Abstract
Here, we provide an overview of the importance of cellular fate in cancer as a group of diseases of abnormal cell growth. Tumor development and progression is a highly dynamic process, with several phases of evolution. The existing evidence about the origin and consequences of cancer cell fate specification (e.g., proliferation, senescence, stemness, dormancy, quiescence, and cell cycle re-entry) in the context of tumor formation and metastasis is discussed. The interplay between these dynamic tumor cell phenotypes, the microenvironment, and the immune system is also reviewed in relation to cancer. We focus on the role of senescence during cancer progression, with a special emphasis on its relationship with stemness and dormancy. Selective interventions on senescence and dormancy cell fates, including the specific targeting of cancer cell populations to prevent detrimental effects in aging and disease, are also reviewed. A new conceptual framework about the impact of synthetic lethal strategies by using senogenics and then senolytics is given, with the promise of future directions on innovative anticancer therapies.
1. Introduction
Natural tumor evolution is a complex process, composed of multiple steps (cell-intrinsic tumorigenesis, tumor growth, invasion, and metastasis), cellular phenotypes, microenvironmental treats, and immune system interplay. Pharmacological treatment just adds more complexity to this evolution by the appearance, selection, and exacerbation of specific phenotypes, including senescent tumor cells, quiescent tumor cells, and cancer stem cells. Among these, a new cellular outcome named dormancy has been proposed. Cells in dormancy may promote a more lethal profile relapse of tumor growth, even after many silent years or decades. There is now a large body of clinical and experimental evidence to accept the existence of tumor cell dormancy; however, there are still a number of questions to be addressed about the nature of this kind of cell, including its origin, evolution, and nature. One of the aims of this review is to attempt to understand the nature of dormant tumor cells through the knowledge that we currently have about other tumor cell phenotypes; in particular, from the state-of-the-art on cancer stem cells, because these two phenotypes share some similar characteristics, and on senescence, because senescence is a primary response to pharmacological treatment in cancer (despite apoptosis) and it strongly influences the regulation of stem-like phenotypes.
Since their discovery, cancer stem cells (CSC) have gained a lot of attention, and extensive research has been focused on CSCs since they are not only highly resistant to conventional chemotherapy, but also possess the capacity to regrow a complete tumor after clinical intervention. This last capacity is due to their intrinsic self-renewal capacity. CSCs exist in a most undifferentiated state within tumors; however, there is no consensus about the origin of CSCs. It is proposed that they arise from normal adult stem cells, obtaining the capacity to grow as a tumor by a mutation on specific genes (reviewed in [1]).
The rapid advances in cellular senescence—a highly relevant phenotype in physiology and disease widely involved in eukaryotic organism physiology—make it difficult to keep up with and integrate many of the key concepts and developments. Depending on the biological context, senescence can be a beneficial or deleterious cellular outcome. Senescence is a natural intrinsic response of cells against stress situations, and its activation avoids the proliferation of potentially malignant cells in an irreversible fashion, so it has been considered a primary tumor suppressor mechanism [2]. Senescence is also associated with the resolution of fibrosis in a mechanism that includes senescent cell recognition by the immune system [3]. In addition, embryonic developmental senescence has been observed to participate in tissue remodeling and the formation of macro structures like limbs or mesonephros (reviewed in [4]). On the other hand, senescence accumulation in tissues promotes a state of chronic inflammation linked with a reduced physiological fitness during aging (reviewed in [5]). This inflammatory microenvironment, in combination with the growth factors produced by senescent cells, may promote the proliferation of non-senescent tumor cells or the acquisition of the most aggressive phenotypes like cancer stemness (reviewed in [6]), or, as we propose, cells with the ability to produce tumor regrowth in cancer patients after years of disease-free survival.
Another non-proliferative but harmful phenotype is quiescence. However, as opposed to senescence, quiescence is characterized by reversible cell cycle arrest, promoting, among other characteristics, a high resistance to toxic stimuli, including cancer therapies [7]. In a tumor context, it has been proposed that this state is the prevalent state in the CSC phenotype and putatively on dormant cells. With respect to this view, it has been proposed that dormant cells are a special case of stem cells in a quiescence state. However, based on the cancer evolution fundament, we propose that senescence could act as a source of dormant tumor cells. Therefore, the general aim of this work is to provide a comprehensive perspective on the definition of the fate of tumor cells (senescent or not) and to highlight the translational potential of therapeutic avenues, primarily based on manipulating cellular senescence.
2. Cancer Stem Cells
Stem cells possess a self-renewal capacity, give rise to progeny capable of differentiating into other cell types [8,9,10], and hold a high cell plasticity emerging from specific pluripotency genetic programs [11,12,13]. Small populations of cells with active pluripotency programs and a high plasticity, known as cancer stem cells (CSCs), exist in tumors [14,15,16,17]. CSCs were characterized for the first time in acute myeloid leukemia (AML), in which cells with the CD34+/CD38− phenotype exhibited proliferative and self-renewal capacities similar to those of primary stem cells [18]. It is now widely accepted that all tumors have CSCs, although it is still unclear whether these cells arise from primary development defects and are tumor initializers [19,20], or if they evolve from a somatic tumor cell that acquires stemness characteristics [21]. For example, a CSC-like phenotype can be acquired by epithelial-mesenchymal transition (EMT) programs [14,22] or by escaping from senescence [23]. Pluripotency and cellular plasticity promote new physiological traits conferring drug resistance [24,25] and/or angiogenic, invasive, and metastatic potentials [26,27,28].
3. Senescence and Stemness Are Relevant in Cancer Evolution
3.1. The Senescent Phenotype
Cellular senescence is a non-proliferative steady state that can be acquired by both primary [2,29,30,31] and malignant cells [32,33,34]. The susceptibility of primary stem cells to enter into a state of senescence was recently described in muscle cells, pituitary tumor cells, and mesenchymal stem cells [35,36,37]. Senescence cell cycle arrest is mainly (but not only) promoted by a sustained DNA damage response (DDR); by oncogenic activation; and by other stress conditions that dramatically change the morphology, gene expression, secretory program [2,34,38,39,40,41]. Senescent cells exhibit a complex secretome (in many cases, a denominated Senescence Associated Secretory Phenotype (SASP)) composed of growth factors, cytokines, chemokines, bioactive lipids, and extracellular matrix remodelers that plays a critical role in the cell cycle, immune response, and tissue remodeling, depending on the physiopathological context [42,43,44].
3.2. Senescence Promotes Intrinsic and Paracrine Stemness
Senescence promotes stem-like reprogramming (a set of pluripotency and stem cell features) in an autocrine and paracrine way. In the last few years, the relationship between senescence and stemness has been intensively explored, including the critical regulation of specific functions of stem cells that drives them into replicative senescence in multiple tissues, including those of the hematopoietic system, intestine, muscle, brain, skin, and germline. Stemness is regulated by a number of signals and genes that influence tumor development (Table 1). It is interesting to note that senescence was described as a physiological mechanism in embryonic development [45,46] and as a mechanism of tissue regeneration [47,48,49]. Likewise, senescent cells from Oncogene-Induced Senescence (OIS) express stem cell-related genes. However, due to their cell cycle arrest, they do not develop clonogenicity or a self-renewal capacity. The SASP, or senescent secretome, promotes an environment in which neighboring cells are favored to express stemness markers [49]. In the same way, SASP can enhance the genetic induction of pluripotency in induced pluripotent stem cell (iPSC) generation in vivo with reprogramming factors [50,51]. Experimental evidence suggests that the accumulation of senescent cells promotes paracrine tumorigenesis through their secretome and favors the appearance of more aggressive stem-like phenotypes [52]. Conditioned media from senescent cells promotes clonogenicity and cancer stemness [53] and the development of subpopulations of cells resistant to chemotherapy [52,54]. As previously mentioned, tumor cells that escape from senescence also acquire preeminent tumorigenicity, drug resistance, and other characteristics of CSCs in a process called senescence-associated stemness (SAS), promoted by the loss of tumor suppressors and the activation of conserved embryonic development pathways as Wnt signaling [23] (see Table 1 for genetic pathways associated with stemness).

Table 1.
Origins and consequences of cancer cell fate: proliferation, senescence, stemness, dormancy, quiescence, and cell cycle re-entry.
3.3. Senescence Induction on Stem Cells and Cancer Stem Cells
Previously, it was believed that only proliferative cells were able to enter in senescence, which was considered an intrinsic barrier preventing reprogramming to a more dynamic state [55,56]; however, it is now well-recognized that primary (muscle) stem cells (low proliferative capacity cells) can shift from quiescence to senescence during aging in a process known as geroconversion (henceforth referred to as senoconversion), which is dependent on the overexpression of the tumor suppressor cyclin-dependent kinase Inhibitor 2A Cdkn2a (p16), a well-characterized senescence biomarker that acts by slowing the progression of the cell cycle from the G1 phase to the S phase [35] (see Figure 1). Likewise, the failure of autophagy caused by aging or genetic mutations in cells from young donors also induces senescence in muscle stem cells [57]. Other putative mechanisms inducing senescence in primary and cancer stem cells include DNA damage by chemotherapeutics [37], hypoxia [58], oncogene activation (oncogenic β-catenin) [36], and the dysregulation of microenvironmental growth factors such as TGF-β (transforming growth factor beta) or BMP7 (bone morphogenetic protein 7) [59,60]. In addition to SASP, other factors might be required to induce senescence. The Notch signaling pathway—a highly conserved system mediated through cell-to-cell contact (juxtacrine)—not only has a different gene transcription signature than primary OIS, but is also an essential driver of secondary senescence, a distinct molecular endpoint from OIS [61]. In summary, tumor suppressor and oncogenic pathways regulate senescence and stemness interplay, influencing the microenvironment and transforming cancer cells in heterogeneous populations, according to their cell cycle state.
