Succinic Semialdehyde Dehydrogenase Deficiency: An Update

 , and
, and 
Abstract
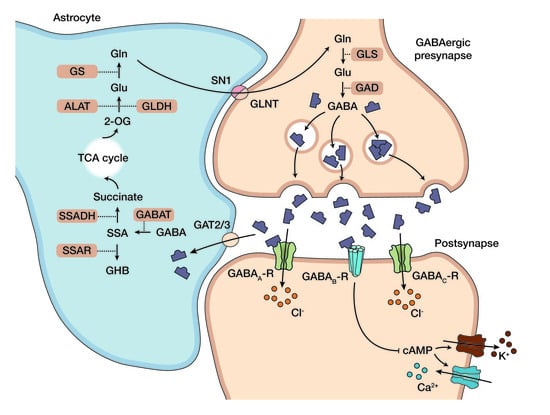
1. Succinic Semialdehyde Dehydrogenase Deficiency: Clinical Phenotype, Genetics, and Standard Care
1.1. Clinical Phenotype and Diagnosis of Succinic Semialdehyde Dehydrogenase Deficiency
1.2. Standard Care and Emerging Picture of the Pathophysiology in SSADH-D
2. The ALDH5A1 Gene and the SSADH Enzyme: Lessons Learned from Disease Models
2.1. ALDH5A1 Gene Splicing Isoforms and Genetic Variants in SSADH-D
2.2. SSADH Enzyme Function and Structure
2.3. Consequences of the Pathogenic Variants Found in SSADH-D
2.4. Disease Models: Cellular Models, Organoids, and the SSADH Knockout Mouse
2.5. Mitochondrial Dysfunction, Redox Imbalance, and Autophagy Defects in SSADH-D
3. Therapy Options for SSADH Deficiency
3.1. Current and Past Clinical Trials in SSADH-D
3.2. Clinical Trials Targeting the Neurotransmitter Receptors
3.3. Further Potential Therapy Options
3.3.1. Enzyme Replacement Therapy: Special Requirements for SSADH-D
3.3.2. Gene Therapy
3.3.3. Small Molecules: Pharmacological Chaperones and Read-Through Drugs
4. Future of SSADH-D Research and Role of Patient Organizations
4.1. Current Tools for Identifying Patients and Patient Registries
4.2. Roles of Patient Advocacy Organizations in Raising Awareness and Supporting Research
4.3. Future Challenges in SSADH-D
Funding
Conflicts of Interest
References
- Pearl, P.L.; Novotny, E.J.; Acosta, M.T.; Jakobs, C.; Gibson, K.M. Succinic semialdehyde dehydrogenase deficiency in children and adults. Ann. Neurol. 2003, 54 (Suppl. 6), S73–S80. [Google Scholar] [CrossRef] [PubMed]
- Gibson, K.M.; Jakobs, C. Disorders of beta- and alpha-amino acids in free and peptide-linked forms. In The Metabolic and Molecular Bases of Inherited Disease, 8th ed.; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Childs, B., Kinzler, K.W., Vogelstein, B., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 2079–2105. [Google Scholar]
- Jakobs, C.; Bojasch, M.; Monch, E.; Rating, D.; Siemes, H.; Hanefeld, F. Urinary excretion of gamma-hydroxybutyric acid in a patient with neurological abnormalities. The probability of a new inborn error of metabolism. Clin. Chim. Acta 1981, 111, 169–178. [Google Scholar] [CrossRef]
- Gibson, K.M.; Sweetman, L.; Nyhan, W.L.; Jakobs, C.; Rating, D.; Siemes, H.; Hanefeld, F. Succinic semialdehyde dehydrogenase deficiency: An inborn error of gamma-aminobutyric acid metabolism. Clin. Chim. Acta 1983, 133, 33–42. [Google Scholar] [CrossRef]
- Chambliss, K.L.; Hinson, D.D.; Trettel, F.; Malaspina, P.; Novelletto, A.; Jakobs, C.; Gibson, K.M. Two exon-skipping mutations as the molecular basis of succinic semialdehyde dehydrogenase deficiency (4-hydroxybutyric aciduria). Am. J. Hum. Genet. 1998, 63, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Benke, D.; Mohler, H. Impact on GABA systems in monogenetic developmental CNS disorders: Clues to symptomatic treatment. Neuropharmacology 2018, 136, 46–55. [Google Scholar] [CrossRef]
- Vogel, K.R.; Pearl, P.L.; Theodore, W.H.; McCarter, R.C.; Jakobs, C.; Gibson, K.M. Thirty years beyond discovery—Clinical trials in succinic semialdehyde dehydrogenase deficiency, a disorder of GABA metabolism. J. Inherit. Metab. Dis. 2013, 36, 401–410. [Google Scholar] [CrossRef]
- Malaspina, P.; Roullet, J.B.; Pearl, P.L.; Ainslie, G.R.; Vogel, K.R.; Gibson, K.M. Succinic semialdehyde dehydrogenase deficiency (SSADHD): Pathophysiological complexity and multifactorial trait associations in a rare monogenic disorder of GABA metabolism. Neurochem. Int. 2016, 99, 72–84. [Google Scholar] [CrossRef]
- Pearl, P.L.; Gibson, K.M.; Cortez, M.A.; Wu, Y.; Carter Snead, O., 3rd; Knerr, I.; Forester, K.; Pettiford, J.M.; Jakobs, C.; Theodore, W.H. Succinic semialdehyde dehydrogenase deficiency: Lessons from mice and men. J. Inherit. Metab. Dis. 2009, 32, 343–352. [Google Scholar] [CrossRef]
- Pearl, P.L.; Gibson, K.M.; Acosta, M.T.; Vezina, L.G.; Theodore, W.H.; Rogawski, M.A.; Novotny, E.J.; Gropman, A.; Conry, J.A.; Berry, G.T.; et al. Clinical spectrum of succinic semialdehyde dehydrogenase deficiency. Neurology 2003, 60, 1413–1417. [Google Scholar] [CrossRef]
- Pearl, P.L.; Hartka, T.R.; Taylor, J. Diagnosis and treatment of neurotransmitter disorders. Curr. Treat. Options Neurol. 2006, 8, 441–450. [Google Scholar] [CrossRef]
- Knerr, I.; Gibson, K.M.; Murdoch, G.; Salomons, G.S.; Jakobs, C.; Combs, S.; Pearl, P.L. Neuropathology in succinic semialdehyde dehydrogenase deficiency. Pediatr. Neurol. 2010, 42, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Gordon, N. Succinic semialdehyde dehydrogenase deficiency (SSADH) (4-hydroxybutyric aciduria, gamma-hydroxybutyric aciduria). Eur. J. Paediatr. Neurol. 2004, 8, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Gibson, K.M.; Christensen, E.; Jakobs, C.; Fowler, B.; Clarke, M.A.; Hammersen, G.; Raab, K.; Kobori, J.; Moosa, A.; Vollmer, B.; et al. The clinical phenotype of succinic semialdehyde dehydrogenase deficiency (4-hydroxybutyric aciduria): Case reports of 23 new patients. Pediatrics 1997, 99, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Bak, L.K.; Schousboe, A.; Waagepetersen, H.S. The glutamate/GABA-glutamine cycle: Aspects of transport, neurotransmitter homeostasis and ammonia transfer. J. Neurochem. 2006, 98, 641–653. [Google Scholar] [CrossRef]
- Kolker, S. Metabolism of amino acid neurotransmitters: The synaptic disorder underlying inherited metabolic diseases. J. Inherit. Metab. Dis. 2018, 41, 1055–1063. [Google Scholar] [CrossRef]
- Bay, T.; Eghorn, L.F.; Klein, A.B.; Wellendorph, P. GHB receptor targets in the CNS: Focus on high-affinity binding sites. Biochem. Pharmacol. 2014, 87, 220–228. [Google Scholar] [CrossRef]
- Tillakaratne, N.J.; Medina-Kauwe, L.; Gibson, K.M. gamma-Aminobutyric acid (GABA) metabolism in mammalian neural and nonneural tissues. Comp. Biochem. Physiol. A Physiol. 1995, 112, 247–263. [Google Scholar] [CrossRef]
- Auteri, M.; Zizzo, M.G.; Serio, R. The GABAergic System and the Gastrointestinal Physiopathology. Curr. Pharm. Des. 2015, 21, 4996–5016. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Sun, S.P.; Zhu, H.S.; Jiao, X.Q.; Zhong, K.; Guo, Y.J.; Zha, G.M.; Han, L.Q.; Yang, G.Y.; Li, H.P. GABA regulates the proliferation and apoptosis of MAC-T cells through the LPS-induced TLR4 signaling pathway. Res. Vet. Sci. 2018, 118, 395–402. [Google Scholar] [CrossRef]
- Petroff, O.A. GABA and glutamate in the human brain. Neuroscientist 2002, 8, 562–573. [Google Scholar] [CrossRef]
- Siucinska, E. Gamma-Aminobutyric acid in adult brain: An update. Behav. Brain. Res. 2019, 376, 112224. [Google Scholar] [CrossRef] [PubMed]
- Grenier, V.; Huppe, G.; Lamarche, M.; Mireault, P. Enzymatic assay for GHB determination in forensic matrices. J. Anal. Toxicol. 2012, 36, 523–528. [Google Scholar] [CrossRef] [PubMed]
- Wernli, C.; Finochiaro, S.; Volken, C.; Andresen-Streichert, H.; Buettler, A.; Gygax, D.; Salomons, G.S.; Jansen, E.E.; Ainslie, G.R.; Vogel, K.R.; et al. Targeted screening of succinic semialdehyde dehydrogenase deficiency (SSADHD) employing an enzymatic assay for gamma-hydroxybutyric acid (GHB) in biofluids. Mol. Genet. Metab. Rep. 2017, 11, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Toriello, H.V. Approach to the genetic evaluation of the child with autism. Pediatr. Clin. N. Am. 2012, 59, 113–128. [Google Scholar] [CrossRef]
- Syndromic Autism Gene Panel. Available online: https://www.centogene.com/science/centopedia/syndromic-autism-gene-panel.html (accessed on 6 December 2019).
