Dynamic Genome Editing Using In Vivo Synthesized Donor ssDNA in Escherichia coli


Abstract

1. Introduction
2. Materials and Methods
2.1. Strains and Culture Conditions
2.2. Determination and Separation of the Circular ssDNA in Vivo
2.3. Iterative Genome Editing Procedure
2.4. Quantitative and Statistical Analysis
3. Results
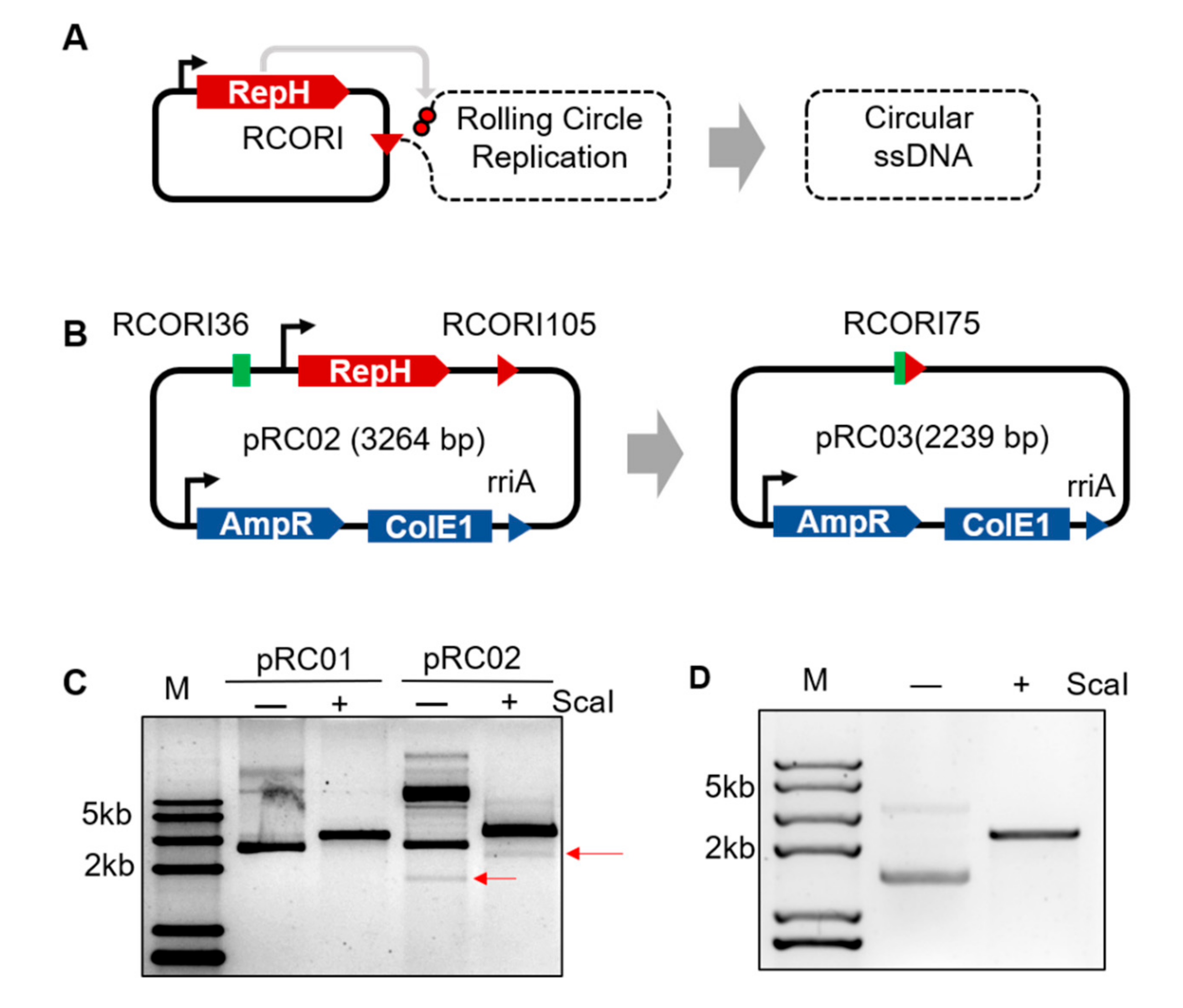
3.1. Engineering Rolling Cycle Replication for in Vivo Circular ssDNA Synthesis
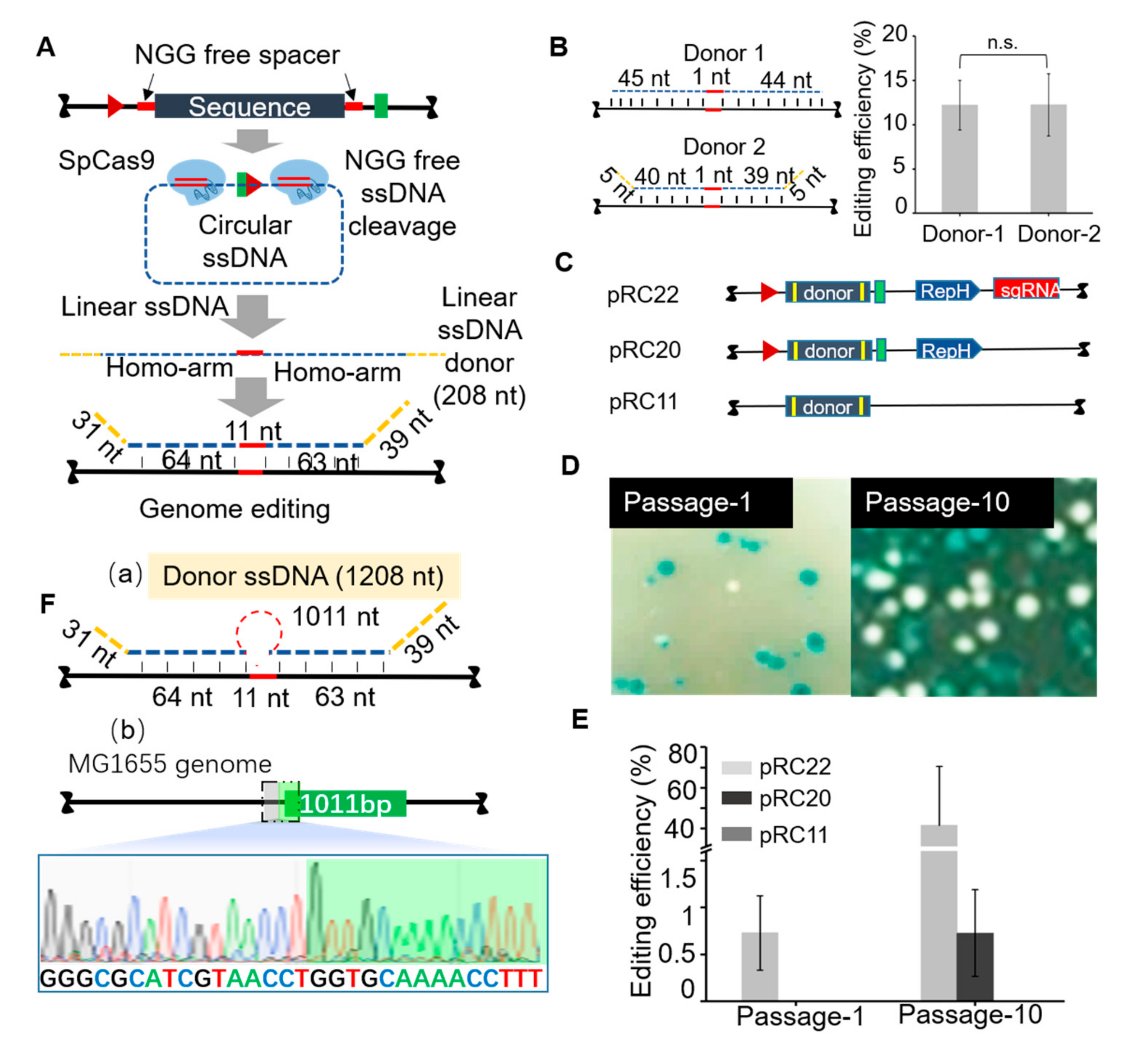
3.2. Homologous Recombination Using in Vivo Synthesized ssDNA Donor
4. Discussion and Conclusions
5. Patents
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A. Supplementary Method
Appendix A.1. Strains and Culture Conditions
Appendix A.2. Construction of Plasmids
Appendix A.3. Quantification of Circular ssDNA
Appendix A.4. Optimization the Cassette for Circular ssDNA Yield
Appendix A.5. Cell Lysis
Appendix A.6. Fluorochromes Probe Binding Assays
Appendix A.7. Purification of Circular ssDNA From Cell Lysis
Appendix A.8. Genome Editing Using a DNA Oligo
Appendix A.9. Genomic DNA Extraction
Appendix B

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence (5’ to 3’) |
|---|---|
| pMR5-F | CCGCTCGAGTGTGAACATCATC |
| pMR5-R | GGAAGATCTAGAGACGCAAGACACTGC |
| 2K-F | ACGCGTCGACTCAAGTCCTCAGCTGCAAG |
| 2K-R | AAGGAAAAAAGCGGCCGCCCTTACCTGGGTCTGTTGTAT |
| G5P-F | TCGATCCGCAGTGTCTTGCGTCTCTCTCGAGAAATCCGCGACCTGCT |
| G5P-R | AAAGATGAACGTGATGATGTTCACACTCGAGCGAATTAAAACGCGATATTTGAAG |
| ty-pET-F | TGTTTAACTTTAAGAAGGAGATATACCATGGGCTGTTATAACATGGAAAAGT |
| ty-pET-R | TATCACGAGGCCCTTTCGTCTTCAAGAATTCAAGCTTGGCAATAGTTAC |
| BglII36-F | GGAAGATCTTTAATTGCGTTGCGCTCAC |
| BglII36-R | GGAAGATCTGGCAATAGTTACCCTTATTATCAAGATAAGAAAGAATGTGAGTTAGCTCACTCAT |
| Lac36-F | CCCTTAAGTTATTGGTATGACTGGTGAGCTCAGACAAGCCCGTCAGG |
| Lac36-R | CTCGGTTGGCAGTGACTCCGTCTCTAAGCTTGGCAATAGTTACCCTTATTAT |
| pMRG53-F | AAGGAAAAAAGCGGCCGCTGTAAAGCCTGGGGTGC |
| pMRG53-R | ACGCGTCGACCATCCAAATCTACAACGTCG |
| 5K-F | ACGCGTCGACTGGAGTGGGCCTACCTC |
| 5K-R | AAGGAAAAAAGCGGCCGCCCTTACCTGGGTCTGTTGTAT |
| Assembly-a | TACTGAGAGTGCACCATATGGTTGGTGTTGTTATGTTGAGAGATCTTAAAGGATTTGAGC |
| Assembly-b | TAGTTACCCTTATTATCAAGATAAGAAAGAAAAGGATTTTTCGCTACGCTCAAATCCTTTAAGATCTCTCAACATA |
| Assembly-c | CTTTTCTTTCTTATCTTGATAATAAGGGTAACTATTGCCGGCGAGGCTAGTTACCCTTAAGTTATTGGTATGACTG |
| Assembly-d | AGGTTAAGGGCAAATCAACACCACCAAACTACCAGTCATACCAATAACTTAAGGGTAAC |
| Assembly-e | TGTTGATTTGCCCTTAACCTCACATACCACCTCGAGGAGCTCGGTACCGTCG |
| Assembly-f | GTTACCCTTATTATCAAGATAAGAAAGAAAAGGATTTTTAAGCTTCTGCAGGTCGACGGTACCGAGCTC |
| Assembly-g | CCTTTTCTTTCTTATCTTGATAATAAGGGTAACTATTGCCGGCGAGGCTAGTTACCCTTAGATCTGTTTACTTT |
| Assembly-h | TGACCATGATTACGCCAAGCTTATGTTCACAATAAAGTAAACAGATCTAAGGGTAACTAGC |
| Assembly-e1 | TGTTGATTTGCCCTTAACCTCACATACCACCTCGAGGAGCTCGGTAC |
| Assembly-f1 | TTATCAAGATAAGAAAGAAAAGCTTGCATGCCTGCAGGTCGACGGTACCGAGCTCCTCGAG |
| Assembly-g1 | CATGCAAGCTTTTCTTTCTTATCTTGATAATAAGGGTAACTATTGCCAGATCTGTTTACTTTATTGTG |
| Assembly-h1 | GACCATGATTACGCCAAGCTTATGTTCACAATAAAGTAAACAGATCTGGCAATAG |
| P19-F | GGTTATCCACAGAATCAGG |
| P19-R | CCTCGTGATACGCCTATTT |
| RepH-F | GATCTGTTTACTTTATTGTGAACATTGTGAACATCATCACGTTC |
| RepH-R | TTATCCCCTGATTCTGTGGATAACCTTTCTTTTGGGTATAGCGTC |
| TY2k-F | GCCTGCAGGTCGACGGTACCGAGCTCCCTTACCTGGGTCTGTTGTAT |
| TY2k-R | ACCTCACATACCACCTCGAGGAGCTCTCAAGTCCTCAGCTGCAAG |
| P19-F1 | GGTTATCCACAGAATCAGG |
| P19-R1 | CCTCGTGATACGCCTATT |
| PlacIq-F | GGGTGGCTCCAGCTTGCCATATGGTACCTTTGTCCTCTT |
| PlacIq-R | CCCTGATTCTGTGGATAACCTTCGTCTTCAAGAATTCTGG |
| EcSSB-R | AAAGAGGACAAAGGTACCATATGGCAAGCTGGAGCCAC |
| EcSSB-F | GACGCTATACCCAAAAGAAACAAAGCCCGCCGAAAGGCGGGCTTTTCTGTTCAGAACGGAATGTCATCATC |
| lacZE1-F | TAATACTAGTCTGTCGTCGTCCCCTCAAACGTTTTAGAGCTAGAAATAGCAAG |
| lacZE1-R | GCTCTAAAACGTTTGAGGGGACGACGACAGACTAGTATTATACCTAGGACTG |
| Sc-gRNA-F | TAATACTAGTATCAACTTCAAACTCACAACGTTTTAGAGCTAGAAATAGCAAG |
| Sc-gRNA-R | GCTCTAAAACGTTGTGAGTTTGAAGTTGATACTAGTATTATACCTAGGACTG |
| Donor-F | ATTTGCCCTTAACCTCACATACCACCTCGAGTTACCTTCACATCCTACCTC |
| Donor-R | CAAGATAAGAAAGAAAAGGATTTTTAAGCTTGGTAAGGCAGGTAGTAGAG |
| gRNA2-F | GAATCAGGGGATAACGCAGGAAAGAACATGTTTGACAGCTAGCTCAGTCC |
| gRNA2-R | CTGGCCTTTTGCTGGCCTTTTGCTCACATGTGGTGCCACTTTTTCAAGTTG |
| PCR-2K-F | GCAGTCAGGCCACAATTAC |
| PCR-2K-R | AACTGTCCTCTGCAGGTG |
| plasmid-F1 | CCAGTCACAGAAAAGCATC |
| plasmid-R1 | ACCAATGCTTAATCAGTGAG |
| ssDNA-F1 | GTAAAACGACGGCCAGTG |
| ssDNA-R1 | GCTGGCGTAATAGCGAAG |
| 16S-F1 | GAAGCTTGCTTCTTTGCT |
| 16S-R1 | GAGCCCGGGGATTTCACAT |
| PCR-LacZ-F | ATGCGTAAGGAGAAAATACC |
| PCR-LacZ-R | ATATGGTGCACTCTCAGTAC |
| PCR-2K-F1 | CATAAAGTGTAAAGCCTGG |
| PCR-2K-R1 | CACCAGTCATACCAATAACT |
| plasmid-F | GGAAGCTAGAGTAAGTAGT |
| plasmid-R | GAATGAAGCCATACCAAAC |
| 16S-F | CATCCACAGAACTTTCCAGAG |