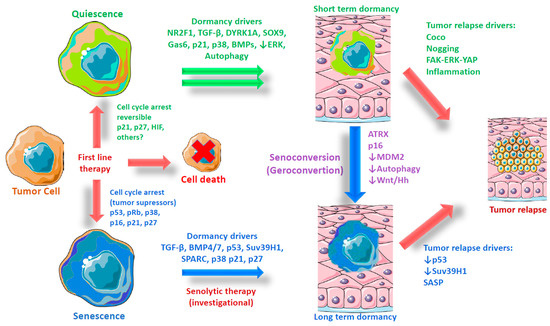
Figure 1.
Origin and evolution of dormant tumor cells. Graphical representation of the evolution of tumor dormancy according to two models—quiescence and senescence—including possible triggers and driver genes. Quiescence is the most accepted model for tumor cell dormancy. This model is based on the observation of some molecular triggers in the microenvironment and final effectors that regulate cell cycling. Dormant cells have been observed to express markers such as a low ERK/ high p38 ratio during the activation of other genes like NR2F1 and DYRK1A. However, in some models of dormancy, senescence markers have been noted. This observation, plus the activation genes during senescence shared with quiescence (i.e., TGF-β, BMPs, p21, and p27), leads us to propose an alternative model of tumor dormancy in which senescence is a major phenotype acquired during long-term dormancy. If this assumption is true, it opens the possibility of the use of senolytics to avoid metastatic recurrence.
4. Role of Tumor Suppressor and Oncogenic Pathways in Senescence
Since senescent cells are non-proliferative, senescence has been considered an intrinsic mechanism of tumor suppression primarily characterized by the activation of “tumor suppressor pathways” p53–p21 or p38–p16–pRb [2,62,63] (Figure 1). Senescent cells are present in premalignant lesions or primary stages of tumorigenesis; however, senescent cells disappear in the late stage of tumor development [31,33,63]. For these reasons, senescence is thought to be essential for complete oncogenic transformation [63,64]. Likewise, the loss of some oncogenes in developed tumors promotes senescence and rapid tumor size regression, while the specific abrogation of some senescence pathways fails to avoid tumoral regrowth [65,66]. For example, in primary mouse embryo astrocytes, Ras activation is not linked to senescence, supporting the possibility that oncogene activation may not be a widespread mechanism [67]. The modulation of SASP in prostate tumors is a promising strategy to elicit tumor suppression. In vivo, Phosphatase and tensin homolog (PTEN)-null senescent cells secrete cytokines through the Jak2/Stat3 pathway; promote the infiltration of immunostimulatory CD8+ T cells, NK cells, and B cells; and decrease the tumor size and invasion capacity [68]. The pharmacological inhibition of Jak2/Stat3 leads to an antitumor immune response that enhances the efficacy of chemotherapy, suggesting that the oncogene PTEN is involved in immunomodulation of the tumor microenvironment through SASP.
5. Therapy-Induced Senescence
Despite the primary induction of apoptosis during cancer treatment and depending on the concentration and time of exposure to pharmacological and physical agents, a senescence phenotype can arise, affecting a large number of cells within the tumor. Treatment with conventional physics methods (i.e., ionizing radiation), conventional chemotherapy that generally produces a DNA damage response, and some target therapies trigger therapy-induced senescence (TIS) [34,69].
TIS is an important outcome since it produces the existence of tumors lacking p53 and pRB; however, senescence induction pathways are redundant in many locations downstream, especially on the final regulators of the cell cycle (i.e., p16, p21, and p27). Due to this, all cancer cells retain the ability to enter on senescence with appropriated stimuli (reviewed in [70]).
Despite the growth suppression of senescence induction, TIS may promote inflammation and angiogenesis and is the cause of some deleterious effects of chemotherapy [71] or even recurrence [6,72,73,74]. Nevertheless, the use of senolytics—molecules that kill specifically senescent cells—has been proven to be effective for avoiding such deleterious effects [71]. In addition, the secretome from primary senescent cells (surrounded or not surrounded by tumor tissue) has major pro-tumorigenic effects [53,75,76], particularly in mesenchymal and stromal cells [77,78,79]; at present, it is not completely clear if these surrounding cells become senescent in response to cancer treatment or other physiological stimuli. Senescence plays a dual role in oncogenesis and tumor suppression, both protective and detrimental, affecting the microenvironment and tissue functionality, and eventually leading to pro-carcinogenic signals. It is necessary to find new senogenic compounds to promote specific senescence on cancer cells and not in primary cells, in order to reduce the negative effects of TIS, and to combine this with senolytics. In this way, a recent report on an experimental treatment for liver cancer cell lines derived xenografts, and described the use of pharmacological screenings to find a novel and specific senogenic and senolytic to promote strong tumor regression [80].
6. Senescence Implications in Metastasis
It is well-established that senescence promotes the appearance of tumor cells with a high stemness and promotes EMT [42,81]. In addition, the senescent secretome from tumor and primary cells (especially from epithelial and stromal cells) promotes the optimal microenvironment for the invasion of adjacent tissues and migration to distant sites. Among the factors secreted by senescent cells, the extracellular matrix remodelers [42,82,83] and angiogenic factors, especially the vascular endothelial growth factor (VEGF) [76,84,85], are the most important for the metastasis process. Because of their large and flattened morphology and due to the lack of a proliferation capacity, it is believed that senescent cells remain motionless and therefore do not display invasive behavior. However, senescent tumor cells are frequently present in the front region of the collective invasion of papillary thyroid carcinoma in patients, as well in lymphatic channels and metastatic foci of lymph nodes [86]. In melanoma cells, exposure to the B-raf inhibitor promotes a senescent-like state (including a high p21 expression, senescence-associated heterochromatic foci (SAHF), Promyelocytic Leukemia (PML) bodies activation, and increased activity of SA-β gal); even so, a subpopulation with a high Wnt5A expression was able to colonize the lungs in in vivo tail-vein colony-forming assays [87], indicating that senescent cells can conserve the malignant capacity to migrate to distant tissues.
Additionally, senescent cells can act as chemo-attractants and lead to proliferative tumor cells through factors included in the senescent secretome [86,88]. Moreover, the increase in senescence-associated aging promotes the emergence of pro-metastatic niches in bone, possibly by the secretion of IL-6. Using antibodies against IL-6 was necessary and sufficient to stop senescence-induced osteoclastogenesis and bone metastasis [89]. These findings suggest that senescent cells are involved in invasion and metastasis and predict that interventions targeting cellular senescence and SASP may improve cancer outcomes and reduce the metastatic process (Table 1).
7. Tumor Cell Dormancy
One of the major challenges in current cancer treatments is cancer relapse and metastatic recurrences. Metastatic recurrences are the resurgence of tumors in different tissues from which primary cancer arose, months, years, or even decades after the eradication of the primary tumor. Metastatic recurrence is indeed the major cause of death in cancer patients [90]. Like all the characteristics of tumor cells used in their favor, the capacity to spend long periods in diapause is a conserved physiological process, and is especially observed in memory T lymphocytes [91,92]. To survive after chemotherapeutic treatments, dormant tumor cells (DoTC) need to possess a different biology from the normal cells and acquire at least the following characteristics: stress and drug resistance, invasion, a dissemination capacity, and the capacity to enter and exit from diapause. Three models of dormancy have been proposed: (a) cellular dormancy, in which a single cell survives in quiescence; (b) angiogenic dormancy, in which cells remain isolated in environments with a low concentration of oxygen and nutrients; and (c) immunosurveillance dormancy, in which the immune system prevents tumoral re-growth [93].