- Attri, S.V.; Singhi, P.; Wiwattanadittakul, N.; Goswami, J.N.; Sankhyan, N.; Salomons, G.S.; Roullett, J.B.; Hodgeman, R.; Parviz, M.; Gibson, K.M.; et al. Incidence and Geographic Distribution of Succinic Semialdehyde Dehydrogenase (SSADH) Deficiency. JIMD Rep. 2017, 34, 111–115. [Google Scholar] [CrossRef]
- Brown, M.; Ashcraft, P.; Arning, E.; Bottiglieri, T.; Roullet, J.B.; Gibson, K.M. Gamma-Hydroxybutyrate content in dried bloodspots facilitates newborn detection of succinic semialdehyde dehydrogenase deficiency. Mol. Genet. Metab. 2019, 128, 109–112. [Google Scholar] [CrossRef]
- Shinka, T.; Ohfu, M.; Hirose, S.; Kuhara, T. Effect of valproic acid on the urinary metabolic profile of a patient with succinic semialdehyde dehydrogenase deficiency. J. Chromatogr. B Anal. Technol. Biomed. Life. Sci. 2003, 792, 99–106. [Google Scholar] [CrossRef]
- Vanadia, E.; Gibson, K.M.; Pearl, P.L.; Trapolino, E.; Mangano, S.; Vanadia, F. Therapeutic efficacy of magnesium valproate in succinic semialdehyde dehydrogenase deficiency. JIMD Rep. 2013, 8, 133–137. [Google Scholar] [CrossRef]
- Pearl, P.L.; Parviz, M.; Vogel, K.; Schreiber, J.; Theodore, W.H.; Gibson, K.M. Inherited disorders of gamma-aminobutyric acid metabolism and advances in ALDH5A1 mutation identification. Dev. Med. Child. Neurol. 2015, 57, 611–617. [Google Scholar] [CrossRef]
- Gropman, A. Vigabatrin and newer interventions in succinic semialdehyde dehydrogenase deficiency. Ann. Neurol. 2003, 54 (Suppl. 6), S66–S72. [Google Scholar] [CrossRef]
- Gibson, K.M.; Gupta, M.; Pearl, P.L.; Tuchman, M.; Vezina, L.G.; Snead, O.C., 3rd; Smit, L.M.; Jakobs, C. Significant behavioral disturbances in succinic semialdehyde dehydrogenase (SSADH) deficiency (gamma-hydroxybutyric aciduria). Biol. Psychiatry 2003, 54, 763–768. [Google Scholar] [CrossRef]
- Ethofer, T.; Seeger, U.; Klose, U.; Erb, M.; Kardatzki, B.; Kraft, E.; Landwehrmeyer, G.B.; Grodd, W.; Storch, A. Proton MR spectroscopy in succinic semialdehyde dehydrogenase deficiency. Neurology 2004, 62, 1016–1018. [Google Scholar] [CrossRef] [PubMed]
- Chambliss, K.L.; Zhang, Y.A.; Rossier, E.; Vollmer, B.; Gibson, K.M. Enzymatic and immunologic identification of succinic semialdehyde dehydrogenase in rat and human neural and nonneural tissues. J. Neurochem. 1995, 65, 851–855. [Google Scholar] [CrossRef] [PubMed]
- Blasi, P.; Boyl, P.P.; Ledda, M.; Novelletto, A.; Gibson, K.M.; Jakobs, C.; Hogema, B.; Akaboshi, S.; Loreni, F.; Malaspina, P. Structure of human succinic semialdehyde dehydrogenase gene: Identification of promoter region and alternatively processed isoforms. Mol. Genet. Metab. 2002, 76, 348–362. [Google Scholar] [CrossRef]
- Kim, K.J.; Pearl, P.L.; Jensen, K.; Snead, O.C.; Malaspina, P.; Jakobs, C.; Gibson, K.M. Succinic semialdehyde dehydrogenase: Biochemical-molecular-clinical disease mechanisms, redox regulation, and functional significance. Antioxid Redox Signal. 2011, 15, 691–718. [Google Scholar] [CrossRef]
- Akaboshi, S.; Hogema, B.M.; Novelletto, A.; Malaspina, P.; Salomons, G.S.; Maropoulos, G.D.; Jakobs, C.; Grompe, M.; Gibson, K.M. Mutational spectrum of the succinate semialdehyde dehydrogenase (ALDH5A1) gene and functional analysis of 27 novel disease-causing mutations in patients with SSADH deficiency. Hum. Mutat. 2003, 22, 442–450. [Google Scholar] [CrossRef]
- Menduti, G.; Biamino, E.; Vittorini, R.; Vesco, S.; Puccinelli, M.P.; Porta, F.; Capo, C.; Leo, S.; Ciminelli, B.M.; Iacovelli, F.; et al. Succinic semialdehyde dehydrogenase deficiency: The combination of a novel ALDH5A1 gene mutation and a missense SNP strongly affects SSADH enzyme activity and stability. Mol. Genet. Metab. 2018, 124, 210–215. [Google Scholar] [CrossRef]
- Pearl, P.L.; Wiwattanadittakul, N.; Roullet, J.B.; Gibson, K.M. Succinic Semialdehyde Dehydrogenase Deficiency. In GeneReviews((R)); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- NCBI SNP Search Results for ALDH5A1. Available online: https://www.ncbi.nlm.nih.gov/snp/?term=ALDH5A1 (accessed on 6 December 2019).
