | CXCL8/IL8 |
| IGFBP1 | FGF7/KGF |
| IGFBP2 | HBEGF |
| IL1B/IL1F2 | PDGF-AA |
| NRG1B1/HRG1B1 | PLAU/uPA |
| PTX3/TSG14 | PRL |
| TYMP/PD-ECGF | VEGFA |
| Anti-angiogenic factors | |
| ADAMTSL1 | |
| TIMP1 | |
| TIMP4 | |
| SERPINF1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leite, M.; Marques, M.S.; Melo, J.; Pinto, M.T.; Cavadas, B.; Aroso, M.; Gomez-Lazaro, M.; Seruca, R.; Figueiredo, C. Helicobacter Pylori Targets the EPHA2 Receptor Tyrosine Kinase in Gastric Cells Modulating Key Cellular Functions. Cells 2020, 9, 513. https://doi.org/10.3390/cells9020513
Leite M, Marques MS, Melo J, Pinto MT, Cavadas B, Aroso M, Gomez-Lazaro M, Seruca R, Figueiredo C. Helicobacter Pylori Targets the EPHA2 Receptor Tyrosine Kinase in Gastric Cells Modulating Key Cellular Functions. Cells. 2020; 9(2):513. https://doi.org/10.3390/cells9020513
Chicago/Turabian StyleLeite, Marina, Miguel S. Marques, Joana Melo, Marta T. Pinto, Bruno Cavadas, Miguel Aroso, Maria Gomez-Lazaro, Raquel Seruca, and Ceu Figueiredo. 2020. "Helicobacter Pylori Targets the EPHA2 Receptor Tyrosine Kinase in Gastric Cells Modulating Key Cellular Functions" Cells 9, no. 2: 513. https://doi.org/10.3390/cells9020513
APA StyleLeite, M., Marques, M. S., Melo, J., Pinto, M. T., Cavadas, B., Aroso, M., Gomez-Lazaro, M., Seruca, R., & Figueiredo, C. (2020). Helicobacter Pylori Targets the EPHA2 Receptor Tyrosine Kinase in Gastric Cells Modulating Key Cellular Functions. Cells, 9(2), 513. https://doi.org/10.3390/cells9020513