Dormancy and Metastatic Recurrence
Dormancy and metastasis are two different concepts, but they are related. Molecular characterization and relevance in disease of the DoTC are currently being extensively reviewed. Both DoTC and metastatic cells present a similar organotropism, but metastatic cells (in their extensive definition) activate proliferation programs in less time [94,95], while DoTC proliferation will take a longer time and may possibly be more sensitive to microenvironmental changes, as determined by its own physiology [96,97,98] (Table 1, oncogenes as the origin of proliferative fate). DoTC are, in fact, a subpopulation of circulating tumor cells (CTC) [99] and a fraction of disseminated tumor cells (DTC) engaged in metastatic niches [100]. The origin of metastatic cells has been associated with the late-stage of tumor development, in which new characteristics are acquired by de novo genetic mutations or selected by chemotherapeutic treatments [94]. However, the best explanation of clinical and preclinical evidence is that metastatic cells are produced and disseminated early on in cancer development [101,102,103]. This situation seems to be no different from DoTC, which develop into lesions prior to the appearance of the primary tumor [92,104,105,106,107]. In this context, it has been demonstrated that dormant cells arise from the primary tumor in specific hypoxic niches [20] by the activation of genes like Nr2f1 and promote heterogenicity (Figure 1). This notion about the existence of cells with long and short diapause is supported by data on D2A1 and D2.0R tumor cell lines derived from murine mammary hyperplastic alveolar nodules. Both cell lines produce lung metastasis, although the D2A1 cell line evolves in a short period of time (1-3 weeks), while D2.0R takes a long time to evolve (4 months), after recipient injection [98,108,109,110]. Dormant pancreatic tumor cells in which oncogenic drivers such as KRAS and c-myc are mutated display increased autocrine IGF1/AKT signaling that controls survival and the pharmacological inhibition of IGF-1R reduces the residual disease burden and cancer recurrence [111]. For reviews on the immune targeting of dormancy and metastasis, see references [97,112].
8. Molecular Mechanisms Underlying Dormancy, Quiescence, and Senescence
Which is the genetic identity of these cellular states? One of the first attempts to reveal DoTC identity compared them with cancer stem cells (CSCs) [113]. As already mentioned above, CSCs [114] and DoTC [90] share similar characteristics (e.g., a resistance to chemotherapy drugs, high migration capacity, and tumor initiation and regrowth ability). CSCs are in a quiescence (or G0)-like state [115] and can restart proliferation with a better disposition and in less stringent conditions than those required by DoTC [116,117,118]. In fact, DoTC expresses some stemness regulation genes related to adult primary stem cells, such as TGF-β and BMP [119,120,121]. Likewise, the dissemination of early breast tumor cells with a DoTC phenotype requires the activation of cellular plasticity pathways (e.g., Wnt and RANK) to promote migration, but not proliferation [107,122]. The pluripotency program is also active in dormancy neck squamous cell carcinoma, in which SOX2, SOX9, OCT4, and NANOG genes are upregulated [123] (see Table 1 for a summary of the genetic origin and relevant trigger signals for proliferation and dormancy fates). Nevertheless, DoTC do not accomplish a complete EMT [124], and upon specific environmental conditions, can restart proliferation [125].
Primary Signaling in Dormancy and its Relevance for Cancer Stem-Like and Senescent Cells
There is a consensus about the existence of dormancy pathways resembling quiescence [90]. Evidence suggests that DoTC have the constitutively active “stress pathway” CDK4/6–p38α/β and inactive “proliferation” pathway Ras–MEK–ERK1/2 [126,127]; whilst other stress pathways can be active during dormancy, like ATFα6-mTor [128]. During adult pluripotency, stem cells activate the Ras–ERK pathway to escape from quiescence and proliferate, in turn suppressing the pluripotency program [129,130], while CSCs can retain some of the stem characteristics after Ras-ERK activation [131,132,133], fitting well with the DoTC profile (Figure 1 shows the involvement of ERK as a driver for quiescence-dormancy conversion and in dormancy-tumor relapse). If pluripotency is genetically induced with Yamanaka’s factors (i.e., Myc Oct4, Sox2, and Klf4), the Ras pathway plays a dual role; in a malignant context, Ras inhibits pluripotency acquisition, while in an oncogenic context, it promotes it [134]. On the other hand, the activation of p38 cellular stress reduces pluripotency and self-renewal in CSCs [135,136,137,138]. Are DoTC and CSCs identical in essence? Both proliferation and stress pathways (Ras–ERK and MKK–p38) have an important role in senescence. As mentioned above, oncogene activation produces senescent cells (OIS) in premalignant lesions, with the Ras mutation being the most common [31,33]. However, once senescence is established, Ras expression does not seem to have direct consequences on the senescent phenotype. Furthermore, its prolonged activation produces negative feedback that reduces the stimulation of the RAS–MEK–ERK pathway [139]. On the other hand, during physiochemical stress situations, primary cells go to senescence by the activation of p38α–p16–pRb [140,141]. We reason that even though DoTC conserve some stem features, they cannot be considered as CSCs in a typical way. The evidence suggests the possible existence of CSC subtypes and perhaps that is also true for DoTC. Puig and cols. [142], used an elegant experimental design to tag colorectal cancer cells (CRC) with the doxycycline-inducible histone H2Be–GFP. After a pulse of doxycycline and some cycles of cellular division, those cells retaining the fluorescence reporter were isolated. The gene expression profile of these slow-cycling cancer cells (SCCCs) was enriched for drug detoxification, stemness, hypoxia, or crosstalk, with the immune system denoting it’s stem cell nature, which might be a starting point for characterizing the underlying molecular signature of putative dormant cells. However, isolated SCCCs from spheroids have the same proliferative behavior in vitro (from single-cell samples) and in vivo than their counterparts (rapid RCCCs), suggesting the different nature of these cells and the relevance of the microenvironment for establishing the signaling of dormancy.
9. Dormant and Senescent Cells: One and the Same or Another Kind?
The elevated activation of p38 in DoTC should make them lose their stem features; however, it is partially conserved. This opens the opportunity to ask relevant questions: Can the DoTC be in a different state to quiescence? Which other diapause state is compatible with a high level of p38 activation and retention of stemness? We must keep in mind that when a cell enters in senescence, it seems to acquire some stem features [23,49], so, it is justified to ask if DoTC are in a senescence-like state or if senescence in tumor cells should be considered a different kind of dormancy. Reports from Kobayashi and cols. [60], Sharma and cols. [143], and Bartosh and cols. [144] (revised further) suggest that, in fact, the state of dormancy is acquired through some kind of senescence. This observation was made using two different models induced by two different mechanisms (including signaling from BMPs and entosis, which is the process whereby cells are internalized into neighboring cells, forming ‘cell-in-cell’ structures), with both occurring putatively in the same site (bone marrow). A study from Kobayashi and cols. [60] also suggests the possibility that they can be in a state of stem cells (quiescence) and senescent at the same time, since DoTC are selected using stem cell markers after exposing them to BMP.
9.1. TGF-β Family Factors in Dormancy and Senescence
A very interesting factor with proven activity during cancer cell dormancy is TGF-β, which is a multifunctional regulatory cytokine. Senescence induction mediated by TGF-β is widely documented in tumor and primary cells [145,146,147,148,149]. High TGF-β2 signaling, as found in bone marrow, promotes the activation of p38α/β stress and a dormancy state in tumor cells by the activation of TGF-βRI yRII receptors, and through the phosphorylation of p27 [120,150]. Similarly, Bone Morphogenic Protein BMP4/7, another TGF-β family member secreted by bone stromal cells, participates in dormancy induction in prostate and triple-negative breast cancers through the activation of p38, p21, p27, and NDRG1 [60,119,151]. Some reports also suggest that TGF-β and BMP induce quiescence in prostate cancer [152] and other solid tumors [153], as well as in primary cells [154]. However, breast and prostate tumor cells can be induced into dormancy by BMP7 secreted from bone stromal cells through BMPRII (BMP receptor II), using β-gal staining as positive markers of senescence and genetic in vivo models of suppression of BMP receptor signaling [60,155]. Prostate tumor cells in senescence show p38 phosphorylation activity and low ERK activity (even in the presence of an epidermal growth factor (EGF), a very well-known ERK activator), proposed as a marker of cells undergoing dormancy. This dormant-senescent equilibrium state is directly related to the activity of N-myc downstream-regulated gene 1 (NDRG1), which may open up new opportunities for targeting as a regulatory mechanism. Interestingly, blocking BMP7 signaling in vitro and in vivo reverses senescence and causes cells to restart proliferation [60]. NDRG1 and BMP4 also induce senescence in other tumor and primary cells [156,157,158,159]. Finally, TGF-β and other members of its family (like BMP6, BMP2, Inhibin A, and GDF15) are fundamental factors present in the senescent secretome [160] essential for the paracrine induction of senescence [43,161,162] and recognized as dormancy drivers (Figure 1). It is also interesting that, during aging, TGF-β-mediated signaling induces senescence in bone-derived mesenchymal stromal/stem cells through p21 activation [59,163].
9.2. CDK Inhibitors in Dormancy, Quiescence, and Senescence
Cell cycle arrest is carried out by CDK inhibitors (CDKi), especially p21 and p27 producing quiescence upon TGF-β-induced dormancy [60,120,164]. However, these signaling pathways controlling the cell cycle during quiescence are overlapped with the senescence induction process [165,166], especially in tumor cells lacking active p16 [167], in which senescence primarily depends on p21 and p27. As mentioned above, there is still controversy on the role of CDK4/6 inhibitor drugs (e.g., Palbociclib or Abemaciclib) in senescence or quiescence induction [168,169,170]. Senescence induction depends not only on the chronicity of treatment, but also on other factors, such as proteasome inactivation [171] and autophagy [172]. DNA-damaging chemotherapeutics induce senescence mainly activated by the p53-p21 pathway, although, if p53 is deactivated, cells can escape from senescence, resuming proliferation and not entering in quiescence (reviewed in [173]). On the other hand, p27 participates in senescence induction, but more importantly, provides the ability to avoid cells to escape from senescence by blocking CDK1, an effect not observed for p21 [174]. These mechanisms might be clinically relevant, since high levels of p27 are correlated with a better prognosis in patients with different types of tumors [175], including prostate cancer [176], which is associated with the longest dormancy periods.