- Kim, Y.G.; Lee, S.; Kwon, O.S.; Park, S.Y.; Lee, S.J.; Park, B.J.; Kim, K.J. Redox-switch modulation of human SSADH by dynamic catalytic loop. EMBO J. 2009, 28, 959–968. [Google Scholar] [CrossRef]
- Chambliss, K.L.; Gibson, K.M. Succinic semialdehyde dehydrogenase from mammalian brain: Subunit analysis using polyclonal antiserum. Int. J. Biochem. 1992, 24, 1493–1499. [Google Scholar] [CrossRef]
- Ryzlak, M.T.; Pietruszko, R. Human brain “high Km” aldehyde dehydrogenase: Purification, characterization, and identification as NAD+ -dependent succinic semialdehyde dehydrogenase. Arch. Biochem. Biophys. 1988, 266, 386–396. [Google Scholar] [CrossRef]
- Akiyama, T.; Osaka, H.; Shimbo, H.; Kuhara, T.; Shibata, T.; Kobayashi, K.; Kurosawa, K.; Yoshinaga, H. SSADH deficiency possibly associated with enzyme activity-reducing SNPs. Brain Dev. 2016, 38, 871–874. [Google Scholar] [CrossRef] [PubMed]
- Struys, E.A.; Verhoeven, N.M.; Salomons, G.S.; Berthelot, J.; Vianay-Saban, C.; Chabrier, S.; Thomas, J.A.; Tsai, A.C.; Gibson, K.M.; Jakobs, C. D-2-hydroxyglutaric aciduria in three patients with proven SSADH deficiency: Genetic coincidence or a related biochemical epiphenomenon? Mol. Genet. Metab. 2006, 88, 53–57. [Google Scholar] [CrossRef] [PubMed]
- Leo, S.; Capo, C.; Ciminelli, B.M.; Iacovelli, F.; Menduti, G.; Funghini, S.; Donati, M.A.; Falconi, M.; Rossi, L.; Malaspina, P. SSADH deficiency in an Italian family: A novel ALDH5A1 gene mutation affecting the succinic semialdehyde substrate binding site. Metab. Brain Dis. 2017, 32, 1383–1388. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Cai, F.; Cao, L.; Wang, Y.; Zou, Q.; Zhao, P.; Wang, C.; Zhang, Y.; Cai, C.; Shu, J. Clinical diagnosis and mutation analysis of four Chinese families with succinic semialdehyde dehydrogenase deficiency. BMC Med. Genet. 2019, 20, 88. [Google Scholar] [CrossRef]
- Puttmann, L.; Stehr, H.; Garshasbi, M.; Hu, H.; Kahrizi, K.; Lipkowitz, B.; Jamali, P.; Tzschach, A.; Najmabadi, H.; Ropers, H.H.; et al. A novel ALDH5A1 mutation is associated with succinic semialdehyde dehydrogenase deficiency and severe intellectual disability in an Iranian family. Am. J. Med. Genet. A 2013, 161A, 1915–1922. [Google Scholar] [CrossRef]
- Hempel, J.; Nicholas, H.; Lindahl, R. Aldehyde dehydrogenases: Widespread structural and functional diversity within a shared framework. Protein Sci. 1993, 2, 1890–1900. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. bioRxiv 2019, 531210. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Knoblich, J.A. Generation of cerebral organoids from human pluripotent stem cells. Nat. Protoc. 2014, 9, 2329–2340. [Google Scholar] [CrossRef]
- Jo, J.; Xiao, Y.; Sun, A.X.; Cukuroglu, E.; Tran, H.D.; Goke, J.; Tan, Z.Y.; Saw, T.Y.; Tan, C.P.; Lokman, H.; et al. Midbrain-like Organoids from Human Pluripotent Stem Cells Contain Functional Dopaminergic and Neuromelanin-Producing Neurons. Cell Stem Cell 2016, 19, 248–257. [Google Scholar] [CrossRef]
- Kadoshima, T.; Sakaguchi, H.; Nakano, T.; Soen, M.; Ando, S.; Eiraku, M.; Sasai, Y. Self-organization of axial polarity, inside-out layer pattern, and species-specific progenitor dynamics in human ES cell-derived neocortex. Proc. Natl. Acad. Sci. USA 2013, 110, 20284–20289. [Google Scholar] [CrossRef]
- Monzel, A.S.; Smits, L.M.; Hemmer, K.; Hachi, S.; Moreno, E.L.; van Wuellen, T.; Jarazo, J.; Walter, J.; Bruggemann, I.; Boussaad, I.; et al. Derivation of Human Midbrain-Specific Organoids from Neuroepithelial Stem Cells. Stem Cell Reports 2017, 8, 1144–1154. [Google Scholar] [CrossRef] [PubMed]
- Hogema, B.M.; Gupta, M.; Senephansiri, H.; Burlingame, T.G.; Taylor, M.; Jakobs, C.; Schutgens, R.B.; Froestl, W.; Snead, O.C.; Diaz-Arrastia, R.; et al. Pharmacologic rescue of lethal seizures in mice deficient in succinate semialdehyde dehydrogenase. Nat. Genet. 2001, 29, 212–216. [Google Scholar] [CrossRef] [PubMed]
- Gibson, E.M.; Geraghty, A.C.; Monje, M. Bad wrap: Myelin and myelin plasticity in health and disease. Dev. Neurobiol. 2018, 78, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Yalcinkaya, C.; Gibson, K.M.; Gunduz, E.; Kocer, N.; Ficicioglu, C.; Kucukercan, I. MRI findings in succinic semialdehyde dehydrogenase deficiency. Neuropediatrics 2000, 31, 45–46. [Google Scholar] [CrossRef]
- Ziyeh, S.; Berlis, A.; Korinthenberg, R.; Spreer, J.; Schumacher, M. Selective involvement of the globus pallidus and dentate nucleus in succinic semialdehyde dehydrogenase deficiency. Pediatr. Radiol. 2002, 32, 598–600. [Google Scholar] [CrossRef]
- Donarum, E.A.; Stephan, D.A.; Larkin, K.; Murphy, E.J.; Gupta, M.; Senephansiri, H.; Switzer, R.C.; Pearl, P.L.; Snead, O.C.; Jakobs, C.; et al. Expression profiling reveals multiple myelin alterations in murine succinate semialdehyde dehydrogenase deficiency. J. Inherit. Metab. Dis. 2006, 29, 143–156. [Google Scholar] [CrossRef]
- Barcelo-Coblijn, G.; Murphy, E.J.; Mills, K.; Winchester, B.; Jakobs, C.; Snead, O.C., 3rd; Gibson, K.M. Lipid abnormalities in succinate semialdehyde dehydrogenase (Aldh5a1-/-) deficient mouse brain provide additional evidence for myelin alterations. Biochim. Biophys. Acta 2007, 1772, 556–562. [Google Scholar] [CrossRef][Green Version]
- Niemi, A.K.; Brown, C.; Moore, T.; Enns, G.M.; Cowan, T.M. Evidence of redox imbalance in a patient with succinic semialdehyde dehydrogenase deficiency. Mol. Genet. Metab. Rep. 2014, 1, 129–132. [Google Scholar] [CrossRef]
- Sgaravatti, A.M.; Sgarbi, M.B.; Testa, C.G.; Durigon, K.; Pederzolli, C.D.; Prestes, C.C.; Wyse, A.T.; Wannmacher, C.M.; Wajner, M.; Dutra-Filho, C.S. Gamma-hydroxybutyric acid induces oxidative stress in cerebral cortex of young rats. Neurochem. Int. 2007, 50, 564–570. [Google Scholar] [CrossRef]