| 16S-R | CCAACATTTCACAACACGAG |
| PCR-PU-F | CAGAACTTTAAAAGTGCTCAT |
| PCR-PU-R | GATTCTCTCCAATATGACGT |
| PCR-vactor-F | GGTTACGATGCGCCCATC |
| PCR-vactor-R | AGTTTGAGGGGACGACGAC |
| TY-1011-F | GATACTGTCGTCGTCCCCTCAAACTATTTGTCCTACTCAGGAGAG |
| TY-1011-R | TGGTGTAGATGGGCGCATCGTAACCTGGTGCAAAACCTTTCGC |
| D-5+2K-F | CTGTTTACTTTATTGTGAACATTGTGAACATCATCACGTTCATCT |
| D-5+2K-R | ACAATGTTCACAATAAAGTAAACAGACCAGTCATACCAATAACTT |
| TY-D-dso-2K-F | TATAAAAATAGGCGTATCACGAGGTCAAGTCCTCAGCTGCAAG |
| TY-D-dso-2K-R | GATGATGTTCACAATGTTCACAATCCTTACCTGGGTCTGTTGTAT |
| TY-D-DSO-F | ATTGTGAACATTGTGAACATCAT |
| TY-D-DSO-R | CCTCGTGATACGCCTATT |
| PCR-MG1655-F | GGTTTATGCAGCAACGAGAC |
| PCR-MG1655-R | GATTCATTAATGCAGCTGG |
| PCR-1011-R | CTAATTCAACAGAATTGGGACAAC |
| TY-gR-F | GAATCAGGGGATAACGCAGGAAAGAACATGTTTGACAGCTAGCTCAGTCCT |
| TY-gR-R | CTGGCCTTTTGCTGGCCTTTTGCTCACATGTAAAAAAACACCGACTCGGTG |
| D-Rep-F | AACATGGTTATCCACAGAATCAGGGGATAACGCAGGAAAGA |
| D-Rep-R | CCCTGATTCTGTGGATAACCATGTTCACAATAAAGTAAACAG |
| TY-RepH-F | TGCAAGCGAGCTTGGCGTAATCATGTGTGAACATCATCACGTTCATC |
| TY-gRNA2-R | TTGCTGGCCTTTTGCTCACATGTTCGGTGCCACTTTTTCAAGTTG |
| Phm01-F | GAACATGTGAGCAAAAGG |
| Phm01-R | CATGATTACGCCAAGCTC |
| Rep-F | CATGCCATGGGCTGTTATAACATGGAAAAGT |
| Rep-R | CCGGGATCCTCAAAGACTTTTTGATTTAT |
| G-F | TTAACTGTGATAAACTACCGCATTAAAGCTTAAATCCGCGACCTGCT |
| G-R | TCATGTTTGACAGCTTATCATCGATAAGCTTCGAATTAAAACGCGATATTTGAAG |
| RCR-F | ATCGATGATAAGCTGTCAAACATGAGAATTCAAAAATCCTTTTCTTTCTTATC |
| RCR-R | TATCACGAGGCCCTTTCGTCTTCAAGAATTCAAGCTTGGCAATAGTTAC |
| Name | Sequence (5’–3’) |
|---|---|
| RCORI 105 | TAAAGGATTTGAGCGTAGCGAAAAATCCTTTTCTTTCTTATCTTGATAATAAGGGTAACTATTGCCGGCGAGGCTAGTTACCCTTAAGTTATTGGTATGACTGGT |
| RCORI 65 | AAAAATCCTTTTCTTTCTTATCTTGATAATAAGGGTAACTATTGCCGGCGAGGCTAGTTACCCTT |
| RCORI 36 | TTCTTTCTTATCTTGATAATAAGGGTAACTATTGCC |
| RCORI 75 | TTCTTTCTTATCTTGATAATAAGGGTAACTATTGCCGGCGAGGCTAGTTACCCTTAAGTTATTGGTATGACTGGT |
| RCORI 85 | AAAAATCCTTTTCTTTCTTATCTTGATAATAAGGGTAACTATTGCCGGCGAGGCTAGTTACCCTTAAGTTATTGGTATGACTGGT |
| pRC03 | AAAAATAAACAAATAGGGGTTCCGCGCACATTTCCCCGAAAAGTGCCACCTGACGTCTAAGAAACCATTATTATCATGACATTAACCTATAAAAATAGGCGTATCACGAGGCCCTTTCGTTGTAAAACGACGGCCAGTCGAACCACGCAATGCGTCTCGATCCGCAGTGTCTTGCGTCTCTAGATCTTTCTTTCTTATCTTGATAATAAGGGTAACTATTGCCGGCGAGGCTAGTTACCCTTAAGTTATTGGTATGACTGGTGAGCTCGGTACCCGGCCATGGTCTAGAGTCGACCTGCAGGCATGCAAGCTTAGAGACGGAGTCACTGCCAACCGAGACGGTCATAGCTGTTTCCTGTGTGCCGCTTCCTCGCTCACTGACTCGCTGCGCTCGGTCGTTCGGCTGCGGCGAGCGGTATCAGCTCACTCAAAGGCGGTAATACGGTTACCCACAGAATCAGGGGATAACGCAGGAAAGAACATGTGAGCAAAAGGCCAGCAAAAGGCCAGGAACCGTAAAAAGGCCGCGTTGCTGGCGTTTTTCCATAGGCTCCGCCCCCCTGACGAGCATCACAAAAATCGACGCTCAAGTCAGAGGTGGCGAAACCCGACAGGACTATAAAGATACCAGGCGTTTCCCCCTGGAAGCTCCCTCGTGCGCTCTCCTGTTCCGACCCTGCCGCTTACCGGATACCTGTCCGCCTTTCTCCCTTCGGGAAGCGTGGCGCTTTCTCATAGCTCACGCTGTAGGTATCTCAGTTCGGTGTAGGTCGTTCGCTCCAAGCTGGGCTGTGTGCACGAACCCCCCGTTCAGCCCGACCGCTGCGCCTTATCCGGTAACTATCGTCTTGAGTCCAACCCGGTAAGACACGACTTATCGCCACTGGCAGCAGCCACTGGTAACAGGATTAGCAGAGCGAGGTATGTAGGCGGTGCTACAGAGTTCTTGAAGTGGTGGCCTAACTACGGCTACACTAGAAGGACAGTATTTGGTATCTGCGCTCTGCTGAAGCCAGTTACCTTCGGAAAAAGAGTTGGTAGCTCTTGATCCGGCAAACAAACCACCGCTGGTAGCGGTGGTTTTTTTGTTTGCAAGCAGCAGATTACGCGCAGAAAAAAAGGATCTCAAGAAGATCCTTTGATCTTTTCTACGGGGTCTGACGCTCAGTGGAACGAAAACTCACGTTAAGGGATTTTGGTCATGAGATTATCAAAAAGGATCTTCACCTAGATCCTTTTAAATTAAAAATGAAGTTTTAAATCAATCTAAAGTATATATGAGTAAACTTGGTCTGACAGTTACCAATGCTTAATCAGTGAGGCACCTATCTCAGCGATCTGTCTATTTCGTTCATCCATAGTTGCCTGACTCCCCGTCGTGTAGATAACTACGATACGGGAGGGCTTACCATCTGGCCCCAGTGCTGCAATAATACCGCGGGACCCACGCTCACCGGCTCCAGATTTATCAGCAATAAACCAGCCAGCCGGAAGGGCCGAGCGCAGAAGTGGTCCTGCAACTTTATCCGCCTCCATCCAGTCTATTAATTGTTGCCGGGAAGCTAGAGTAAGTAGTTCGCCAGTTAATAGTTTGCGCAACGTTGTTGCCATCGCTACAGGCATCGTGGTATCACGCTCGTCGTTTGGTATGGCTTCATTCAGCTCCGGTTCCCAACGATCAAGGCGAGTTACATGATCCCCCATGTTGCGCAAAAAAGCGGTTAGCTCCTTCGGTCCTCCGATCGTTGTCAGAAGTAAGTTGGCCGCCGTGTTATCACTCATGGTTATGGCAGCACTACATAATTCTCTTACTGTCATGCCATCCGTAAGATGCTTTTCTGTGACTGGTGAGTACTCAACCAAGTCATTCTGAGAATAGTGTATGCGGCGACCGAGTTGCTCTTGCCCGGCGTCAATACGGGATAATACCGCGCCACATAGCAGAACTTTAAAAGTGCTCATCATTGGAAAACGTTCTTCGGGGCGAAAACTCTCAAGGATCTTACCGCTGTTGAGATCCAGTTCGATGTAACCCACTCGTGCACCCAACTGATCTTCAGCATCTTTTACTTTCACCAGCGTTTCTGGGTGAGCAAAAACAGGAAGGCAAAATGCCGCAAAAAAGGGAATAAGGGCGACACGGAAATGTTGAATACTCATACTCTTCCTTTTTCAATATTATTGAAGCATTTATCAGGGTTATTGTCTCATGAGCGGATACATATTTGAATGTATTTAG |
| pRC06- circular ssDNA | TTCTTTCTTATCTTGATAATAAGGGTAACTATTGCCGGCGAGGCTAGTTACCCTTAAGTTATTGGTATGACTGGTGAGCTCAGATCTCCGACGTTGTAGATTTGGATGAGACAAGCCCGTCAGGGCGCGTCAGCGGGTGTTGGCGGGTGTCGGGGCTGGCTTAACTATGCGGCATCAGAGCAGATTGTACTGAGAGTGCACCATATGCGGTGTGAAATACCGCACAGATGCGTAAGGAGAAAATACCGCATCAGGCGCCATTCGCCATTCAGGCTGCGCAACTGTTGGGAAGGGCGATCGGTGCGGGCCTCTTCGCTATTACGCCAGCTGGCGAAAGGGGGATGTGCTGCAAGGCGATTAAGTTGGGTAACGCCAGGGTTTTCCCAGTCACGACGTTGTAAAACGACGGCCAGTGAATTCGAGCTCGGTACCCGGGGATCCTCTAGAGTCGACCTGCAGGCATGCAAGCTTGGCGTAATCATGGTCATAGCTGTTTCCTGTGTGAAATTGTTATCCGCTCACAATTCCACACAACATACGAGCCGGAAGCATAAAGTGTAAAGCCTGGGGTGCCTAATGAG |