9.3. Emerging Molecular Mechanisms Controlling Quiescent or Senescent Fate
If we consider as a premise that not only are DoTC not senescent, but that both senescence and quiescence can be reversed, we can ask which mechanisms are responsible for avoiding regrowth, as well as what specific signals promote quiescence instead of senescence. New research is necessary to address these fundamental questions. An example of an effort to generate new knowledge is provided by focusing on the DREAM protein complex as a mechanism that can act as a switch regulating cell fate [177]. DREAM consists of the retinoblastoma protein (p130 or p107), gene repressor E2F (E2F4 or E2F5), and Multi-vulval class B (MuvB) core of proteins [178]. It functions by suppressing the expression of genes related to cell cycle progression through the induction of senescence or quiescence in a mechanism directly dependent on the initial stimuli. DREAM activity is also regulated by two specific kinases DYRK1A and DYRK1B, highly phosphorylated in tumor cells to promote both cell cycle arrest and high survival [179,180]. Moreover, it has been shown that the specific inhibition of DYRK1 kinases reduces cell viability in cells under dormancy isolated from ovarian spheroids [181]. In the same way that the transforming growth factor-beta (TGF-beta)/bone morphogenic protein (BMP) is involved in cell differentiation, the presence of secreted protein, acidic and rich in cysteine (SPARC), in the bone marrow microenvironment induces DoTC positive for senescence markers, including β-gal staining [143]. Additionally, we must consider the specific genetic background of tumor cells as a major factor influencing the cellular fate. Kovatcheva and cols. [170,182] showed that after CDKi treatment (a well-known senescence inducer), liposarcoma cells can enter in senescence or quiescence, depending on the level of expression of ATRX (Figure 1). In the presence of ATRX, CDKi promotes the formation of senescence-associated heterochromatic foci (SAHF) by recruiting HIRA, HP1, and PML. Additionally, cells that enter in quiescence after CDKi treatment can be senoconverted to senescent cells by downregulating MDM2. Interestingly, the senoconversion process (or the specific conversion from quiescence to senescence) is observed in normal muscle quiescent stem cells and can be promoted by increasing the p16 expression [35] or by genetically and pharmacologically blocking autophagy with bafilomycin A1 [57]. In the cancer context, Buczacki and cols. [183] have shown that senoconversion can be achieved in DoTC cells using small molecules. In this study, putative dormant quiescent CRC cells were induced to senescence through the inhibition of Wnt and Hedgehog pathways by using itraconazole antifungal treatment. This study emphasizes the feasibility of using pharmacological agents to induce senescence in quiescent DoTC and opens the possibility of exploiting synthetic lethality by combining seroconversion agents with other drugs, such as senolytics.
Altogether, these examples highlight the existence of pivotal and additional molecular mechanisms that define the fate of DoTC. To completely understand this process, one must explore deeper to discover the underlying regulatory molecular mechanisms that may be eventually translated into novel therapeutic interventions.
10. Microenvironment Influences Dormancy and Senescence
10.1. Microenvironment and Dormancy Induction
Many physiological processes are intimately related to the microenvironment, especially relevant due to its role in regulating dormancy control and reawaking tumor cells. Specific tumor cells are prompted to enter dormancy in different niches, but long-term dormancy can be acquired in niches from (a) bone marrow, (b) the lung, (c) perivascular tissue, and (d) the “inside of tumor”. There are specialized spaces within these tissues capable of sheltering stem cells, and it has been proposed that DoTC share the same niches. Additionally, all niches are characterized by an elevated concentration of members of TGF-β/BMP family factors secreted by either cells neighboring the niche or by tumor cells. From these four niches, the most prolonged dormancy seems to be induced by the perivascular and bone marrow niche (reviewed in [72,184]) (see Table 1 for a summary of microenvironment trigger signals associated with dormancy and senescence). These stem cell niches are primarily occupied by mesenchymal stem/stromal cells (MSCs) [185]. MCS exhibits a physiological secretome mainly composed of HGF, TGF-β, CCL2/5, IL-6, VEGF, TSG-6, PGE2, and galectins 1/9 ([186] and reviewed in [187]), an anti-inflammatory environment protective against pro-inflammatory stimuli like INF-γ [188] or in 3D conditions [189]. During dissemination, the prostate DoTC target and occupy the hematopoietic stem cell (HSC) niche in the bone marrow [190]. In the osteoblastic niche, tumor cells require the expression of the Axl receptor to activate it in response to microenvironmental growth-arrest specific 6 (GAS6) to maintain the dormancy state. The Gas6-Axl axis is also necessary for TGF-β2-mediated quiescence. Interestingly, low Axl expression is correlated with longer survival in prostate cancer patients since it is not observed in tumor cells in either primary or in metastatic lesions [121,150,191]. Activation of the axis Gas6-Axl in primary vascular cells delays senescence through PI3K/Akt/FoxO signaling [192,193], although more research is required to understand if this mechanism is relevant in tumor evolution.
10.2. Senescent Secretome in a Dormancy Context
As previously mentioned, senescent cells possess the capacity to remodel and reorganize the microenvironment through SASP, and although they share many mutual components with the MCS secretome, they have more prominent pro-inflammatory factors [53,86,194,195]. It is therefore justifiable to believe that there is a relationship between senescence and dormancy at many levels, despite the possibility that senescence could be considered another kind of dormancy. In this way, an interesting report demonstrated that dormancy is induced in triple-negative breast cancer cells through entosis of a coculture with MSC in a 3D format [144]. These results suggest that tumor cells swallow MSC and then acquire a dormant-senescent phenotype, characterized by cell cycle arrest; a high resistance to starvation; and, interestingly, a secretome similar to SASP that includes CSF3 (granulocyte colony-stimulating factor), PTGS2 (COX2), TNF-α, IL1-α, IL1-β, IL-6, IL-8, CXCL1, CXCL2, CXCL10 (IP10), and CCL20, as well as the antiapoptotic factor IFI6 and the tumor suppressor EGR1. Similarly, p53 wild type breast tumor cells induced to senescence with doxorubicin activate phagocytosis and macrophage-like programs promoting a higher survival capacity during long dormancy periods after engulfing proliferative neighbors [196]. Factors present in the microenvironment can modulate senescence induction. In this context, prostate dormant cells isolated in vivo secrete high levels of protein acidic and rich in cysteine (SPARC). SPARC induces the expression of BMP in bone stromal cells and promotes a reversible senescence state characterized by high levels of p38 and p21 (see Table 1). High SPARC promoter methylation negatively correlates with the disease-free survival of prostate cancer patients. On the other hand, Noggin expression in tumor cells or its presence in the microenvironment suppresses BMP7-BMPR2 signaling and supports cancer cells to avoid dormancy [143].
10.3. Immune Recognition and Clearance of Dormant and Senescent Cells
The interaction between the human immune system and cancer is complex and highly regulated. The immune system is able to identify specific surface receptors (e.g., major histocompatibility complex (MHC) molecules class I and II) and specifically eliminate cancer cells. Immunotherapy by immune checkpoint blockade targeting tumor-specific neoantigens, by monoclonal antibodies, and by T-cell transfer therapy is now a reality for patients with solid and liquid tumors [197,198,199]. Patients undergoing immunosuppressive treatments have a higher incidence of cancer [199], which suggests that cells prone to tumor development might be in a dormant state; an event observed in bone marrow from patients with breast cancer and accompanied by over-activated immune cells, including natural killer (NK) cells and T lymphocytes [199]. In healthy individuals (and not in melanoma patients), melanoma-specific T lymphocytes displaying a strong reactivity against peptides of melanoma antigens Tyrosinase–MAGEA3–Melan-A/Mart-1–Pmel 17gp100 and NY-ESO-1 have been identified [200]. These data support the existence of endogenous autoimmunity against melanoma, preserving tumor dormancy and protecting from malignant cell growth. T lymphocytes and NK cells are relevant as they trigger cytotoxic responses to regulate the equilibrium between metastatic dormant cells and the immune system. Moreover, dormancy of immune cells promotes cancer cell growth arrest and angiogenic control. Immunotherapeutic interventions against dormancy and senescence cell fates can be considered suitable approaches for targeting primary tumors and metastasis. Coupled to these observations, evidence suggests that immune clues on the microenvironment promote dormancy on tumor cells. Immune cells present in some potential dormant niches directly impact dormancy physiology through the secretion of specific factors (reviewed in [201,202]). For example, CD8+ T cells can induce tumor cell dormancy via the production of IFN-γ, and CD4+ T cells produce CXCL9 and CXCL10, avoiding micro angiogenesis and promoting hypoxic-induced dormancy. Finally, it has been observed that perforin secreted by natural killers induces a long period of dormancy in vitro and in vivo. It is therefore predictable that this kind of microenvironment allows dormancy to be maintained for long periods of time.