- Silva, A.R.; Ruschel, C.; Helegda, C.; Brusque, A.M.; Wannmacher, C.M.; Wajner, M.; Dustra-Filho, C.S. Inhibition of rat brain lipid synthesis in vitro by 4-hydroxybutyric acid. Metab. Brain Dis. 1999, 14, 157–164. [Google Scholar] [CrossRef]
- Pearl, P.L.; Gibson, K.M.; Quezado, Z.; Dustin, I.; Taylor, J.; Trzcinski, S.; Schreiber, J.; Forester, K.; Reeves-Tyer, P.; Liew, C.; et al. Decreased GABA-A binding on FMZ-PET in succinic semialdehyde dehydrogenase deficiency. Neurology 2009, 73, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Okun, J.G.; Horster, F.; Farkas, L.M.; Feyh, P.; Hinz, A.; Sauer, S.; Hoffmann, G.F.; Unsicker, K.; Mayatepek, E.; Kolker, S. Neurodegeneration in methylmalonic aciduria involves inhibition of complex II and the tricarboxylic acid cycle, and synergistically acting excitotoxicity. J. Biol. Chem. 2002, 277, 14674–14680. [Google Scholar] [CrossRef] [PubMed]
- Chandler, R.J.; Zerfas, P.M.; Shanske, S.; Sloan, J.; Hoffmann, V.; DiMauro, S.; Venditti, C.P. Mitochondrial dysfunction in mut methylmalonic acidemia. FASEB J. 2009, 23, 1252–1261. [Google Scholar] [CrossRef] [PubMed]
- Wajner, M.; Brites, E.C.; Dutra, J.C.; Buchalter, M.S.; Pons, A.H.; Pires, R.F.; Wannmacher, L.E.; Rosa Junior, A.; Trindade, V.M.; Wannmacher, C.M. Diminished concentrations of ganglioside N-acetylneuraminic acid (G-NeuAc) in cerebellum of young rats receiving chronic administration of methylmalonic acid. J. Neurol. Sci. 1988, 85, 233–238. [Google Scholar] [CrossRef]
- Schnaar, R.L. Brain gangliosides in axon-myelin stability and axon regeneration. FEBS Lett. 2010, 584, 1741–1747. [Google Scholar] [CrossRef]
- Yu, R.K.; Nakatani, Y.; Yanagisawa, M. The role of glycosphingolipid metabolism in the developing brain. J. Lipid Res. 2009, 50 (Suppl), S440–S445. [Google Scholar] [CrossRef]
- Latini, A.; Scussiato, K.; Leipnitz, G.; Gibson, K.M.; Wajner, M. Evidence for oxidative stress in tissues derived from succinate semialdehyde dehydrogenase-deficient mice. J. Inherit. Metab. Dis. 2007, 30, 800–810. [Google Scholar] [CrossRef]
- Murphy, T.C.; Amarnath, V.; Gibson, K.M.; Picklo, M.J., Sr. Oxidation of 4-hydroxy-2-nonenal by succinic semialdehyde dehydrogenase (ALDH5A). J. Neurochem 2003, 86, 298–305. [Google Scholar] [CrossRef]
- Picklo, M.J.; Montine, T.J.; Amarnath, V.; Neely, M.D. Carbonyl toxicology and Alzheimer’s disease. Toxicol. Appl. Pharmacol. 2002, 184, 187–197. [Google Scholar] [CrossRef]
- Lakhani, R.; Vogel, K.R.; Till, A.; Liu, J.; Burnett, S.F.; Gibson, K.M.; Subramani, S. Defects in GABA metabolism affect selective autophagy pathways and are alleviated by mTOR inhibition. EMBO Mol. Med. 2014, 6, 551–566. [Google Scholar] [CrossRef]
- Menzies, F.M.; Fleming, A.; Rubinsztein, D.C. Compromised autophagy and neurodegenerative diseases. Nat. Rev. Neurosci. 2015, 16, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Khandia, R.; Dadar, M.; Munjal, A.; Dhama, K.; Karthik, K.; Tiwari, R.; Yatoo, M.I.; Iqbal, H.M.N.; Singh, K.P.; Joshi, S.K.; et al. A Comprehensive Review of Autophagy and Its Various Roles in Infectious, Non-Infectious, and Lifestyle Diseases: Current Knowledge and Prospects for Disease Prevention, Novel Drug Design, and Therapy. Cells 2019, 8, 674. [Google Scholar] [CrossRef] [PubMed]
- Vogel, K.R.; Ainslie, G.R.; Jansen, E.E.; Salomons, G.S.; Gibson, K.M. Torin 1 partially corrects vigabatrin-induced mitochondrial increase in mouse. Ann. Clin. Transl. Neurol. 2015, 2, 699–706. [Google Scholar] [CrossRef]
- Vogel, K.R.; Ainslie, G.R.; Jansen, E.E.; Salomons, G.S.; Gibson, K.M. Therapeutic relevance of mTOR inhibition in murine succinate semialdehyde dehydrogenase deficiency (SSADHD), a disorder of GABA metabolism. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 33–42. [Google Scholar] [CrossRef]
- Shoji-Kawata, S.; Sumpter, R.; Leveno, M.; Campbell, G.R.; Zou, Z.; Kinch, L.; Wilkins, A.D.; Sun, Q.; Pallauf, K.; MacDuff, D.; et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature 2013, 494, 201–206. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Codogno, P.; Levine, B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat. Rev. Drug Discov. 2012, 11, 709–730. [Google Scholar] [CrossRef]
- Chauhan, S.; Ahmed, Z.; Bradfute, S.B.; Arko-Mensah, J.; Mandell, M.A.; Won Choi, S.; Kimura, T.; Blanchet, F.; Waller, A.; Mudd, M.H.; et al. Pharmaceutical screen identifies novel target processes for activation of autophagy with a broad translational potential. Nat. Commun. 2015, 6, 8620. [Google Scholar] [CrossRef]
- Petcherski, A.; Chandrachud, U.; Butz, E.S.; Klein, M.C.; Zhao, W.N.; Reis, S.A.; Haggarty, S.J.; Ruonala, M.O.; Cotman, S.L. An Autophagy Modifier Screen Identifies Small Molecules Capable of Reducing Autophagosome Accumulation in a Model of CLN3-Mediated Neurodegeneration. Cells 2019, 8, 1531. [Google Scholar] [CrossRef] [PubMed]
- Vogel, K.R.; Ainslie, G.R.; Walters, D.C.; McConnell, A.; Dhamne, S.C.; Rotenberg, A.; Roullet, J.B.; Gibson, K.M. Succinic semialdehyde dehydrogenase deficiency, a disorder of GABA metabolism: An update on pharmacological and enzyme-replacement therapeutic strategies. J. Inherit. Metab. Dis. 2018, 41, 699–708. [Google Scholar] [CrossRef]
- Froestl, W.; Gallagher, M.; Jenkins, H.; Madrid, A.; Melcher, T.; Teichman, S.; Mondadori, C.G.; Pearlman, R. SGS742: The first GABA(B) receptor antagonist in clinical trials. Biochem. Pharmacol. 2004, 68, 1479–1487. [Google Scholar] [CrossRef] [PubMed]
- Cortez, M.A.; Wu, Y.; Gibson, K.M.; Snead, O.C., 3rd. Absence seizures in succinic semialdehyde dehydrogenase deficient mice: A model of juvenile absence epilepsy. Pharmacol. Biochem. Behav. 2004, 79, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Phase 2 Clinical Trial of SGS-742 Therapy in Succinic Semialdehyde Dehydrogenase Deficiency. Available online: https://clinicaltrials.gov/ct2/show/NCT02019667 (accessed on 8 December 2019).