| pRC08- circular ssDNA | TTCTTTCTTATCTTGATAATAAGGGTAACTATTGCCGGCGAGGCTAGTTACCCTTAAGTTATTGGTATGACTGGTGAGCTCAGATCTCCGACGTTGTAGATTTGGATGGTCGACTCAAGTCCTCAGCTGCAAGAATATCTGAATGTCTTTTGGAGTGTTAGAGTCCTCTGTGTCTTAGAAATTTTGAAAAGAAAAACAAATCTCAATATTAATGTTGATTAGTTTCTCTGAGCCAATTGGGGAAAATAAACGTCCTTCACCTCAAAGGTTTAAGTGACACCGAAGGGTAGCCACCAGTGTCTCGGCCACTGAAGCCTCATGCATGCTCTCACTACCAGTTTGATTTGCAGCCCCATAGTTGTGTTGTACTAAATATTCTTTCCTCTGGCCTTGTCCAGTGAACACGGTTCACATGGCTAACACCACTTCTTGAGATGCGAGCACCATGCAAAGCTGAGAACGGATTGGGTTTTGTGACCATTGTGCCTCCTCCTCACCTGAGAGGCCCATTTTTCCTGGTTGATTCATTAAGTGTATTGGTGCTGTCAGTCGCCTCTGGACAATTGAAATGACAAGTGGCTGTTGATTCATAAAGAAAATGAAGGCTTTAGATGTGAAACCCTCGTTTTCTCTTGTCCTTCTCTTAGGTGAAAGATTTTATTTTTTTCAAAAGGCTACATACTGGTATCCCAGCAGGTGTAGTGTGAGAACTGGCATATGTTAGGCTATGGTGTCAGTGTGGATGGGCAATTCTTCAAGATGGAAAACCAAGTCTCACTGAGTTGCTGGAGCCACACTGACCTTTCTCCACATCCCCCACCATGGGCTTTCACTTTTATCCTGTGCTTGAATTTTTTTCACATACAAATTCTTTATACACACACACAGACACACACACACATATCTCACTCTGTCAATGCAGTGGCTGAATCATGGGTCACTGCATCTTCAAATTCTTAGGCTCCAGTGATGCTTTCAAATCAGCCTCTCAAGTAGCTGGGACTACAGGCATGCAAAGCTACACCCAGACAATTTTTAAATATTTTTCTAGAGACTGAGCCTACTTATGTTGCTCAGACTCGTCTTGCACTCCTGGGATCAAACGATAATCCCACCTTGACCACCCAAAGTGTTTAGATTACAGGTGTGAGCTAGCACTCTCAGCAAAAATATATTTTAAAGAACCGTTACAACCAAATTATGAGTTATCATTATGCCACTGCCCTCCACCCTGGGCACCAGAACAAGACCTTGTATCCAAAAACTAAGCAAAACTAAACAAGAACAAAAAAAAAAAAACTTATAAATAAACTTTGAAGATTGTGTCATCTGTGTCCTTCCCTGCCCTCCAAGCTATCAATGTTAAATATAATGGTTATTGAGAAAATGGTTAGATATTATTAAGAAATTTCTATATATCTTCCAGCTGAGAATAGGTATTCTGTTGTGGCCCAAATATTTTCTCACCGCTACCTTCAGGGTCTAAACTAGCAAATCAGGACACCTGCAGAGGACAGTTGGCCGTTTTCAAATAGAAAGAGAAATACCCCCGTTCATGAGAGTAATCCAGTGATTTTCAAAAAGACAAGTCAGACTGACATGCAGCGCAGTCAGGCCACAATTACCCTGGAATAATCACTTCACACAGAATGGTTGAGGAGACTTTCTAAGATGAGCAAATTTGGGCAGCATAATCCTTGCTTATTTATTCCCAGCCCCCACTGCCCGCCTGATTCCTAATGGCTACCCTACAATGTGGTCAGCAGTGGGATGTAGCGTGGTGAGAGAGGGGCTCAGGGACGGGATGAAGGTCTTTCCTGCATTATCAAAATGCAGGTTAAAAAGTTGTTAAAAAGATGTCCAAATGTTCTAATTCCTACTGTTAAATAGCTGCTAAGATGCATTATACAACAGACCCAGGTAAGGGCGGCCGCTGTAAAGCCTGGGGTGCCTAATGAG |
| pRC09- circular ssDNA | ATAATAAGGGTAACTATTGCCGGCGAGGCTAGTTACCCTTAAGTTATTGGTATGACTGGTGAGCTCAGATCTCCGACGTTGTAGATTTGGATGGTCGACTGGAGTGGGCCTACCTCCCATCAAGCTGTGTCTCCACAGCTGACCCTTGTAACCAGGAGGTGTTTTACAACATGTGCAAGGCAGTGAGCTCCATCAGCTGTGTGGCATTCAACACTCACTTCAACTCGGACATCTCACCAGAAAGCAGTGGGGACTGGCCAATGCAGAAGCCTGCAAAGTGGAACAGAGCGTCATGGGGTGGGGGATGTGGGGCCTGCCTGCTCATCTGAGCACTGCTCCCTGAGGGTGTGATCTGCAGGCTTCCTGAAGGAGGGCTGTGAGCTCTTCTGCGAGGCCCTGAGCCTGTGGAACATAGCTGAGGCCAAGCCCATGGGGATTTGTGTCTACTTGCACCTCCTTGCTCATCTCAGTACACTACAGGTGACTGTGCCGAGGTGGGCCTTGAGCATCCCCTGGGCTGTGTCAGCAAATGGCTCTGGGCCTGGCCTGGCATTGAGGGATGGCAAAAAAGGAGCCTGGGGTTGCATTGTCATCCCCTATGGTAGCATAAAATGAGAGAGTCCAGACCTGCAGGACTGGAACCCTAACAAAGGGGTTAGGAGACTGCTCACTTTCCCTCAGGAACCCATGTGGAGGAGCTGAGGGAGGTTAAGGAGACCCTAGGGACTCACTTGTTCTGTCTGGGCTTCCCCCTGCTCCATCGTTTGATGACCATTTTCTGGGAAGAGCTCAGGAACCTCCTGTGCTCTAGTGAGACGGGGCCTCCCCTCACAGGGTATTCTGAGACTGTGAGTGAGAAGCTAACACAGTGCCTTGCAATACTCATGGGAGCTGTCATCCTCTGTGACCATCACGTGGCCTTGTAGTGTTCAGACTGCCTGGCCTGCCTGGGGTTTGGTGAGGCTGTTTTGTGGTCAGGTGCTTTAGAAGCTCACTTTCTCTGCAATCAAACAGTGACTGTTTTCATGTCTGTTTATGGGTTTAAAAAATCCTAATATTTCCTTTATAGTAGTTCACCTTGTATGTGTTTATTTGTATAAATTTTATTAGAGTAAAGAGAGCTTAAGACAATAGCATTTTAAGGTCTTAATGAGGCATAGACTTTCATGTCACAACAGCTAATGTTGACCTCCTTTTGCTGCCTTTGTGTAAATTACACATAAAAAGTGCAGCCAGAGGTGACTAGAGCTGAGCTGCTTGGGCTTGCTTGCTGGCCTGCAGTCAGGTGGACTCTGGCTGTGAGGCAGTGCCCACCCTGGATCTACATCCCCCACCCCCTCTCCTTAGTCCCTGAGTAACCAACAAGGCCGTGCTAATGAGAGGGCGAGTGATGGGCATCGGGCACCCCCATATTATCCGGGAAGATTTGAATGCCATCTGGGCTGGAGCTGTTGGGATTAGGGGCTGAGGCTGTCTTGGCTTGTCATGGTGCCACCCACAGATGTGCCTGCCCTGTGCTGCTTCTCCAGAAGCCGGCTGCCCATGGCCCTGAGCCTGTCACACCATGCTTGCTACCTCATGCTGCTTGTGTTTGAAAAACCCATCCCGAGATGACGCTGCTGGATGTAAGTCCTGAAAAGGGGGCATCACCTTTGTCCTGGGGGATTAGGAGCTGACCAGATTCCTCTTGACTCCCTCCCAGAACAAGTGGGGCAGGTGCTGCAATTAATGTTGCCCCCTAGAAGATGTGTTTGCACTGGCTGAGCAAATATACGATGCAGAGACCTAAATGAAGACACGTGAATGGGGTGTGTGGACATCAGTTAGTAGCTGGGAAACAGGTGCCTCTCAGGCGTCTCGTGTTCCAGCAAGTGTGGAATATGCCTGTGCCCATGAGTGTAGACATCTGAAGTGTATACATTTGGCTGCTGCTTTTGCTGCCACTATTCCCAGGCCCAACCTGGCTTAAAGTCCAGGTTTTAAGTAAAAAGTAGGAGGCTTTTTGCCATACAGCTACTTGAGAGGCTGAGGTGAAAGCATCACTGGAGCCTAAGAGATTGAGGCTGCAGTGACCCATGATTCAGCCACTGCACTGACACAGTGAGACCTGCGTGTGCCCTTCTACAGAGAATAGCTCTGGGGCATTTGGGGATCCCTACAGTCCCGGACCCTCCCTGTCCCCTGCTGCCTGTGCTCCTTTCCTTGCCTGCTGTCAGAGCCTAACATGGAGGCGGTTGCCACCCTGTGAGCCTGAGGGAGCTGTGTCTGACTGGAACTTCTGTCTGAGGTTTTGCGAAGTCTTACTTATGAATATGGTCTGTCCAGATACCTTGTTTCAAAGGAAGTGAGCATGAGCTAGCAAGTGTAGCCACCCCACAGCTGATAAACAACTTTGTCTTGTTTTTAAATCATCAATCTTCATTTCACATTGGAATAAAGTAAGTGAAACCTGCTACCCGAGCCTCGCCCGTGTGTTCTGTAACCCAGACTCATGTGGTTGTGTGGGCTGTTGTCAGAAATGTTATAAAAAGGTTATGCATAAATTAGATCAAATATAAAATTATGCTTATAATGTCACTTGAGTGGGAGGTAAGAGGGTAGAGTCACAGGAAATCTGTTGGGGTTTACACCCCTGCTACTTACCAAGCTCATGAGAGTGTGGCACTGGTGACCATCACCTGACATTGGTGACAGAAGAGAAAAGGTCGAAGTGAAGGCCAGGTAGGAGAGAGGTGCCAGGCTGTGGGGCCAGGCCCTGCGCATGCTGGGCCTGTTAGGTCACTGAACATCTAACTACCCGGGAACCAGCTCTTTTCACATCATTTGAGGTAAGACGATGGGGGAGCACTCTCCAGAAGTCACACTGCGCTGGGAGAATGGAGGAGAGTCTACATACCGCCATCTTAGGGTAGGTTTTAGATTGAGCTGAACTGTCTTGGAGAGCTAATGAGATGGGAGGAAGACAGTCCCCCAGGTGCACCTAACAGCCAGAGCCTATGAAGTTAGGGGGGTTGTGTGGGGGTGGCCTTTCCCTATAAGAGGAGGAGCTTAAAGCTCTTAAAGCTGGTGGCTGCTGCTCTGCCATCCCTCTACAGAGCAGTCAAGTCCTCAGCTGCAAGAATATCTGAATGTCTTTTGGAGTGTTAGAGTCCTCTGTGTCTTAGAAATTTTGAAAAGAAAAACAAATCTCAATATTAATGTTGATTAGTTTCTCTGAGCCAATTGGGGAAAATAAACGTCCTTCACCTCAAAGGTTTAAGTGACACCGAAGGGTAGCCACCAGTGTCTCGGCCACTGAAGCCTCATGCATGCTCTCACTACCAGTTTGATTTGCAGCCCCATAGTTGTGTTGTACTAAATATTCTTTCCTCTGGCCTTGTCCAGTGAACACGGTTCACATGGCTAACACCACTTCTTGAGATGCGAGCACCATGCAAAGCTGAGAACGGATTGGGTTTTGTGACCATTGTGCCTCCTCCTCACCTGAGAGGCCCATTTTTCCTGGTTGATTCATTAAGTGTATTGGTGCTGTCAGTCGCCTCTGGACAATTGAAATGACAAGTGGCTGTTGATTCATAAAGAAAATGAAGGCTTTAGATGTGAAACCCTCGTTTTCTCTTGTCCTTCTCTTAGGTGAAAGATTTTATTTTTTTCAAAAGGCTACATACTGGTATCCCAGCAGGTGTAGTGTGAGAACTGGCATATGTTAGGCTATGGTGTCAGTGTGGATGGGCAATTCTTCAAGATGGAAAACCAAGTCTCACTGAGTTGCTGGAGCCACACTGACCTTTCTCCACATCCCCCACCATGGGCTTTCACTTTTATCCTGTGCTTGAATTTTTTTCACATACAAATTCTTTATACACACACACAGACACACACACACATATCTCACTCTGTCAATGCAGTGGCTGAATCATGGGTCACTGCATCTTCAAATTCTTAGGCTCCAGTGATGCTTTCAAATCAGCCTCTCAAGTAGCTGGGACTACAGGCATGCAAAGCTACACCCAGACAATTTTTAAATATTTTTCTAGAGACTGAGCCTACTTATGTTGCTCAGACTCGTCTTGCACTCCTGGGATCAAACGATAATCCCACCTTGACCACCCAAAGTGTTTAGATTACAGGTGTGAGCTAGCACTCTCAGCAAAAATATATTTTAAAGAACCGTTACAACCAAATTATGAGTTATCATTATGCCACTGCCCTCCACCCTGGGCACCAGAACAAGACCTTGTATCCAAAAACTAAGCAAAACTAAACAAGAACAAAAAAAAAAAAACTTATAAATAAACTTTGAAGATTGTGTCATCTGTGTCCTTCCCTGCCCTCCAAGCTATCAATGTTAAATATAATGGTTATTGAGAAAATGGTTAGATATTATTAAGAAATTTCTATATATCTTCCAGCTGAGAATAGGTATTCTGTTGTGGCCCAAATATTTTCTCACCGCTACCTTCAGGGTCTAAACTAGCAAATCAGGACACCTGCAGAGGACAGTTGGCCGTTTTCAAATAGAAAGAGAAATACCCCCGTTCATGAGAGTAATCCAGTGATTTTCAAAAAGACAAGTCAGACTGACATGCAGCGCAGTCAGGCCACAATTACCCTGGAATAATCACTTCACACAGAATGGTTGAGGAGACTTTCTAAGATGAGCAAATTTGGGCAGCATAATCCTTGCTTATTTATTCCCAGCCCCCACTGCCCGCCTGATTCCTAATGGCTACCCTACAATGTGGTCAGCAGTGGGATGTAGCGTGGTGAGAGAGGGGCTCAGGGACGGGATGAAGGTCTTTCCTGCATTATCAAAATGCAGGTTAAAAAGTTGTTAAAAAGATGTCCAAATGTTCTAATTCCTACTGTTAAATAGCTGCTAAGATGCATTATACAACAGACCCAGGTAAGGGCGGCCGCTGTAAAGCCTGGGGTGCCTAATGAGTGAGCTAACTCACATTCTTTCTTATCTTG |
| pRC17- circular ssDNA | ATAATAAGGGTAACTATTGCCGGCGAGGCTAGTTACCCTTAAGTTATTGGTATGACTGGTAGTTTGGTGGTGTTGATTTGCCCTTAACCTCACATACCACCTCGAGGAGCTCTCAAGTCCTCAGCTGCAAGAATATCTGAATGTCTTTTGGAGTGTTAGAGTCCTCTGTGTCTTAGAAATTTTGAAAAGAAAAACAAATCTCAATATTAATGTTGATTAGTTTCTCTGAGCCAATTGGGGAAAATAAACGTCCTTCACCTCAAAGGTTTAAGTGACACCGAAGGGTAGCCACCAGTGTCTCGGCCACTGAAGCCTCATGCATGCTCTCACTACCAGTTTGATTTGCAGCCCCATAGTTGTGTTGTACTAAATATTCTTTCCTCTGGCCTTGTCCAGTGAACACGGTTCACATGGCTAACACCACTTCTTGAGATGCGAGCACCATGCAAAGCTGAGAACGGATTGGGTTTTGTGACCATTGTGCCTCCTCCTCACCTGAGAGGCCCATTTTTCCTGGTTGATTCATTAAGTGTATTGGTGCTGTCAGTCGCCTCTGGACAATTGAAATGACAAGTGGCTGTTGATTCATAAAGAAAATGAAGGCTTTAGATGTGAAACCCTCGTTTTCTCTTGTCCTTCTCTTAGGTGAAAGATTTTATTTTTTTCAAAAGGCTACATACTGGTATCCCAGCAGGTGTAGTGTGAGAACTGGCATATGTTAGGCTATGGTGTCAGTGTGGATGGGCAATTCTTCAAGATGGAAAACCAAGTCTCACTGAGTTGCTGGAGCCACACTGACCTTTCTCCACATCCCCCACCATGGGCTTTCACTTTTATCCTGTGCTTGAATTTTTTTCACATACAAATTCTTTATACACACACACAGACACACACACACATATCTCACTCTGTCAATGCAGTGGCTGAATCATGGGTCACTGCATCTTCAAATTCTTAGGCTCCAGTGATGCTTTCAAATCAGCCTCTCAAGTAGCTGGGACTACAGGCATGCAAAGCTACACCCAGACAATTTTTAAATATTTTTCTAGAGACTGAGCCTACTTATGTTGCTCAGACTCGTCTTGCACTCCTGGGATCAAACGATAATCCCACCTTGACCACCCAAAGTGTTTAGATTACAGGTGTGAGCTAGCACTCTCAGCAAAAATATATTTTAAAGAACCGTTACAACCAAATTATGAGTTATCATTATGCCACTGCCCTCCACCCTGGGCACCAGAACAAGACCTTGTATCCAAAAACTAAGCAAAACTAAACAAGAACAAAAAAAAAAAAACTTATAAATAAATTAAACTTTGAAGATTGTGTCATCTGTGTCCTTCCCTGCCCTCCAAGCTATCAATGTTAAATATAATGGTTATTGAGAAAATGGTTAGATATTATTAAGAAATTTCTATATATCTTCCAGCTGAGAATAGGTATTCTGTTGTGGCCCAAATATTTTCTCACCGCTACCTTCAGGGTCTAAACTAGCAAATCAGGACACCTGCAGAGGACAGTTGGCCGTTTTCAAATAGAAAGAGAAATACCCCCGTTCATGAGAGTAATCCAGTGATTTTCAAAAAGACAAGTCAGACTGACATGCAGCGCAGTCAGGCCACAATTACCCTGGAATAATCACTTCACACAGAATGGTTGAGGAGACTTTCTAAGATGAGCAAATTTGGGCAGCATAATCCTTGCTTATTTATTCCCAGCCCCCACTGCCCGCCTGATTCCTAATGGCTACCCTACAATGTGGTCAGCAGTGGGATGTAGCGTGGTGAGAGAGGGGCTCAGGGACGGGATGAAGGTCTTTCCTGCATTATCAAAATGCAGGTTAAAAAGTTGTTAAAAAGATGTCCAAATGTTCTAATTCCTACTGTTAAATAGCTGCTAAGATGCATTATACAACAGACCCAGGTAAGGGAGCTCGGTACCGTCGACCTGCAGAAGCTTAAAAATCCTTTTCTTTCTTATCTTG |