On the other hand, senescence limits tumor growth and affects immunosurveillance. Evidence suggests that in early premalignant lesions, an increased number of senescent cells are found, revealing the intrinsic tumor suppression nature of senescence. Through their secretome, oncogene-induced senescent hepatocytes in vivo are capable of activating the immune system for its own clearance and this depends on the intact CD4(+) T-cell-mediated adaptive immune response and the tuned activity of CCR2+ myeloid cells. On the contrary, the impairment of immune senescent surveillance results in the growth of hepatocellular carcinoma tumors in murine models, and it is a poor prognosis marker for patients [203,204].
If senescent cells are well-recognized and cleared by immune cells, there are situations in which the immune system is incapable of recognizing them. For example, in individuals with impaired cell cytotoxicity, the accumulation of senescent cells in tissues is recognized and accompanied by signs of premature aging [204]. Moreover, in aged organisms, dermal senescent fibroblasts expressing high levels of HLA-E can evade immune clearance. These MHC surface molecules interact with inhibitory receptor NKG2A from NK and highly differentiated CD8+ T, inhibiting the immune response against them [205].
In the cancer context, some of the mechanisms that senescent cells use for immune evasion have been identified. One of them (shared with normal senescent cells) includes NKG2D, which, like Matrix metalloproteinases (MMPs) (a factor included in the senescent secretome), disrupts immunosurveillance by paracrine action in the microenvironment [206]. Moreover, the senescent secretome from Pten-null tumors can establish an immunosuppressive microenvironment by the activation of Jak2/Stat3 and downregulation of PTPN11/SHP2 pathways [68].
The senescent secretome is a complex issue, and its composition depends on cellular types and senogenic stimuli [207,208]; however, target therapy, specifically the use of CDK inhibitors, promotes the awaking of cytotoxic T-cell immunity directed by the production of type III interferons induced by the production of double-chain RNA molecules inside the tumor senescent cell [209]. Furthermore, the use of CDKi plus immune activators (e.g., kinase inhibitors, PD-1 inhibitors) increases the survival of treated human xenograft models [210,211,212]. This combination (CDKi abemaciclib with anti PD-1 antibody pembrolizumab) is currently being used in clinical trials to test the efficacy in glioblastoma (NCT04118036) and in HR+ and HER2- metastatic breast cancer (NCT02779751) [212].
10.4. The Awaking of Dormant Cells is Primarily Promoted by the Microenvironment
Finally, a fundamental question remains to be addressed: what are the main pathways that trigger the awaking of DoTC? A potential explanation is the action of specific signaling molecules such as Noggin. Another interesting example is the activity of Coco in lung metastases. Coco functions in a similar way to Noggin as a BMP inhibitor that promotes the reawaking of dormant breast tumor cells, leading to a state of high stemness by activating the expression of Nanog, Sox2, and Taz. Conversely, BMP4 completely suppresses the stemness profile expression [119]. Another possibility causative of DoTC reawaking is inflammation, specifically if disbalance [212] between anti-inflammatory and pro-inflammatory factors occurs in the dormant niche. In D2A1 slow-cycling cells, in vivo isolated dormant cells were exposed to Lipopolysaccharides (LPS) endotoxins. The inflammation produced was sufficient to promote proliferation in DoTC and increase the number of recurrences of lung tumors [125]. Similarly, D2.0R dormant cells exposed to tobacco smoke or LPS inflammation stimuli are characterized by the formation of neutrophil extracellular traps (NETs) and subsequent matrix remodeling by MMP9 and NR (Neutrophil elastase). The NET-mediated proteolytic remodeling of laminin revealed an epitope sensed by DoTC through integrin α3β1, leading to proliferation by the activation of FAK/ERK/MLCK/YAP signaling [213]. Finally, a study performed in the Mcf7 cell line induced to dormancy by culturing in the presence of FGF-2 revealed the possible action of pro-inflammatory cytokines such as IL-6, IL-8, and TGF-β1. Furthermore, dormant cells co-cultured with SASP from bone marrow stromal cells (e.g., oxidation, hypoxia, or estrogen deprivation) proliferate after 6 days in vitro [214]. Consistent with this evidence, it has been proposed that the capacity of senescent cells to escape from cell cycle arrest is due to exposition to SASP, especially tumor senescent cells exposed to different chemotherapeutics [6,73,215] which retain the capacity to regrow tumors in vivo [74]. This kind of recurrence occurs after long periods of time. During aging, TGF-β signaling induces senescence in bone-derived mesenchymal stromal/stem cells accompanied by the activation of p21 [59,163], and in long-term recurrences, aging could be the major risk factor of recurrence associated with a chronic inflammation state called inflammation [216].
11. Two Models of Tumor Dormancy
Up to this point, considering the existence of more than one kind of dormancy is not an unwarranted idea. Based on the existing evidence, one can divide cancer cell dormancy into two different phenotypes with regards to evolution: (1) Short-term dormancy, putatively caused by quiescent cells and coinciding with clinic evidence of relapse after months or even a few years of disease-free survival, and (2) long-term dormancy, putatively caused by senescent cells and coinciding with clinical evidence of relapse after 10 years or more of disease-free survival. The incidence of one or another sort of dormancy could also depend on two main factors: the origin of the primary tumor and the niche in which the dormant cells engage (Figure 1 depicts the factors involved in short-term and long-term dormancy). Under this classification, a clear example of senescent-induced dormancy is presented by prostate tumor cells, which present a long time period of dormancy, target bone marrow niches, and are positive for SA-β gal staining. In the same manner, colorectal cancer tumor cells could be relevant as a model for quiescent dormant cells due to their rapid regrowth capacity. Evidence from lung, ovarian, and breast tumors suggests that both short-term and long-term phenotypes could be acquired in this kind of cell, depending on the microenvironment and the regulatory pathways within the cells. According to this notion, BMP and TGF-β signaling backed by p53 (or other tumor suppressors) promote preferential senescence; as opposed to NR2F1 and DREAM, which drive quiescence. To test this working hypothesis, we propose focusing efforts on the systematic analysis of clinical observations and patient samples in an unbiased manner. Knowledge of underlying molecular mechanisms regulating cancer cell assignments to types and states would provide opportunities to develop novel therapeutic strategies to target cancer outcomes.
12. Senolytic Therapy Is Beneficial in Cancer
Senolytic therapy is defined by the selective clearance of senescent cells in aging and diseased tissues [217]. Senescent cells accumulate during aging in different tissues, promote inflammation, and often elicit deleterious effects [218]. The selective elimination of senescent cells promotes an improvement in physiological performance in aging-related diseases [219,220,221,222]. Cells from transgenic mice overexpressing the tumor suppressor p16INK4a are sensitive to pharmacological elimination. Chemotherapy not only attenuated aging-associated disorders, but also extended the lifespan [219]. In cancer, an increase of senescent cells in normal tissue is normally observed after treatment with chemotherapeutics. Treatment with senolytic compounds targeting cell death regulator caspases (e.g., BCL-2 and BCL-XL) improves the health span, decreases metastasis, and increases the rate of disease-free survival after chemotherapy [220,222]. This data highlights the possibility of using senolytic therapy not just as secondary treatment, but as a “first-line” treatment in combination with senogenic treatment using a two-hit strategy targeting TIS cancer cells: irreversible senescence induction plus the selective elimination of senescent cells ([223,224,225] and reviewed in [226,227,228]).
12.1. Experimental Evidence Highlights the Efficacy of Senolytics as a Cancer Treatment
The selective elimination of senescent cells extends the survival of mice bearing TIS tumors, but not of those mice with a tumor resistant to senescence [223]. In this context, specifically targeting senescent human melanoma cells (i.e., SK-MEL-103) in xenografted tumors demonstrated antitumoral efficacy. The combination of senogenic compound Palbociclib with doxorubicin or navitoclax formulated in 6-mer galactooligosaccharides (referred to as GalNP) nanoparticles shows a remarkable advantage over each treatment alone at reducing tumor growth, and secondary effects like cardiotoxicity and thrombocytopenia [229]. By taking advantage of this approach, a synthetic lethality approach can be used to eliminate epithelial ovarian cancer and Triple Negative Breast Cancer (TNBC) cells transiently induced to senescence with poly ADP ribose polymerase (PARP) inhibitors and a panel of senolytics [230].
In studies from our laboratory, cardiac glycosides (CG) acting as sodium-potassium pump blockers were identified as a new broad class of senolytics. Combination treatment with senogenic gemcitabine and CGs of non-small lung carcinoma xenografted tumors show a significant efficacy at decreasing tumor volume in comparison with each agent alone. This two-hit combination therapy was efficient at diminishing the size of patient-derived xenograft (PDX) tumors from triple-negative breast cancer using doxorubicin as a senogenic and digoxin as a senolytic, offering a clear advantage over each compound alone [231]. Moreover, another group in an independent manner also identified GCs as senolytics. As an intrinsic tumor suppressor mechanism, normal cells go to senescence after oncogenic stimuli, but remain in tissues as preneoplastic cells. Treating mice with ouabain reduced the occurrence of cancer by the direct elimination of senescent cells in two different models of genetic carcinogenesis, opening the possibility of also using senolytics as a cancer prophylactic treatment [232].