- Knerr, I.; Gibson, K.M.; Jakobs, C.; Pearl, P.L. Neuropsychiatric morbidity in adolescent and adult succinic semialdehyde dehydrogenase deficiency patients. CNS Spectr. 2008, 13, 598–605. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.; Hogema, B.M.; Grompe, M.; Bottiglieri, T.G.; Concas, A.; Biggio, G.; Sogliano, C.; Rigamonti, A.E.; Pearl, P.L.; Snead, O.C., 3rd; et al. Murine succinate semialdehyde dehydrogenase deficiency. Ann. Neurol. 2003, 54 (Suppl. 6), S81–S90. [Google Scholar] [CrossRef] [PubMed]
- Saronwala, A.; Tournay, A.E.; Gargus, J.J. Genetic inborn error of metabolism provides unique window into molecular mechanisms underlying taurine therapy. Taurine Health Dis. 2012, 661, 357–369. [Google Scholar]
- Schreiber, J.M.; Pearl, P.L.; Dustin, I.; Wiggs, E.; Barrios, E.; Wassermann, E.M.; Gibson, K.M.; Theodore, W.H. Biomarkers in a Taurine Trial for Succinic Semialdehyde Dehydrogenase Deficiency. JIMD Rep. 2016, 30, 81–87. [Google Scholar] [CrossRef]
- Mehta, A.K.; Gould, G.G.; Gupta, M.; Carter, L.P.; Gibson, K.M.; Ticku, M.K. Succinate semialdehyde dehydrogenase deficiency does not down-regulate gamma-hydroxybutyric acid binding sites in the mouse brain. Mol. Genet. Metab. 2006, 88, 86–89. [Google Scholar] [CrossRef]
- Vogel, K.R.; Ainslie, G.R.; McConnell, A.; Roullet, J.B.; Gibson, K.M. Toxicologic/transport properties of NCS-382, a gamma-hydroxybutyrate (GHB) receptor ligand, in neuronal and epithelial cells: Therapeutic implications for SSADH deficiency, a GABA metabolic disorder. Toxicol. In Vitro 2018, 46, 203–212. [Google Scholar] [CrossRef]
- Vogel, K.R.; Ainslie, G.R.; Roullet, J.B.; McConnell, A.; Gibson, K.M. In vitro toxicological evaluation of NCS-382, a high-affinity antagonist of gamma-hydroxybutyrate (GHB) binding. Toxicol. In Vitro 2017, 40, 196–202. [Google Scholar] [CrossRef]
- Gupta, M.; Greven, R.; Jansen, E.E.; Jakobs, C.; Hogema, B.M.; Froestl, W.; Snead, O.C.; Bartels, H.; Grompe, M.; Gibson, K.M. Therapeutic intervention in mice deficient for succinate semialdehyde dehydrogenase (gamma-hydroxybutyric aciduria). J. Pharmacol. Exp. Ther. 2002, 302, 180–187. [Google Scholar] [CrossRef]
- Divry, P.; Baltassat, P.; Rolland, M.O.; Cotte, J.; Hermier, M.; Duran, M.; Wadman, S.K. A new patient with 4-hydroxybutyric aciduria, a possible defect of 4-aminobutyrate metabolism. Clin. Chim. Acta 1983, 129, 303–309. [Google Scholar] [CrossRef]
- Vogel, K.R.; Ainslie, G.R.; Gibson, K.M. mTOR inhibitors rescue premature lethality and attenuate dysregulation of GABAergic/glutamatergic transcription in murine succinate semialdehyde dehydrogenase deficiency (SSADHD), a disorder of GABA metabolism. J. Inherit. Metab. Dis. 2016, 39, 877–886. [Google Scholar] [CrossRef] [PubMed]
- Nylen, K.; Velazquez, J.L.; Likhodii, S.S.; Cortez, M.A.; Shen, L.; Leshchenko, Y.; Adeli, K.; Gibson, K.M.; Burnham, W.M.; Snead, O.C., 3rd. A ketogenic diet rescues the murine succinic semialdehyde dehydrogenase deficient phenotype. Exp. Neurol. 2008, 210, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Nylen, K.; Velazquez, J.L.; Sayed, V.; Gibson, K.M.; Burnham, W.M.; Snead, O.C., 3rd. The effects of a ketogenic diet on ATP concentrations and the number of hippocampal mitochondria in Aldh5a1(-/-) mice. Biochim. Biophys. Acta 2009, 1790, 208–212. [Google Scholar] [CrossRef]
- Matern, D.; Lehnert, W.; Gibson, K.M.; Korinthenberg, R. Seizures in a boy with succinic semialdehyde dehydrogenase deficiency treated with vigabatrin (gamma-vinyl-GABA). J. Inherit. Metab. Dis. 1996, 19, 313–318. [Google Scholar] [CrossRef]
- Schulz, A.; Ajayi, T.; Specchio, N.; de Los Reyes, E.; Gissen, P.; Ballon, D.; Dyke, J.P.; Cahan, H.; Slasor, P.; Jacoby, D.; et al. Study of Intraventricular Cerliponase Alfa for CLN2 Disease. N. Engl. J. Med. 2018, 378, 1898–1907. [Google Scholar] [CrossRef]
- Borgwardt, L.; Guffon, N.; Amraoui, Y.; Dali, C.I.; De Meirleir, L.; Gil-Campos, M.; Heron, B.; Geraci, S.; Ardigo, D.; Cattaneo, F.; et al. Efficacy and safety of Velmanase alfa in the treatment of patients with alpha-mannosidosis: Results from the core and extension phase analysis of a phase III multicentre, double-blind, randomised, placebo-controlled trial. J. Inherit. Metab. Dis. 2018, 41, 1215–1223. [Google Scholar] [CrossRef]
- Bellotti, A.S.; Andreoli, L.; Ronchi, D.; Bresolin, N.; Comi, G.P.; Corti, S. Molecular Approaches for the Treatment of Pompe Disease. Mol. Neurobiol. 2019. [Google Scholar] [CrossRef]
- Pastores, G.M.; Turkia, H.B.; Gonzalez, D.E.; Ida, H.; Tantawy, A.A.; Qin, Y.; Qiu, Y.; Dinh, Q.; Zimran, A. Development of anti-velaglucerase alfa antibodies in clinical trial-treated patients with Gaucher disease. Blood Cells Mol. Dis. 2016, 59, 37–43. [Google Scholar] [CrossRef]
- Bell, S.M.; Wendt, D.J.; Zhang, Y.; Taylor, T.W.; Long, S.; Tsuruda, L.; Zhao, B.; Laipis, P.; Fitzpatrick, P.A. Formulation and PEGylation optimization of the therapeutic PEGylated phenylalanine ammonia lyase for the treatment of phenylketonuria. PLoS ONE 2017, 12, e0173269. [Google Scholar] [CrossRef]
- Del Grosso, A.; Galliani, M.; Angella, L.; Santi, M.; Tonazzini, I.; Parlanti, G.; Signore, G.; Cecchini, M. Brain-targeted enzyme-loaded nanoparticles: A breach through the blood-brain barrier for enzyme replacement therapy in Krabbe disease. Sci. Adv. 2019, 5, eaax7462. [Google Scholar] [CrossRef] [PubMed]