| pCSHI-PB- circular ssDNA | CAGTGAGCGCAACGCAATTAATGTGAGTTAGCTCACTCATTAGGCACCCCAGGCTTTACACTTTATGCTTCCGGCTCGTATGTTGTGTGGAATTGTGAGCGGATAACAATTTCACACAGGAAACAGCTCTGTTGAGATCCAGTTCGATGTAACCCACTTCTGCTCTGATGCCGCATAGCAGTCAGGCCACAATTACCCTGGAATAATCGAAAGGGCCTCGTGATACGCCTATTTTTATAGGTTAATGTCATGATAATAATGGTTTCTTATTAACAGAGTAACCTCCTCAAAGTAATGAGCCTAACGCTCAGCAATTCCCACTTAGACGTCAGGTGGCACTTTTCGGGGAAATGTGCGCGGAACCCCTATTTGTTTATTTTTCTAAATACATTCAAATATGTATCCGCTCATGAGACAATAACCCTGATAAATGCTTCAATAATATTGAAAAAGGAAGAGTATGAGTATTCAACATTTCCGTGTCGCCCTTATTCCCTTTTTTGCGGCATTTTGCCTTCCTGTTTTTGCTCACCCAGAAACGCTGGTGAAAGTAAAAGATGCTGAAGATCAGTTGGGTGCACGAGTGGGTTACATCGAACTGGATCTCAACAGCGGTAAGATCCTTGAGAGTTTTCGCCCCGAAGAACGTTTTCCAATGATGAGCACTTTTAAAGTTCTGCTATGTGGCGCGGTATTATCCCGTATTGACGCCGGGCAAGAGCAACTCGGTCGCCGCATACACTATTCTCAGAATGACTTGGTTGAGTACTCACCAGTCACAGAAAAGCATCTTACGGATGGCATGACAGTAAGAGAATTATGCAGTGCTGCCATAACCATGAGTGATAACACTGCGGCCAACTTACTTCTGACAACGATCGGAGGACCGAAGGAGCTAACCGCTTTTTTGCACAACATGGGGGATCATGTAACTCGCCTTGATCGTTGGGAACCGGAGCTGAATGAAGCCATACCAAACGACGAGCGTGACACCACGATGCCTGTAGCAATGGCAACAACGTTGCGCAAACTATTAACTGGCGAACTACTTACTCTAGCTTCCCGGCAACAATTAATAGACTGGATGGAGGCGGATAAAGTTGCAGGACCACTTCTGCGCTCGGCCCTTCCGGCTGGCTGGTTTATTGCTGATAAATCTGGAGCCGGTGAGCGTGGGTCTCGCGGTATCATTGCAGCACTGGGGCCAGATGGTAAGCCCTCCCGTATCGTAGTTATCTACACGACGGGGAGTCAGGCAACTATGGATGAACGAAATAGACAGATCGCTGAGATAGGTGCCTCACTGATTAAGCATTGGTAACTGTCAGACCAAGTTTACTCATATATACTTTAGATTGATTTAAAACTTCATTTTTAATTTAAAAGGATCTAGGTGAAGATCCTTTTTGATAATCTCATGACCAAAATCCCTTAACGTGAGTTTTCGTTCCACTGAGCGTCAGACCCCGTAGAAAAGATCAAAGGATCTTCTTGAGATCCTTTTTTTCTGCGCGTAATCTGCTGCTTGCAAACAAAAAAACCACCGCTACCAGCGGTGGTTTGTTTGCCGGATCAAGAGCTACCAACTCTTTTTCCGAAGGTAACTGGCTTCAGCAGAGCGCAGATACCAAATACTGTTCTTCTAGTGTAGCCGTAGTTAGGCCACCACTTCAAGAACTCTGTAGCACCGCCTACATACCTCGCTCTGCTAATCCTGTTACCAGTGGCTGCTGCCAGTGGCGATAAGTCGTGTCTTACCGGGTTGGACTCAAGACGATAGTTACCGGATAAGGCGCAGCGGTCGGGCTGAACGGGGGGTTCGTGCACACAGCCCAGCTTGGAGCGAACGACCTACACCGAACTGAGATACCTACAGCGTGAGCTATGAGAAAGCGCCACGCTTCCCGAAGGGAGAAAGGCGGACAGGTATCCGGTAAGCGGCAGGGTCGGAACAGGAGAGCGCACGAGGGAGCTTCCAGGGGGAAACGCCTGGTATCTTTATAGTCCTGTCGGGTTTCGCCACCTCTGACTTGAGCGTCGATTTTTGTGATGCTCGTCAGGGGGGCGGAGCCTATGGAAAAACGCCAGCAACGCGGCCTTTTTACGGTTCCTGGCCTTTTGCTGGCCTTTTGCTCACATGTTCTTTCCTGCGTTATCCCCTGATTCTGTGGATAACCG |
| P rhaBAD | TGTGAACATCATCACGTTCATCTTTCCCTGGTTGCCAATGGCCCATTTTCCTGTCAGTAACGAGAAGGTCGCGAATTCAGGCGCTTTTTAGACTGGTCGTA |
| PlacIq | TGGTGCAAAACCTTTCGCGGTATGGCATGATAGCGCCCGGAAGAGAGTCAATTCAGG |
| Probe 1 | FAM- GATTATTCCAGGGTAATTGTGGCCTGACTG |
| Probe 2 | ROX- AGTGGGTTACATCGAACTGGATCTCAACAG |
| Probe 3 | ROX-CAGTCAGGCCACAATTACCCTGGAATAATC |
| Donor 1 | A*T*GATTACGGATTCACTGGCCGTCGTTTTACAACGTCGTGACTGAGAAAACCCTGGCGTTACCCAACTTAATCGCCTTGCAGCACATC*C*C |
| Donor 2 | G*G*GGGTACGGATTCACTGGCCGTCGTTTTACAACGTCGTGACTGAGAAAACCCTGGCGTTACCCAACTTAATCGCCTTGCAGCACTTT*T*T |
| gRNA2 | GTTGTGAGTTTGAAGTTGATGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTG |
| circular ssDNA for 11 nts substitution | AAAAATCCTTTTCTTTCTTATCTTGATAATAAGGGTAACTATTGCCGGCGAGGCTAGTTACCCTTAAGTTATTGGTATGACTGGTAGTTTGGTGGTGTTGATTTGCCCTTAACCTCACATACCACCTCGAGTTACCTTCACATCCTACCTCGAATTCAAAAAAAAAAATCAACTTCAAACTCACAACAAAAAAAAAAAGATCTAAGCTTATCGATGTGCCGGAAAGCTGGCTGGAGTGCGATCTTCCTGAGGCCGATACTGTCGTCGTCCCCTCAAACTAGTAGTAGTAGGGTTACGATGCGCCCATCTACACCAACGTGACCTATCCCATTACGGTCAATCCGCCGTTTGTTTCTAGACTGCAGAAAAAAAAAAATCAACTTCAAACTCACAACAAAAAAAAAAGAATTCACTCTACTACCTGCCTTACCAAGCTT |
| circular ssDNA for 1011 nts insertion | AAAAATCCTTTTCTTTCTTATCTTGATAATAAGGGTAACTATTGCCGGCGAGGCTAGTTACCCTTAAGTTATTGGTATGACTGGTAGTTTGGTGGTGTTGATTTGCCCTTAACCTCACATACCACCTCGAGTTACCTTCACATCCTACCTCGAATTCAAAAAAAAAAATCAACTTCAAACTCACAACAAAAAAAAAAAGATCTAAGCTTATCGATGTGCCGGAAAGCTGGCTGGAGTGCGATCTTCCTGAGGCCGATACTGTCGTCGTCCCCTCAAACTATTTGTCCTACTCAGGAGAGCGTTCACCGACAAACAACAGATAAAACGAAAGGCCCAGTCTTTCGACTGAGCCTTTCGTTTTATTTGATGCCTCTAGCACGCGTACCATGGGATCCCCCATCAAGCTTATTAAACTCGTCTTCTGTTCGCCTGGCCCGCGTTCAGCATAAAGGTCGGCACTTCGTTGGTGTTTTCTTCGTTTTTGTTCGCCGCTTTGTATAGTTCATCCATGCCATGTGTAATCCCAGCAGCTGTTACAAACTCAAGAAGGACCATGTGGTCTCTCTTTTCGTTGGGATCTTTCGAAAGGGCAGATTGTGTGGACAGGTAATGGTTGTCTGGTAAAAGGACAGGGCCATCGCCAATTGGAGTATTTTGTTGATAATGGTCTGCTAGTTGAACGCTTCCATCTTCAATGTTGTGTCTAATTTTGAAGTTAACTTTGATTCCATTCTTTTGTTTGTCTGCCATGATGTATACATTGTGTGAGTTATAGTTGTATTCCAATTTGTGTCCAAGAATGTTTCCATCTTCTTTAAAATCAATACCTTTTAACTCGATTCTATTAACAAGGGTATCACCTTCAAACTTGACTTCAGCACGTGTCTTGTAGTTCCCGTCATCTTTGAAAAATATAGTTCTTTCCTGTACGTAACCTTCGGGCATGGCACTCTTGAAAAAGTCATGCTGTTTCATATGATCTGGGTATCTCGCAAAGCATTGAACACCATAACCGAAAGTAGTGACAAGTGTTGGCCATGGAACAGGTAGTTTTCCAGTAGTGCAAATAAATTTAAGGGTAAGTTTTCCGTATGTTGCATCACCTTCACCCTCTCCACTGACAGAAAATTTGTGCCCATTAACATCACCATCTAATTCAACAAGAATTGGGACAACTCCAGTGAAAAGTTCTTCTCCTTTACGCATGGTACCTTTGTCCTCTTTAATGAATTCCCTGAATTGACTCTCTTCCGGGCGCTATCATGCCATACCGCGAAAGGTTTTGCACCAGGTTACGATGCGCCCATCTACACCAACGTGACCTATCCCATTACGGTCAATCCGCCGTTTGTTTCTAGACTGCAGAAAAAAAAAAATCAACTTCAAACTCACAACAAAAAAAAAAGAATTCACTCTACTACCTGCCTTACCAAGCTT |
| Donor ssDNA for 11 nts substitution | AACAAAAAAAAAAAGATCTAAGCTTATCGATGTGCCGGAAAGCTGGCTGGAGTGCGATCTTCCTGAGGCCGATACTGTCGTCGTCCCCTCAAACTAGTAGTAGTAGGGTTACGATGCGCCCATCTACACCAACGTGACCTATCCCATTACGGTCAATCCGCCGTTTGTTTCTAGACTGCAGAAAAAAAAAAATCAACTTCAAACTCAC |