A recent study from Wang and cols. [80], using kinome-focused genetic screening, identified a DNA-replication kinase CDC7 as a target for senescence induction in p53 null liver cancer. The pharmacological inhibition of CDC7 induces senescence in solid tumor models. Moreover, using chemical screening, they found that the pharmacological inhibition of mTOR promotes senolysis in CDC7-inhibited senescent cells. This combination showed beneficial effects in a xenograft and in immune-competent, somatic mouse models of hepatocellular carcinoma by promoting tumor regression and the highest overall survival.
As these studies indicate, this therapeutic strategy offers an enhanced efficacy during and after treatment, with special relevance for tumors inherently resistant to conventional chemotherapy and with a high rate of metastasis and recurrence. In the shreds of evidence reviewed, we can list another point in favor of this approach. Treatment with senolytics with suitable timing can ensure the elimination of cells, with the possibility of preventing senescence and acquiring the most aggressive stem-like phenotype (autocrine stemness) [23], by avoiding the rise of subpopulations with a high stemness through SASP from tumor cells and non-malignant cells from tumor-surrounded tissues [52,53,54,233]. In the same way, clearing senescent cells existing in tumors can prevent migration and possibly metastasis [86,88]. Quiescent DoTC can be possibly senoconverted to senescent cells, which may make them sensitive to senolytics [183]. However, it will be necessary to develop tools and methodologies to identify new pharmacological senoconverters that can facilitate such differentiation. The low rate of recurrence after chemotherapy observed in these studies suggests that targeting the senescent-dormant phenotype with senolytics might be a front-line strategy to reduce the metastatic recurrence. We hypothesize that a combination of senogenic and senolytic treatments might have a beneficial effect on cancer evolution by preventing metastasis. To test this hypothesis, efforts should be directed to demonstrating the broad efficacy of the selective elimination of senescent-dormant cells on invasion and metastasis progression from dormant solid tumors, for example, by targeting dormancy drivers (Figure 1).
12.2. Senolytics in Clinical Development
The first in-human trial of senolytic drugs in patients with idiopathic pulmonary fibrosis (IPF) data is encouraging and indicates the feasibility of larger clinical trials [234]. In this pilot study, investigators enrolled 14 adults diagnosed with stable, primarily mild-to-moderate IPF. Each participant received a combination of senolytic drugs, dasatinib, and quercetin (DQ), U.S. FDA-approved for other indications. The mobility of participants in a six-minute walk distance test was improved by an average of 21.5 m. DQ treatment has shown efficacy at eliminating senescent cells originating from different cell types and improving pathology in animal models of Alzheimer’s disease. Indeed, in a second study performed in nine patients with diabetes-related kidney disease, it has been demonstrated that DQ senolytic treatment not only removed senescent cells from the body, but also alleviated insulin resistance, cell dysfunction, and other processes that cause disease progression and complications [235]. Although these preliminary data are promising, they should be interpreted with caution and further study is required due to limitations in terms of the sample size and lack of a placebo group. The identification of safe senolytics within the approved drugs may facilitate the development of such studies in larger randomized and controlled trials.
13. Outlook
The overall scope of this review was to collect relevant evidence on the state-of-the-art of cancer cell fate specification in development and progression. The development and translational investigation of new therapies or treatments for cancer can be expedited by the latest precision medicine and artificial intelligence approaches. This can facilitate drug discovery with a direct input into the regulatory science and industrial technological innovation pipeline. Understanding the continuum of proliferation, senescence, stemness, dormancy, quiescence, and cell cycle re-entry states and contexts of tumor formation and metastasis in relevant 3D models and organ-/lab-on-a-chip might provide a platform to increase the success rate in translating new solutions (e.g., diagnostics, treatment, and follow up) into real clinical innovative approaches that ultimately benefit patients.
Author Contributions
F.T.-M. and E.D. conceived of the presented idea. M.I.L. developed the theory, and encouraged the investigation of specific aspects, supervised the concepts of this work. All authors discussed the results and contributed to the final manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
Consolidation and structuring 2017 NETWORKS-REGID Galician Research and Drug Development Network. Ministry of Culture, Education and University Planning of Galician Government.
Acknowledgments
F.T.-M. want to thanks to CONACYT-Mexico (cvu 268632).
Conflicts of Interest
All authors declare no competing interests.
Abbreviations
| AML | Acute myeloid leukemia |
| BMP | Bone morphogenetic protein |
| BMPR | Bone morphogenetic protein receptor |
| CCL | chemokine (C-C motif) ligand |
| CDK | Cyclin-dependent kinase |
| CSC | Cancer stem cells |
| CSF3 | Granulocyte colony-stimulating factor |
| CTC | Circulating tumor cells |
| CXCL | C-X-C motif chemokine |
| DDR | DNA damage response |
| DoTC | Dormant tumor cells |
| DREAM | Dimerization partner, RB-like, E2F, and multi-vulval class B |
| DTC | Disseminated tumor cells |
| DYRK | Dual specificity tyrosine-phosphorylation-regulated kinase |
| EGF | Epidermal growth factor |
| EMT | Epithelial-mesenchymal transition |
| ERK | Extracellular signal–regulated kinase |
| FAK | Focal adhesion kinase |
| Gas6 | Growth-arrest specific 6 |
| GDF | Growth/differentiation factor |
| HGF | Hepatocyte growth factor |
| IGF | Insulin-like growth factor 1 |
| IGFR | Insulin-like growth factor receptor |
| iPS | Induced pluripotent stem cell |
| MSC | Mesenchymal stem/stromal cells |
| MuvB | Multi-vulval class B |
| MYLK | Myosin light-chain kinase |
| NR2F1 | Nuclear Receptor subfamily 2, group F, member 1 |
| OIS | Oncogene-Induced Senescence |
| RANK | Receptor activator of nuclear factor κ B |
| RCCC | Rapid-cycling cancer cells |
| SAHF | Senescence-associated heterochromatin foci |
| SASP | Senescence associated secretory phenotype |
| SCCC | Slow-cycling cancer cells |
| SPARC | Secreted protein acidic and rich in cysteine |
| TGF-β | Transforming growth factor beta |
| TGS | Tumor necrosis factor-inducible gene |
| TIS | Therapy induced senescence |
| TNF-α | Tumor necrosis factor alpha |
| VEGF | Vascular endothelial growth factor |
| YAP | Yes-associated protein |
References
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef]
- Krizhanovsky, V.; Yon, M.; Dickins, R.A.; Hearn, S.; Simon, J.; Miething, C.; Yee, H.; Zender, L.; Lowe, S.W. Senescence of Activated Stellate Cells Limits Liver Fibrosis. Cell 2008, 134, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Da Silva-Álvarez, S.; Picallos-Rabina, P.; Antelo-Iglesias, L.; Triana-Martínez, F.; Barreiro-Iglesias, A.; Sánchez, L.; Collado, M. The development of cell senescence. Exp. Gerontol. 2019, 128, 110742. [Google Scholar] [CrossRef] [PubMed]
- McHugh, D.; Gil, J. Senescence and aging: Causes, consequences, and therapeutic avenues. J. Cell Biol. 2018, 217, 65–77. [Google Scholar] [CrossRef]