- Tosi, G.; Duskey, J.T.; Kreuter, J. Nanoparticles as carriers for drug delivery of macromolecules across the blood-brain barrier. Expert. Opin. Drug Deliv. 2019, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, I.; Boje, K.M. GHB (gamma-hydroxybutyrate) carrier-mediated transport across the blood-brain barrier. J. Pharmacol. Exp. Ther. 2004, 311, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Lyon, R.C.; Johnston, S.M.; Watson, D.G.; McGarvie, G.; Ellis, E.M. Synthesis and catabolism of gamma-hydroxybutyrate in SH-SY5Y human neuroblastoma cells: Role of the aldo-keto reductase AKR7A2. J. Biol. Chem. 2007, 282, 25986–25992. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.; Jansen, E.E.; Senephansiri, H.; Jakobs, C.; Snead, O.C.; Grompe, M.; Gibson, K.M. Liver-directed adenoviral gene transfer in murine succinate semialdehyde dehydrogenase deficiency. Mol. Ther. 2004, 9, 527–539. [Google Scholar] [CrossRef] [PubMed]
- Ziccardi, L.; Cordeddu, V.; Gaddini, L.; Matteucci, A.; Parravano, M.; Malchiodi-Albedi, F.; Varano, M. Gene Therapy in Retinal Dystrophies. Int. J. Mol. Sci. 2019, 20, 5722. [Google Scholar] [CrossRef]
- Keeler, A.M.; Flotte, T.R. Recombinant Adeno-Associated Virus Gene Therapy in Light of Luxturna (and Zolgensma and Glybera): Where Are We, and How Did We Get Here? Annu. Rev. Virol. 2019, 6, 601–621. [Google Scholar] [CrossRef]
- Wang, D.; Tai, P.W.L.; Gao, G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019, 18, 358–378. [Google Scholar] [CrossRef]
- Wang, L.; Bell, P.; Somanathan, S.; Wang, Q.; He, Z.; Yu, H.; McMenamin, D.; Goode, T.; Calcedo, R.; Wilson, J.M. Comparative Study of Liver Gene Transfer With AAV Vectors Based on Natural and Engineered AAV Capsids. Mol. Ther. 2015, 23, 1877–1887. [Google Scholar] [CrossRef]
- Wang, L.; Wang, H.; Bell, P.; McCarter, R.J.; He, J.; Calcedo, R.; Vandenberghe, L.H.; Morizono, H.; Batshaw, M.L.; Wilson, J.M. Systematic evaluation of AAV vectors for liver directed gene transfer in murine models. Mol. Ther. 2010, 18, 118–125. [Google Scholar] [CrossRef]
- Ginocchio, V.M.; Ferla, R.; Auricchio, A.; Brunetti-Pierri, N. Current Status on Clinical Development of Adeno-Associated Virus-Mediated Liver-Directed Gene Therapy for Inborn Errors of Metabolism. Hum. Gene. Ther. 2019, 30, 1204–1210. [Google Scholar] [CrossRef] [PubMed]
- Baruteau, J.; Waddington, S.N.; Alexander, I.E.; Gissen, P. Delivering efficient liver-directed AAV-mediated gene therapy. Gene. Ther. 2017, 24, 263–264. [Google Scholar] [CrossRef] [PubMed]
- Nagel-Wolfrum, K.; Moller, F.; Penner, I.; Baasov, T.; Wolfrum, U. Targeting Nonsense Mutations in Diseases with Translational Read-Through-Inducing Drugs (TRIDs). BioDrugs 2016, 30, 49–74. [Google Scholar] [CrossRef] [PubMed]
- Liguori, L.; Monticelli, M.; Allocca, M.; Hay Mele, B.; Lukas, J.; Cubellis, M.V.; Andreotti, G. Pharmacological Chaperones: A Therapeutic Approach for Diseases Caused by Destabilizing Missense Mutations. Int. J. Mol. Sci. 2020, 21, 489. [Google Scholar] [CrossRef]
- Leidenheimer, N.J. Pharmacological Chaperones: Beyond Conformational Disorders. Handb Exp. Pharmacol. 2018, 245, 135–153. [Google Scholar] [CrossRef]
- Boyd, R.E.; Lee, G.; Rybczynski, P.; Benjamin, E.R.; Khanna, R.; Wustman, B.A.; Valenzano, K.J. Pharmacological chaperones as therapeutics for lysosomal storage diseases. J. Med. Chem. 2013, 56, 2705–2725. [Google Scholar] [CrossRef]
- Banning, A.; Gulec, C.; Rouvinen, J.; Gray, S.J.; Tikkanen, R. Identification of Small Molecule Compounds for Pharmacological Chaperone Therapy of Aspartylglucosaminuria. Sci. Rep. 2016, 6, 37583. [Google Scholar] [CrossRef]
- Banning, A.; Schiff, M.; Tikkanen, R. Amlexanox provides a potential therapy for nonsense mutations in the lysosomal storage disorder Aspartylglucosaminuria. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 668–675. [Google Scholar] [CrossRef]
- Shin, M.H.; Lim, H.S. Screening methods for identifying pharmacological chaperones. Mol. Biosyst. 2017, 13, 638–647. [Google Scholar] [CrossRef]
- Mort, M.; Ivanov, D.; Cooper, D.N.; Chuzhanova, N.A. A meta-analysis of nonsense mutations causing human genetic disease. Hum. Mutat. 2008, 29, 1037–1047. [Google Scholar] [CrossRef]
- Barton-Davis, E.R.; Cordier, L.; Shoturma, D.I.; Leland, S.E.; Sweeney, H.L. Aminoglycoside antibiotics restore dystrophin function to skeletal muscles of mdx mice. J. Clin. Investig. 1999, 104, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Howard, M.; Frizzell, R.A.; Bedwell, D.M. Aminoglycoside antibiotics restore CFTR function by overcoming premature stop mutations. Nat. Med. 1996, 2, 467–469. [Google Scholar] [CrossRef] [PubMed]
- Clancy, J.P.; Rowe, S.M.; Bebok, Z.; Aitken, M.L.; Gibson, R.; Zeitlin, P.; Berclaz, P.; Moss, R.; Knowles, M.R.; Oster, R.A.; et al. No detectable improvements in cystic fibrosis transmembrane conductance regulator by nasal aminoglycosides in patients with cystic fibrosis with stop mutations. Am. J. Respir. Cell Mol. Biol. 2007, 37, 57–66. [Google Scholar] [CrossRef]
- Politano, L.; Nigro, G.; Nigro, V.; Piluso, G.; Papparella, S.; Paciello, O.; Comi, L.I. Gentamicin administration in Duchenne patients with premature stop codon. Preliminary results. Acta Myol. 2003, 22, 15–21. [Google Scholar] [PubMed]
- Wilschanski, M.; Yahav, Y.; Yaacov, Y.; Blau, H.; Bentur, L.; Rivlin, J.; Aviram, M.; Bdolah-Abram, T.; Bebok, Z.; Shushi, L.; et al. Gentamicin-induced correction of CFTR function in patients with cystic fibrosis and CFTR stop mutations. N. Engl. J. Med. 2003, 349, 1433–1441. [Google Scholar] [CrossRef] [PubMed]
- Woodley, D.T.; Cogan, J.; Hou, Y.; Lyu, C.; Marinkovich, M.P.; Keene, D.; Chen, M. Gentamicin induces functional type VII collagen in recessive dystrophic epidermolysis bullosa patients. J. Clin. Investig. 2017, 127, 3028–3038. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Karasawa, T.; Steyger, P.S. Aminoglycoside-Induced Cochleotoxicity: A Review. Front. Cell Neurosci. 2017, 11, 308. [Google Scholar] [CrossRef]