| Donor ssDNA for 1011 nts insertion | AACAAAAAAAAAAAGATCTAAGCTTATCGATGTGCCGGAAAGCTGGCTGGAGTGCGATCTTCCTGAGGCCGATACTGTCGTCGTCCCCTCAAACTATTTGTCCTACTCAGGAGAGCGTTCACCGACAAACAACAGATAAAACGAAAGGCCCAGTCTTTCGACTGAGCCTTTCGTTTTATTTGATGCCTCTAGCACGCGTACCATGGGATCCCCCATCAAGCTTATTAAACTCGTCTTCTGTTCGCCTGGCCCGCGTTCAGCATAAAGGTCGGCACTTCGTTGGTGTTTTCTTCGTTTTTGTTCGCCGCTTTGTATAGTTCATCCATGCCATGTGTAATCCCAGCAGCTGTTACAAACTCAAGAAGGACCATGTGGTCTCTCTTTTCGTTGGGATCTTTCGAAAGGGCAGATTGTGTGGACAGGTAATGGTTGTCTGGTAAAAGGACAGGGCCATCGCCAATTGGAGTATTTTGTTGATAATGGTCTGCTAGTTGAACGCTTCCATCTTCAATGTTGTGTCTAATTTTGAAGTTAACTTTGATTCCATTCTTTTGTTTGTCTGCCATGATGTATACATTGTGTGAGTTATAGTTGTATTCCAATTTGTGTCCAAGAATGTTTCCATCTTCTTTAAAATCAATACCTTTTAACTCGATTCTATTAACAAGGGTATCACCTTCAAACTTGACTTCAGCACGTGTCTTGTAGTTCCCGTCATCTTTGAAAAATATAGTTCTTTCCTGTACGTAACCTTCGGGCATGGCACTCTTGAAAAAGTCATGCTGTTTCATATGATCTGGGTATCTCGCAAAGCATTGAACACCATAACCGAAAGTAGTGACAAGTGTTGGCCATGGAACAGGTAGTTTTCCAGTAGTGCAAATAAATTTAAGGGTAAGTTTTCCGTATGTTGCATCACCTTCACCCTCTCCACTGACAGAAAATTTGTGCCCATTAACATCACCATCTAATTCAACAAGAATTGGGACAACTCCAGTGAAAAGTTCTTCTCCTTTACGCATGGTACCTTTGTCCTCTTTAATGAATTCCCTGAATTGACTCTCTTCCGGGCGCTATCATGCCATACCGCGAAAGGTTTTGCACCAGGTTACGATGCGCCCATCTACACCAACGTGACCTATCCCATTACGGTCAATCCGCCGTTTGTTTCTAGACTGCAGAAAAAAAAAAATCAACTTCAAACTCAC |
| Seq-11 nts substitution | TCCGTGCGCTCTCCGCCACATATCCTGATCTTCCAGATAACTGCCGTCACTCCAGCGCAGCACCATCACCGCGAGGCGGTTTTCTCCGGCGCGTAAAAATGCGCTCAGGTCAAATTCAGACGGCAAACGACTGTCCTGGCCGTAACCGACCCAGCGCCCGTTGCACCACAGATGAAACGCCGAGTTAACGCCATCAAAAATAATTCGCGTCTGGCCTTCCTGTAGCCAGCTTTCATCAACATTAAATGTGAGCGAGTAACAACCCGTCGGATTCTCCGTGGGAACAAACGGCGGATTGACCGTAATGGGATAGGTCACGTTGGTGTAGATGGGCGCATCGTAACCCTACTACTACTAGTTTGAGGGGACGACGACAGTATCGGCCTCAGGAAGATCGCACTCCAGCCAGCTTTCCGGCACCGCTTCTGGTGCCGGAAACCAGGCAAAGCGCCATTCGCCATTCAGGCTGCGCAACTGTTGGGAAGGGCGATCGGTGCGGGCCTCTTCGCTATTACGCCAGCTGGCGAAAGGGGGATGTGCTGCAAGGCGATTAAGTTGGGTAACGCCAGGGTTTTCCCAGTCACGACGTTGTAAAACGACGGCCAGTGAATCCGTAATCATGGTCATAGCTGTTTCCTGTGTGAAATTGTTATCCGCTCACAATTCCACACAACATACGAGCCGGAAGCATAAAGTGTAAAGCCTGGGGTGCCTAATGAGTGAGCTAACTCACATTAATTGCGTTGCGCTCACTGCCCGCTTTCCAGTCGGGAAACCTGTCGTGCCAGCTGTTATTATTGGAATCAAA |
| Seq-1011 nts insertion | CCGGACCGCCGCTTATCCGCCACTTATCCTGATCTTCCAGATAACTGCCGTCACTCCAGCGCAGCACCATCACCGCGAGGCGGTTTTCTCCGGCGCGTAAAAATGCGCTCAGGTCAAATTCAGACGGCAAACGACTGTCCTGGCCGTAACCGACCCAGCGCCCGTTGCACCACAGATGAAACGCCGAGTTAACGCCATCAAAAATAATTCGCGTCTGGCCTTCCTGTAGCCAGCTTTCATCAACATTAAATGTGAGCGAGTAACAACCCGTCGGATTCTCCGTGGGAACAAACGGCGGATTGACCGTAATGGGATAGGTCACGTTGGTGTAGATGGGCGCATCGTAACCTGTTGCAAAACCTTTCGCGGTATGGCATGATAGCGCCCGGAAGAGAGTCAATTCAGGGAATTCATTAAAGAGGACAAAGGTACCATGCGTAAAGGAGAAGAACTTTTCACTGGAGTTGTCCCATTTCTTTGTTGAA |
References
- Urnov, F.D. Genome Editing BC (before CRISPR): lasting lessons from the “old testament”. Cris. J. 2018, 1, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Knott, G.J.; Doudna, J.A. CRISPR-Cas guides the future of genetic engineering. Science 2018, 361, 866–869. [Google Scholar] [CrossRef] [PubMed]
- Glass, Z.; Lee, M.; Li, Y.; Xu, Q. Engineering the delivery system for CRISPR-based genome editing. Trends Biotechnol. 2018, 36, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Hegge, J.W.; Swarts, D.C.; van der Oost, J. Prokaryotic Argonaute proteins: novel genome-editing tools? Nat. Rev. Microbiol. 2018, 16, 5. [Google Scholar] [CrossRef] [PubMed]
- Codner, G.F.; Mianné, J.; Caulder, A.; Loeffler, J.; Fell, R.; King, R.; Allan, A.J.; Mackenzie, M.; Pike, F.J.; McCabe, C.V. Application of long single-stranded DNA donors in genome editing: Generation and validation of mouse mutants. BMC Biol. 2018, 16, 70. [Google Scholar] [CrossRef]
- Zhang, J.-P.; Li, X.-L.; Li, G.-H.; Chen, W.; Arakaki, C.; Botimer, G.D.; Baylink, D.; Zhang, L.; Wen, W.; Fu, Y.-W. Efficient precise knockin with a double cut HDR donor after CRISPR/Cas9-mediated double-stranded DNA cleavage. Genome Biol. 2017, 18, 35. [Google Scholar] [CrossRef]
- Li, Y.; Lin, Z.; Huang, C.; Zhang, Y.; Wang, Z.; Tang, Y.J.; Chen, T.; Zhao, X. Metabolic engineering of Escherichia coli using CRISPR-Cas9 meditated genome editing. Metab. Eng. 2015, 31, 13–21. [Google Scholar] [CrossRef]
- Rong, Z.; Zhu, S.; Xu, Y.; Fu, X. Homologous recombination in human embryonic stem cells using CRISPR/Cas9 nickase and a long DNA donor template. Protein Cell 2014, 5, 258–260. [Google Scholar] [CrossRef]
- Richardson, C.D.; Ray, G.J.; DeWitt, M.A.; Curie, G.L.; Corn, J.E. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat. Biotechnol. 2016, 34, 339–344. [Google Scholar] [CrossRef]
- Chen, F.; Pruett-Miller, S.M.; Huang, Y.; Gjoka, M.; Duda, K.; Taunton, J.; Collingwood, T.N.; Frodin, M.; Davis, G.D. High-frequency genome editing using ssDNA oligonucleotides with zinc-finger nucleases. Nat. Methods 2011, 8, 753–755. [Google Scholar] [CrossRef]