- Saleh, T.; Tyutynuk-Massey, L.; Cudjoe, E.K.; Idowu, M.O.; Landry, J.W.; Gewirtz, D.A. Non-Cell Autonomous Effects of the Senescence-Associated Secretory Phenotype in Cancer Therapy. Front. Oncol. 2018, 8, 164. [Google Scholar] [CrossRef]
- Schober, M.; Fuchs, E. Tumor-initiating stem cells of squamous cell carcinomas and their control by TGF-β and integrin/focal adhesion kinase (FAK) signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 10544–10549. [Google Scholar] [CrossRef]
- Spangrude, G.; Heimfeld, S.; Weissman, I. Purification and characterization of mouse hematopoietic stem cells. Science 1988, 241, 58–62. [Google Scholar] [CrossRef]
- Morrison, S.J.; Weissman, I.L. The long-term repopulating subset of hematopoietic stem cells is deterministic and isolatable by phenotype. Immunity 1994, 1, 661–673. [Google Scholar] [CrossRef]
- Wu, J.; Izpisua Belmonte, J.C. Dynamic Pluripotent Stem Cell States and Their Applications. Cell Stem Cell 2015, 17, 509–525. [Google Scholar] [CrossRef]
- Chaffer, C.L.; Brueckmann, I.; Scheel, C.; Kaestli, A.J.; Wiggins, P.A.; Rodrigues, L.O.; Brooks, M.; Reinhardt, F.; Su, Y.; Polyak, K.; et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc. Natl. Acad. Sci. USA 2011, 108, 7950–7955. [Google Scholar] [CrossRef] [PubMed]
- Ramalho-Santos, M.; Melton, D.A.; McMahon, A.P. Hedgehog signals regulate multiple aspects of gastrointestinal development. Development 2000, 127, 2763–2772. [Google Scholar] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Taipale, J.; Beachy, P.A. The Hedgehog and Wnt signalling pathways in cancer. Nature 2001, 411, 349–354. [Google Scholar] [CrossRef]
- Reya, T.; O’Riordan, M.; Okamura, R.; Devaney, E.; Willert, K.; Nusse, R.; Grosschedl, R. Wnt Signaling Regulates B Lymphocyte Proliferation through a LEF-1 Dependent Mechanism. Immunity 2000, 13, 15–24. [Google Scholar] [CrossRef]
- Abad, M.; Mosteiro, L.; Pantoja, C.; Cañamero, M.; Rayon, T.; Ors, I.; Graña, O.; Megías, D.; Domínguez, O.; Martínez, D.; et al. Reprogramming in vivo produces teratomas and iPS cells with totipotency features. Nature 2013, 502, 340–345. [Google Scholar] [CrossRef]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef]
- Ishizawa, K.; Rasheed, Z.A.; Karisch, R.; Wang, Q.; Kowalski, J.; Susky, E.; Pereira, K.; Karamboulas, C.; Moghal, N.; Rajeshkumar, N.V.; et al. Tumor-Initiating Cells Are Rare in Many Human Tumors. Cell Stem Cell 2010, 7, 279–282. [Google Scholar] [CrossRef]
- Fluegen, G.; Avivar-Valderas, A.; Wang, Y.; Padgen, M.R.; Williams, J.K.; Nobre, A.R.; Calvo, V.; Cheung, J.F.; Bravo-Cordero, J.J.; Entenberg, D.; et al. Phenotypic heterogeneity of disseminated tumour cells is preset by primary tumour hypoxic microenvironments. Nat. Cell Biol. 2017, 19, 120–132. [Google Scholar] [CrossRef]
- Rycaj, K.; Tang, D.G. Cell-of-Origin of Cancer versus Cancer Stem Cells: Assays and Interpretations. Cancer Res. 2015, 75, 4003–4011. [Google Scholar] [CrossRef]
- Morel, A.-P.; Lièvre, M.; Thomas, C.; Hinkal, G.; Ansieau, S.; Puisieux, A. Generation of Breast Cancer Stem Cells through Epithelial-Mesenchymal Transition. PLoS ONE 2008, 3, e2888. [Google Scholar] [CrossRef]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Däbritz, J.H.M.; Zhao, Z.; Yu, Y.; Dörr, J.R.; Dimitrova, L.; Lenze, D.; Monteiro Barbosa, I.A.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2018, 553, 96–100. [Google Scholar] [CrossRef]
- Todaro, M.; Alea, M.P.; Di Stefano, A.B.; Cammareri, P.; Vermeulen, L.; Iovino, F.; Tripodo, C.; Russo, A.; Gulotta, G.; Medema, J.P.; et al. Colon Cancer Stem Cells Dictate Tumor Growth and Resist Cell Death by Production of Interleukin-4. Cell Stem Cell 2007, 1, 389–402. [Google Scholar] [CrossRef]
- Sullivan, J.P.; Minna, J.D.; Shay, J.W. Evidence for self-renewing lung cancer stem cells and their implications in tumor initiation, progression, and targeted therapy. Cancer Metastasis Rev. 2010, 29, 61–72. [Google Scholar] [CrossRef]
- Merlos-Suárez, A.; Barriga, F.M.; Jung, P.; Iglesias, M.; Céspedes, M.V.; Rossell, D.; Sevillano, M.; Hernando-Momblona, X.; da Silva-Diz, V.; Muñoz, P.; et al. The Intestinal Stem Cell Signature Identifies Colorectal Cancer Stem Cells and Predicts Disease Relapse. Cell Stem Cell 2011, 8, 511–524. [Google Scholar] [CrossRef]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct Populations of Cancer Stem Cells Determine Tumor Growth and Metastatic Activity in Human Pancreatic Cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar] [CrossRef]
- Baccelli, I.; Schneeweiss, A.; Riethdorf, S.; Stenzinger, A.; Schillert, A.; Vogel, V.; Klein, C.; Saini, M.; Bäuerle, T.; Wallwiener, M.; et al. Identification of a population of blood circulating tumor cells from breast cancer patients that initiates metastasis in a xenograft assay. Nat. Biotechnol. 2013, 31, 539–544. [Google Scholar] [CrossRef]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef]
- Michaloglou, C.; Vredeveld, L.C.W.; Soengas, M.S.; Denoyelle, C.; Kuilman, T.; van der Horst, C.M.A.M.; Majoor, D.M.; Shay, J.W.; Mooi, W.J.; Peeper, D.S. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 2005, 436, 720–724. [Google Scholar] [CrossRef]
- Chang, B.D.; Broude, E.V.; Dokmanovic, M.; Zhu, H.; Ruth, A.; Xuan, Y.; Kandel, E.S.; Lausch, E.; Christov, K.; Roninson, I.B. A senescence-like phenotype distinguishes tumor cells that undergo terminal proliferation arrest after exposure to anticancer agents. Cancer Res. 1999, 59, 3761–3767. [Google Scholar]
- Collado, M.; Gil, J.; Efeyan, A.; Guerra, C.; Schuhmacher, A.J.; Barradas, M.; Benguría, A.; Zaballos, A.; Flores, J.M.; Barbacid, M.; et al. Senescence in premalignant tumours. Nature 2005, 436, 642. [Google Scholar] [CrossRef]
- Roninson, I.B. Tumor cell senescence in cancer treatment. Cancer Res. 2003, 63, 2705–2715. [Google Scholar]
- Sousa-Victor, P.; Gutarra, S.; García-Prat, L.; Rodriguez-Ubreva, J.; Ortet, L.; Ruiz-Bonilla, V.; Jardí, M.; Ballestar, E.; González, S.; Serrano, A.L.; et al. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature 2014, 506, 316–321. [Google Scholar] [CrossRef]
- Gonzalez-Meljem, J.M.; Haston, S.; Carreno, G.; Apps, J.R.; Pozzi, S.; Stache, C.; Kaushal, G.; Virasami, A.; Panousopoulos, L.; Mousavy-Gharavy, S.N.; et al. Stem cell senescence drives age-attenuated induction of pituitary tumours in mouse models of paediatric craniopharyngioma. Nat. Commun. 2017, 8, 1819. [Google Scholar] [CrossRef]
- Xia, W.; Hou, M. Macrophage migration inhibitory factor rescues mesenchymal stem cells from doxorubicin-induced senescence though the PI3K-Akt signaling pathway. Int. J. Mol. Med. 2017, 41, 1127–1137. [Google Scholar] [CrossRef]
- Di Leonardo, A.; Linke, S.P.; Clarkin, K.; Wahl, G.M. DNA damage triggers a prolonged p53-dependent G1 arrest and long-term induction of Cip1 in normal human fibroblasts. Genes Dev. 1994, 8, 2540–2551. [Google Scholar] [CrossRef]
- Dumont, P.; Balbeur, L.; Remacle, J.; Toussaint, O. Appearance of biomarkers of in vitro ageing after successive stimulation of WI-38 fibroblasts with IL-1alpha and TNF-alpha: Senescence associated beta-galactosidase activity and morphotype transition. J. Anat. 2000, 197, 529–537. [Google Scholar] [CrossRef]
- Di Fagagna, F.D.A.; Reaper, P.M.; Clay-Farrace, L.; Fiegler, H.; Carr, P.; von Zglinicki, T.; Saretzki, G.; Carter, N.P.; Jackson, S.P. A DNA damage checkpoint response in telomere-initiated senescence. Nature 2003, 426, 194–198. [Google Scholar] [CrossRef]
- Nakamura, A.J.; Chiang, Y.J.; Hathcock, K.S.; Horikawa, I.; Sedelnikova, O.A.; Hodes, R.J.; Bonner, W.M. Both telomeric and non-telomeric DNA damage are determinants of mammalian cellular senescence. Epigenetics Chromatin 2008, 1, 6. [Google Scholar] [CrossRef]