- Randjelovic, P.; Veljkovic, S.; Stojiljkovic, N.; Sokolovic, D.; Ilic, I. Gentamicin nephrotoxicity in animals: Current knowledge and future perspectives. EXCLI J. 2017, 16, 388–399. [Google Scholar] [CrossRef]
- Baradaran-Heravi, A.; Niesser, J.; Balgi, A.D.; Choi, K.; Zimmerman, C.; South, A.P.; Anderson, H.J.; Strynadka, N.C.; Bally, M.B.; Roberge, M. Retraction for Baradaran-Heravi et al., Gentamicin B1 is a minor gentamicin component with major nonsense mutation suppression activity. Proc. Natl. Acad. Sci. USA 2018, 115, E11885. [Google Scholar] [CrossRef]
- Friesen, W.J.; Johnson, B.; Sierra, J.; Zhuo, J.; Vazirani, P.; Xue, X.; Tomizawa, Y.; Baiazitov, R.; Morrill, C.; Ren, H.; et al. The minor gentamicin complex component, X2, is a potent premature stop codon readthrough molecule with therapeutic potential. PLoS ONE 2018, 13, e0206158. [Google Scholar] [CrossRef]
- Welch, E.M.; Barton, E.R.; Zhuo, J.; Tomizawa, Y.; Friesen, W.J.; Trifillis, P.; Paushkin, S.; Patel, M.; Trotta, C.R.; Hwang, S.; et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature 2007, 447, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Bushby, K.; Finkel, R.; Wong, B.; Barohn, R.; Campbell, C.; Comi, G.P.; Connolly, A.M.; Day, J.W.; Flanigan, K.M.; Goemans, N.; et al. Ptc124-Gd-007-Dmd Study, G. Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve 2014, 50, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Finkel, R.S.; Flanigan, K.M.; Wong, B.; Bonnemann, C.; Sampson, J.; Sweeney, H.L.; Reha, A.; Northcutt, V.J.; Elfring, G.; Barth, J.; et al. Phase 2a study of ataluren-mediated dystrophin production in patients with nonsense mutation Duchenne muscular dystrophy. PLoS ONE 2013, 8, e81302. [Google Scholar] [CrossRef] [PubMed]
- Kerem, E.; Konstan, M.W.; De Boeck, K.; Accurso, F.J.; Sermet-Gaudelus, I.; Wilschanski, M.; Elborn, J.S.; Melotti, P.; Bronsveld, I.; Fajac, I.; et al. Cystic Fibrosis Ataluren Study, G. Ataluren for the treatment of nonsense-mutation cystic fibrosis: A randomised, double-blind, placebo-controlled phase 3 trial. Lancet Respir. Med. 2014, 2, 539–547. [Google Scholar] [CrossRef]
- McDonald, C.M.; Campbell, C.; Torricelli, R.E.; Finkel, R.S.; Flanigan, K.M.; Goemans, N.; Heydemann, P.; Kaminska, A.; Kirschner, J.; Muntoni, F.; et al. Ataluren in patients with nonsense mutation Duchenne muscular dystrophy (ACT DMD): A multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1489–1498. [Google Scholar] [CrossRef]
- Keeling, K.M.; Wang, D.; Conard, S.E.; Bedwell, D.M. Suppression of premature termination codons as a therapeutic approach. Crit. Rev. Biochem. Mol. Biol. 2012, 47, 444–463. [Google Scholar] [CrossRef]
- Gonzalez-Hilarion, S.; Beghyn, T.; Jia, J.; Debreuck, N.; Berte, G.; Mamchaoui, K.; Mouly, V.; Gruenert, D.C.; Deprez, B.; Lejeune, F. Rescue of nonsense mutations by amlexanox in human cells. Orphanet J. Rare Dis. 2012, 7, 58. [Google Scholar] [CrossRef]
- Greer, R.O., Jr.; Lindenmuth, J.E.; Juarez, T.; Khandwala, A. A double-blind study of topically applied 5% amlexanox in the treatment of aphthous ulcers. J. Oral Maxillofac. Surg. 1993, 51, 243–248, discussion 248–249. [Google Scholar] [CrossRef]
- Atanasova, V.S.; Jiang, Q.; Prisco, M.; Gruber, C.; Pinon Hofbauer, J.; Chen, M.; Has, C.; Bruckner-Tuderman, L.; McGrath, J.A.; Uitto, J.; et al. Amlexanox Enhances Premature Termination Codon Read-Through in COL7A1 and Expression of Full Length Type VII Collagen: Potential Therapy for Recessive Dystrophic Epidermolysis Bullosa. J. Investig. Dermatol. 2017, 137, 1842–1849. [Google Scholar] [CrossRef]
- Benhabiles, H.; Gonzalez-Hilarion, S.; Amand, S.; Bailly, C.; Prevotat, A.; Reix, P.; Hubert, D.; Adriaenssens, E.; Rebuffat, S.; Tulasne, D.; et al. Optimized approach for the identification of highly efficient correctors of nonsense mutations in human diseases. PLoS ONE 2017, 12, e0187930. [Google Scholar] [CrossRef] [PubMed]
- Friesen, W.J.; Trotta, C.R.; Tomizawa, Y.; Zhuo, J.; Johnson, B.; Sierra, J.; Roy, B.; Weetall, M.; Hedrick, J.; Sheedy, J.; et al. The nucleoside analog clitocine is a potent and efficacious readthrough agent. RNA 2017, 23, 567–577. [Google Scholar] [CrossRef] [PubMed]
- Opladen, T.; Cortes-Saladelafont, E.; Mastrangelo, M.; Horvath, G.; Pons, R.; Lopez-Laso, E.; Fernandez-Ramos, J.A.; Honzik, T.; Pearson, T.; Friedman, J.; et al. International Working Group on Neurotransmitter related, d. The International Working Group on Neurotransmitter related Disorders (iNTD): A worldwide research project focused on primary and secondary neurotransmitter disorders. Mol. Genet. Metab. Rep. 2016, 9, 61–66. [Google Scholar] [CrossRef] [PubMed]
- MetabERN: European Reference Network for Hereditary Metabolis Disorders. Available online: https://metab.ern-net.eu/ (accessed on 11 December 2019).
- Natural History Study of Patients With Succinic Semialdehyde Dehydrogenase (SSADH) Deficiency. Available online: https://clinicaltrials.gov/ct2/show/NCT03758521 (accessed on 11 December 2019).
- Stoller, J.K. The Challenge of Rare Diseases. Chest 2018, 153, 1309–1314. [Google Scholar] [CrossRef] [PubMed]
- Orphanet. Available online: www.orpha.net (accessed on 11 December 2019).
- EURORDIS: Rare Diseases Europe. Available online: www.eurordis.org (accessed on 11 December 2019).
- NORD: National Organization for Rare Diseases. Available online: www.rarediseases.org (accessed on 11 December 2019).
- SSADH-Defizit e.V. Available online: www.ssadh.de (accessed on 11 December 2019).
- De Neu. Available online: www.deneu.org (accessed on 11 December 2019).
- SSADH Association. Available online: www.ssadh.net (accessed on 11 December 2019).