- Ellis, H.M.; Yu, D.; DiTizio, T. High efficiency mutagenesis, repair, and engineering of chromosomal DNA using single-stranded oligonucleotides. Proc. Natl. Acad. Sci. USA 2001, 98, 6742–6746. [Google Scholar] [CrossRef] [PubMed]
- Sharan, S.K.; Thomason, L.C.; Kuznetsov, S.G.; Court, D.L. Recombineering: a homologous recombination-based method of genetic engineering. Nat. Protoc. 2009, 4, 206–223. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.H. Programming cells by multiplex genome engineering and accelerated evolution. Nature 2009, 460, 894–898. [Google Scholar] [CrossRef] [PubMed]
- Chung, M.E.; Yeh, I.H.; Sung, L.Y.; Wu, M.Y.; Chao, Y.P.; Ng, I.S.; Hu, Y.C. Enhanced integration of large DNA into E. coli chromosome by CRISPR/Cas9. Biotechnol. Bioeng. 2017, 114, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Xi, A.G.; Hunter, C.P. Efficient Homologous Recombination in Mice Using Long Single Stranded DNA and CRISPR Cas9 Nickase. G3: GenesGenomesGenet. 2019, 9, 281–286. [Google Scholar] [CrossRef]
- Miura, H.; Gurumurthy, C.B.; Sato, T.; Sato, M.; Ohtsuka, M. CRISPR/Cas9-based generation of knockdown mice by intronic insertion of artificial microRNA using longer single-stranded DNA. Sci. Rep. 2015, 5, 12799. [Google Scholar] [CrossRef]
- Quadros, R.M.; Miura, H.; Harms, D.W.; Akatsuka, H.; Sato, T.; Aida, T.; Redder, R.; Richardson, G.P.; Inagaki, Y.; Sakai, D. Easi-CRISPR: a robust method for one-step generation of mice carrying conditional and insertion alleles using long ssDNA donors and CRISPR ribonucleoproteins. Genome Biol. 2017, 18, 92. [Google Scholar] [CrossRef]
- Li, H.; Beckman, K.A.; Pessino, V.; Huang, B.; Weissman, J.S.; Leonetti, M.D. Design and specificity of long ssDNA donors for CRISPR-based knock-in. BioRxiv 2017. [Google Scholar] [CrossRef]
- Costantino, N. Enhanced levels of λ Red-mediated recombinants in mismatch repair mutants. Proc. Natl. Acad. Sci. USA 2003, 100, 15748–15753. [Google Scholar] [CrossRef] [PubMed]
- Veneziano, R.; Shepherd, T.R.; Ratanalert, S.; Bellou, L.; Tao, C.; Bathe, M. In vitro synthesis of gene-length single-stranded DNA. Sci Reports 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Kishi, J.Y.; Schaus, T.E.; Gopalkrishnan, N.; Xuan, F.; Yin, P. Programmable autonomous synthesis of single-stranded DNA. Nat. Chem. 2018, 10, 155. [Google Scholar] [CrossRef]
- Zhao, W.; Gao, Y.; Brook, M.A.; Li, Y. Wrapping single-walled carbon nanotubes with long single-stranded DNA molecules produced by rolling circle amplification. Chem. Commun. 2006, 34, 3582–3584. [Google Scholar] [CrossRef] [PubMed]
- Praetorius, F.; Kick, B.; Behler, K.L.; Honemann, M.N.; Weuster-Botz, D.; Dietz, H. Biotechnological mass production of DNA origami. Nature 2017, 552, 84. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Dong, Q.; Ma, R.; Chen, Y.; Yang, J.; Sun, L.-Z.; Huang, C. Rapid isolation of highly pure single-stranded DNA from phagemids. Anal. Biochem. 2009, 389, 177–179. [Google Scholar] [CrossRef] [PubMed]
- Nafisi, P.M.; Aksel, T.; Douglas, S.M. Construction of a novel phagemid to produce custom DNA origami scaffolds. Synthetic Biology 2018, 3, ysy015. [Google Scholar] [CrossRef]
- Shepherd, T.R.; Du, R.R.; Huang, H.; Wamhoff, E.-C.; Bathe, M. Bioproduction of pure, kilobase-scale single-stranded DNA. Sci. Rep. 2019, 9, 6121. [Google Scholar] [CrossRef]
- Horinouchi, S.; Weisblum, B. Nucleotide sequence and functional map of pE194, a plasmid that specifies inducible resistance to macrolide, lincosamide, and streptogramin type B antibodies. J. Bacteriol. 1982, 150, 804–814. [Google Scholar] [CrossRef]
- Horinouchi, S.; Weisblum, B. Nucleotide sequence and functional map of pC194, a plasmid that specifies inducible chloramphenicol resistance. J. Bacteriol. 1982, 150, 815–825. [Google Scholar] [CrossRef]
- Khan, S.A.; Novick, R.P. Complete nucleotide sequence of pT181, a tetracycline-resistance plasmid from Staphylococcus aureus. Plasmid 1983, 10, 251–259. [Google Scholar] [CrossRef]
- McKenzie, T.; Hoshino, T.; Tanaka, T.; Sueoka, N. The nucleotide sequence of pUB110: some salient features in relation to replication and its regulation. Plasmid 1986, 15, 93–103. [Google Scholar] [CrossRef]
- Projan, S.J.; Kornblum, J.; Moghazeh, S.L.; Edelman, I.; Gennaro, M.L.; Novick, R.P. Comparative sequence and functional analysis of pT181 and pC221, cognate plasmid replicons from Staphylococcus aureus. Mol. Gen. Genet. Mgg 1985, 199, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.A. Plasmid rolling-circle replication: highlights of two decades of research. Plasmid 2005, 53, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Goze, A.; Ehrlich, S. Replication of plasmids from Staphylococcus aureus in Escherichia coli. Proc. Natl. Acad. Sci. USA 1980, 77, 7333–7337. [Google Scholar] [CrossRef] [PubMed]
- Te Riele, H.; Michel, B.; Ehrlich, S.D. Are single-stranded circles intermediates in plasmid DNA replication? Embo J. 1986, 5, 6. [Google Scholar] [CrossRef]
- Gros, M.F. Replication origin of a single-stranded DNA plasmid pC194. EMBO J. 1989, 8. [Google Scholar] [CrossRef]
- Farzadfard, F.; Lu, T.K. Genomically encoded analog memory with precise in vivo DNA writing in living cell populations. Science 2014, 346, 1256272. [Google Scholar] [CrossRef]
- Gros, M.F.; Te Riele, H.; Ehrlich, S. D Rolling circle replication of single-stranded DNA plasmid pC194. Embo J. 1987, 6, 6. [Google Scholar] [CrossRef]
- Enbo, M.; Harrington, L.B.; O’Connell, M.R.; Kaihong, Z.; Doudna, J.A. Single-Stranded DNA Cleavage by Divergent CRISPR-Cas9 Enzymes. Mol. Cell 2015, 60, 398–407. [Google Scholar] [CrossRef]
- Briner, A.E.; Donohoue, P.D.; Gomaa, A.A.; Selle, K.; Slorach, E.M.; Nye, C.H.; Haurwitz, R.E.; Beisel, C.L.; May, A.P.; Barrangou, R. Guide RNA functional modules direct Cas9 activity and orthogonality. Mol. Cell 2014, 56, 333–339. [Google Scholar] [CrossRef]
- Dirks, R.M.; Bois, J.S.; Schaeffer, J.M.; Winfree, E.; Pierce, N.A. Thermodynamic Analysis of Interacting Nucleic Acid Strands. Siam Rev. 2007, 49, 65–88. [Google Scholar] [CrossRef]
- Bruand, C.; Ehrlich, S.D. UvrD-dependent replication of rolling-circle plasmids in Escherichia coli. Mol. Microbiol. 2000, 35, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Lohman, T.M.; Ferrari, M.E. Escherichia coli single-stranded DNA-binding protein: multiple DNA-binding modes and cooperativities. Annu. Rev. Biochem. 1994, 63, 527–570. [Google Scholar] [CrossRef] [PubMed]
- Elbaz, J.; Yin, P.; Voigt, C.A. Genetic encoding of DNA nanostructures and their self-assembly in living bacteria. Nat. Commun. 2016, 7, 11179. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Huang, J.; Yang, X.; He, X.; Quan, K.; Xie, N.; Ou, M.; Wang, K. Gold nanoparticle based hairpin-locked-DNAzyme probe for amplified miRNA imaging in living cells. Anal. Chem. 2017, 89, 5850–5856. [Google Scholar] [CrossRef]
- Morrison, D.; Rothenbroker, M.; Li, Y. DNAzymes: selected for applications. Small Methods 2018, 2, 1700319. [Google Scholar] [CrossRef]
- Chen, J.S.; Ma, E.; Harrington, L.B.; Da Costa, M.; Tian, X.; Palefsky, J.M.; Doudna, J.A. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science 2018, 360, 436–439. [Google Scholar] [CrossRef]
- Li, S.-Y.; Cheng, Q.-X.; Liu, J.-K.; Nie, X.-Q.; Zhao, G.-P.; Wang, J. CRISPR-Cas12a has both cis-and trans-cleavage activities on single-stranded DNA. Cell Res. 2018, 28, 491. [Google Scholar] [CrossRef]
- Hughes, R.A.; Ellington, A.D. Synthetic DNA synthesis and assembly: putting the synthetic in synthetic biology. Cold Spring Harbor Perspect. Biol. 2017, 9, a023812. [Google Scholar] [CrossRef]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef]
- Berti, M.; Vindigni, A. Replication stress: Getting back on track. Nat. Struct. Mol. Biol. 2016, 23, 103–109. [Google Scholar] [CrossRef]
- Inoue, H.; Nojima, H.; Okayama, H. High efficiency transformation of Escherichia coli with plasmids. Gene 1990, 96, 23–28. [Google Scholar] [CrossRef]
- Gerling, T.; Wagenbauer, K.F.; Neuner, A.M.; Dietz, H. Dynamic DNA devices and assemblies formed by shape-complementary, non-base pairing 3D components. Science 2015, 347, 1446–1452. [Google Scholar] [CrossRef] [PubMed]
- Simpson, R.J. Large-scale extraction of recombinant proteins from bacteria. Cold Spring Harbor Protocols 2010, 2010, pdb prot5484. [Google Scholar] [CrossRef]
- Jiang, Y.-X.; Wu, J.-G.; Yu, K.-Q.; Ai, C.-X.; Zou, F.; Zhou, H.-W. Integrated lysis procedures reduce extraction biases of microbial DNA from mangrove sediment. J. Biosci. Bioeng. 2011, 111, 153–157. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hao, M.; Wang, Z.; Qiao, H.; Yin, P.; Qiao, J.; Qi, H. Dynamic Genome Editing Using In Vivo Synthesized Donor ssDNA in Escherichia coli. Cells 2020, 9, 467. https://doi.org/10.3390/cells9020467
Hao M, Wang Z, Qiao H, Yin P, Qiao J, Qi H. Dynamic Genome Editing Using In Vivo Synthesized Donor ssDNA in Escherichia coli. Cells. 2020; 9(2):467. https://doi.org/10.3390/cells9020467
Chicago/Turabian StyleHao, Min, Zhaoguan Wang, Hongyan Qiao, Peng Yin, Jianjun Qiao, and Hao Qi. 2020. "Dynamic Genome Editing Using In Vivo Synthesized Donor ssDNA in Escherichia coli" Cells 9, no. 2: 467. https://doi.org/10.3390/cells9020467
APA StyleHao, M., Wang, Z., Qiao, H., Yin, P., Qiao, J., & Qi, H. (2020). Dynamic Genome Editing Using In Vivo Synthesized Donor ssDNA in Escherichia coli. Cells, 9(2), 467. https://doi.org/10.3390/cells9020467