- Stehr, F.; Forkel, M. Funding resources for rare disease research. Biochim. Biophys. Acta 2013, 1832, 1910–1912. [Google Scholar] [CrossRef][Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolite | Urine (mmol/mol Creatinine) | Plasma (µM) | CSF (µM) |
|---|---|---|---|
| GHB | 34–514 (Ref. < 10) 1 | 26–533 (Ref. < 3) | 116–1110 (Ref. < 3) |
| GABA | n.d. 2 | n.d. | 13.6–22.4 (Ref. < 12) |
| SSA | 3–20 (Ref. < 2) | n.d. | 1280–2570 nmol/L (Ref. < 10 nmol/L) |
| D-2-hydroxyglutaric acid | 22–102 (Ref. < 18) | n.d. | 04–4.7 (Ref. < 0.3) |
| Homocarnosine | n.d. | 3.1–7.6 (Ref. < 1) | 14.8–41 (Ref. < 10) |
| Guanodinobutyrate | 4.6–35 (Ref. < 5.8) | n.d. | 0.04–0.32 (Ref. < 0.03) |
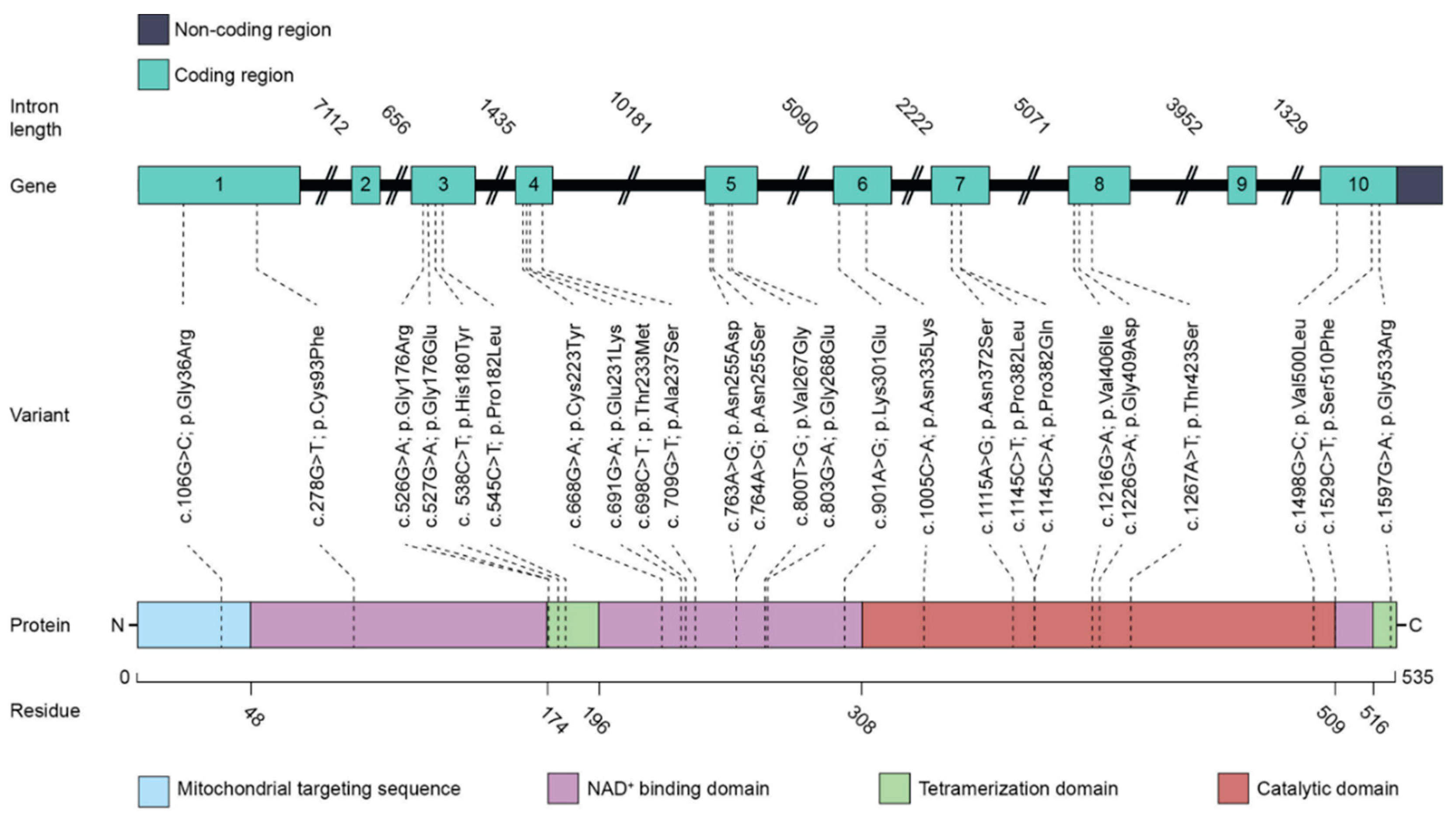
| Genetic Variant | Amino Acid Change | Domain 1 | Remark, Severity 2 | Ref |
|---|---|---|---|---|
| c.106G>C | Gly36Arg | MTS | Mild | [36,38] |
| c.278G>T | Cys93Phe | NADB | Several families, severe | [38] |
| c.526G>A | Gly176Arg | Oligom. | Severe, conserved residue | [39] |
| c.527G>A | Gly176Glu | Oligom. | Conserved residue | [48] |
| c. 538C>T | His180Tyr | Oligom. | Mild, exacerbating | [36,38,39] |
| c.545C>T | Pro182Leu | Oligom. | Mild, exacerbating | [36,38] |
| c.691G>A | Glu231Lys | NADB | Conserved residue | [48] |
| c.668G>A | Cys223Tyr | NADB | Severe | [38] |
| c. 709G>T | Ala237Ser | NADB | Mild, exacerbating? | [36,38,39] |
| c.698C>T | Thr233Met | NADB | Severe | [38] |
| c.763A>G | Asn255Asp | NADB | Several families, severe | |
| c.764A>G | Asn255Ser | NADB | Intermediate | [38] |
| c.800T>G | Val267Gly | NADB | Conserved residue | [48] |
| c.803G>A | Gly268Glu | NADB | Cofactor binding, severe | [38,42] |
| c.901A>G | Lys301Glu | NADB | Cofactor binding | [49] |
| c.1005C>A | Asn335Lys | Catal. | Dynamic loop, severe | [38,42] |
| c.1115A>G | Asn372Ser | Catal. | Not characterized | [36] |
| c.1145C>T | Pro382Leu | Catal. | Severe | [38] |
| c.1145C>A | Pro382Gln | Catal. | Not characterized | [38] |
| c.1216G>A | Val406Ile | Catal. | Not characterized | [36] |
| c.1226G>A | Gly409Asp | Catal. | Severe | [38,47] |
| c.1267A>T | Thr423Ser | Catal. | Mild, exacerbating? | [39] |
| c.1498G>C | Val500Leu | Catal. | Substrate binding, severe | [47] |
| c.1529C>T | Ser510Phe | NADB | Not characterized | [48] |
| c.1597G>A | Gly533Arg | Oligom. | Severe | [38] |
| Intervention | Primary Target | Mode of Action | Outcome in Preclinical Models | Clinical Trial and Outcome |
|---|---|---|---|---|
| SGS-742 [85] CGP-35348 [86] | GABAB receptor | Antagonism | Improvement of epileptiform activity, reduced absence seizures in Aldh5a-/- mice | Completed, phase 2, 19 patients enrolled [87] |
| Taurine [88] | Diffuse GABAA/B modulatory receptor effects | Resuscitative effect of an antagonist | Improves survival of Aldh5a−/− mice [89] | 1 patient, reversal of MRI-documented lesions [90], no effect in TMS [91] |
| NCS-382 [92,93,94] | GHB receptor | Antagonism | Improves survival of Aldh5a−/− mice [95], MDCK cells [93] | - |
| Vigabatrin | GABA transaminase | Inhibition | Improves survival of Aldh5a−/− mice [56] | Effective in 1/3 of patients. Side effect: narrowing of the visual field [7] |
| Valproic acid [29,96] | SSADH | Inhibition | - | Increased level of GHB in urine with valproic acid [29] |
| Rapamycin, Torin, XL-765 [74,78,79,97] | mTORC1/2 | mTORC inhibition, induction of autophagy | Improves survival of Aldh5a−/− mice | - |
| Tat-Bec1 [97] | Beclin 1 | mTORC independent induction of autophagy | Improves survival of Aldh5a−/− mice, induces modest weight gain | - |
| Ketogenic diet [98,99] | Neuroprotective effects | Improves survival of Aldh5a−/− mice [98,99] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Didiasova, M.; Banning, A.; Brennenstuhl, H.; Jung-Klawitter, S.; Cinquemani, C.; Opladen, T.; Tikkanen, R. Succinic Semialdehyde Dehydrogenase Deficiency: An Update. Cells 2020, 9, 477. https://doi.org/10.3390/cells9020477
Didiasova M, Banning A, Brennenstuhl H, Jung-Klawitter S, Cinquemani C, Opladen T, Tikkanen R. Succinic Semialdehyde Dehydrogenase Deficiency: An Update. Cells. 2020; 9(2):477. https://doi.org/10.3390/cells9020477
Chicago/Turabian StyleDidiasova, Miroslava, Antje Banning, Heiko Brennenstuhl, Sabine Jung-Klawitter, Claudio Cinquemani, Thomas Opladen, and Ritva Tikkanen. 2020. "Succinic Semialdehyde Dehydrogenase Deficiency: An Update" Cells 9, no. 2: 477. https://doi.org/10.3390/cells9020477
APA StyleDidiasova, M., Banning, A., Brennenstuhl, H., Jung-Klawitter, S., Cinquemani, C., Opladen, T., & Tikkanen, R. (2020). Succinic Semialdehyde Dehydrogenase Deficiency: An Update. Cells, 9(2), 477. https://doi.org/10.3390/cells9020477