- Coppé, J.-P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.-Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, e301. [Google Scholar]
- Acosta, J.C.; O’Loghlen, A.; Banito, A.; Guijarro, M.V.; Augert, A.; Raguz, S.; Fumagalli, M.; Da Costa, M.; Brown, C.; Popov, N.; et al. Chemokine Signaling via the CXCR2 Receptor Reinforces Senescence. Cell 2008, 133, 1006–1018. [Google Scholar] [CrossRef]
- Rodier, F.; Coppé, J.-P.; Patil, C.K.; Hoeijmakers, W.A.M.; Muñoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 2009, 11, 973–979. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Cañamero, M.; Maraver, A.; Gómez-López, G.; Contreras, J.; Murillo-Cuesta, S.; Rodríguez-Baeza, A.; Varela-Nieto, I.; Ruberte, J.; Collado, M.; et al. Programmed Cell Senescence during Mammalian Embryonic Development. Cell 2013, 155, 1104–1118. [Google Scholar] [CrossRef]
- Storer, M.; Mas, A.; Robert-Moreno, A.; Pecoraro, M.; Ortells, M.C.; Di Giacomo, V.; Yosef, R.; Pilpel, N.; Krizhanovsky, V.; Sharpe, J.; et al. Senescence Is a Developmental Mechanism that Contributes to Embryonic Growth and Patterning. Cell 2013, 155, 1119–1130. [Google Scholar] [CrossRef]
- Yun, M.H.; Davaapil, H.; Brockes, J.P. Recurrent turnover of senescent cells during regeneration of a complex structure. Elife 2015, 4. [Google Scholar] [CrossRef]
- Demaria, M.; Ohtani, N.; Youssef, S.A.; Rodier, F.; Toussaint, W.; Mitchell, J.R.; Laberge, R.-M.; Vijg, J.; Van Steeg, H.; Dollé, M.E.T.; et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell 2014, 31, 722–733. [Google Scholar] [CrossRef]
- Ritschka, B.; Storer, M.; Mas, A.; Heinzmann, F.; Ortells, M.C.; Morton, J.P.; Sansom, O.J.; Zender, L.; Keyes, W.M. The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev. 2017, 31, 172–183. [Google Scholar] [CrossRef]
- Mosteiro, L.; Pantoja, C.; Alcazar, N.; Marión, R.M.; Chondronasiou, D.; Rovira, M.; Fernandez-Marcos, P.J.; Muñoz-Martin, M.; Blanco-Aparicio, C.; Pastor, J.; et al. Tissue damage and senescence provide critical signals for cellular reprogramming in vivo. Science 2016, 354, aaf4445. [Google Scholar] [CrossRef]
- Chiche, A.; Le Roux, I.; von Joest, M.; Sakai, H.; Aguín, S.B.; Cazin, C.; Salam, R.; Fiette, L.; Alegria, O.; Flamant, P.; et al. Injury-Induced Senescence Enables In Vivo Reprogramming in Skeletal Muscle. Cell Stem Cell 2017, 20, 407.e4–414.e4. [Google Scholar] [CrossRef]
- Castro-Vega, L.J.; Jouravleva, K.; Ortiz-Montero, P.; Liu, W.-Y.; Galeano, J.L.; Romero, M.; Popova, T.; Bacchetti, S.; Vernot, J.P.; Londoño-Vallejo, A. The senescent microenvironment promotes the emergence of heterogeneous cancer stem-like cells. Carcinogenesis 2015, 36, 1180–1192. [Google Scholar] [CrossRef]
- Cahu, J.; Bustany, S.; Sola, B. Senescence-associated secretory phenotype favors the emergence of cancer stem-like cells. Cell Death Dis. 2012, 3, e446. [Google Scholar] [CrossRef]
- Canino, C.; Mori, F.; Cambria, A.; Diamantini, A.; Germoni, S.; Alessandrini, G.; Borsellino, G.; Galati, R.; Battistini, L.; Blandino, R.; et al. SASP mediates chemoresistance and tumor-initiating-activity of mesothelioma cells. Oncogene 2012, 31, 3148–3163. [Google Scholar] [CrossRef]
- Banito, A.; Rashid, S.T.; Acosta, J.C.; Li, S.; Pereira, C.F.; Geti, I.; Pinho, S.; Silva, J.C.; Azuara, V.; Walsh, M.; et al. Senescence impairs successful reprogramming to pluripotent stem cells. Genes Dev. 2009, 23, 2134–2139. [Google Scholar] [CrossRef]
- Koch, C.M.; Reck, K.; Shao, K.; Lin, Q.; Joussen, S.; Ziegler, P.; Walenda, G.; Drescher, W.; Opalka, B.; May, T.; et al. Pluripotent stem cells escape from senescence-associated DNA methylation changes. Genome Res. 2013, 23, 248–259. [Google Scholar] [CrossRef]
- García-Prat, L.; Martínez-Vicente, M.; Perdiguero, E.; Ortet, L.; Rodríguez-Ubreva, J.; Rebollo, E.; Ruiz-Bonilla, V.; Gutarra, S.; Ballestar, E.; Serrano, A.L.; et al. Autophagy maintains stemness by preventing senescence. Nature 2016, 529, 37–42. [Google Scholar] [CrossRef]
- Xing, J.; Ying, Y.; Mao, C.; Liu, Y.; Wang, T.; Zhao, Q.; Zhang, X.; Yan, F.; Zhang, H. Hypoxia induces senescence of bone marrow mesenchymal stem cells via altered gut microbiota. Nat. Commun. 2018, 9, 2020. [Google Scholar] [CrossRef]
- Kawamura, H.; Nakatsuka, R.; Matsuoka, Y.; Sumide, K.; Fujioka, T.; Asano, H.; Iida, H.; Sonoda, Y. TGF-β Signaling Accelerates Senescence of Human Bone-Derived CD271 and SSEA-4 Double-Positive Mesenchymal Stromal Cells. Stem Cell Rep. 2018, 10, 920–932. [Google Scholar] [CrossRef]
- Kobayashi, A.; Okuda, H.; Xing, F.; Pandey, P.R.; Watabe, M.; Hirota, S.; Pai, S.K.; Liu, W.; Fukuda, K.; Chambers, C.; et al. Bone morphogenetic protein 7 in dormancy and metastasis of prostate cancer stem-like cells in bone. J. Exp. Med. 2011, 208, 2641–2655. [Google Scholar] [CrossRef]
- Teo, Y.V.; Rattanavirotkul, N.; Olova, N.; Salzano, A.; Quintanilla, A.; Tarrats, N.; Kiourtis, C.; Müller, M.; Green, A.R.; Adams, P.D.; et al. Notch Signaling Mediates Secondary Senescence. Cell Rep. 2019, 27, 997.e5–1007.e5. [Google Scholar] [CrossRef]
- Lowe, S.W.; Cepero, E.; Evan, G. Intrinsic tumour suppression. Nature 2004, 432, 307–315. [Google Scholar] [CrossRef]
- Chen, Z.; Trotman, L.C.; Shaffer, D.; Lin, H.-K.; Dotan, Z.A.; Niki, M.; Koutcher, J.A.; Scher, H.I.; Ludwig, T.; Gerald, W.; et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005, 436, 725–730. [Google Scholar] [CrossRef]
- Braig, M.; Lee, S.; Loddenkemper, C.; Rudolph, C.; Peters, A.H.F.M.; Schlegelberger, B.; Stein, H.; Dörken, B.; Jenuwein, T.; Schmitt, C.A. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature 2005, 436, 660–665. [Google Scholar] [CrossRef]
- Wu, C.-H.; van Riggelen, J.; Yetil, A.; Fan, A.C.; Bachireddy, P.; Felsher, D.W. Cellular senescence is an important mechanism of tumor regression upon c-Myc inactivation. Proc. Natl. Acad. Sci. USA 2007, 104, 13028–13033. [Google Scholar] [CrossRef]
- Rakhra, K.; Bachireddy, P.; Zabuawala, T.; Zeiser, R.; Xu, L.; Kopelman, A.; Fan, A.C.; Yang, Q.; Braunstein, L.; Crosby, E.; et al. CD4+ T Cells Contribute to the Remodeling of the Microenvironment Required for Sustained Tumor Regression upon Oncogene Inactivation. Cancer Cell 2010, 18, 485–498. [Google Scholar] [CrossRef]
- Seoane, M.; Costoya, J.A.; Arce, V.M. Uncoupling Oncogene-Induced Senescence (OIS) and DNA Damage Response (DDR) triggered by DNA hyper-replication: Lessons from primary mouse embryo astrocytes (MEA). Sci. Rep. 2017, 7, 12991. [Google Scholar] [CrossRef]
- Toso, A.; Revandkar, A.; Di Mitri, D.; Guccini, I.; Proietti, M.; Sarti, M.; Pinton, S.; Zhang, J.; Kalathur, M.; Civenni, G.; et al. Enhancing Chemotherapy Efficacy in Pten -Deficient Prostate Tumors by Activating the Senescence-Associated Antitumor Immunity. Cell Rep. 2014, 9, 75–89. [Google Scholar] [CrossRef]
- Fan, D.N.Y.; Schmitt, C.A. Detecting Markers of Therapy-Induced Senescence in Cancer Cells. In Oncogene-Induced Senescence; Humana Press: New York, NY, USA, 2017; Volume 1534, pp. 41–52. [Google Scholar]
- Ewald, J.A.; Desotelle, J.A.; Wilding, G.; Jarrard, D.F. Therapy-induced senescence in cancer. J. Natl. Cancer Inst. 2010, 102, 1536–1546. [Google Scholar] [CrossRef]
- Demaria, M.; O’Leary, M.N.; Chang, J.; Shao, L.; Liu, S.; Alimirah, F.; Koenig, K.; Le, C.; Mitin, N.; Deal, A.M.; et al. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov. 2017, 7, 165–176. [Google Scholar] [CrossRef]
- Prunier, C.; Baker, D.; Ten Dijke, P.; Ritsma, L. TGF-β Family Signaling Pathways in Cellular Dormancy. Trends Cancer 2019, 5, 66–78. [Google Scholar] [CrossRef]
- Saleh, T.; Tyutyunyk-Massey, L.; Murray, G.F.; Alotaibi, M.R.; Kawale, A.S.; Elsayed, Z.; Henderson, S.C.; Yakovlev, V.; Elmore, L.W.; Toor, A.; et al. Tumor cell escape from therapy-induced senescence. Biochem. Pharmacol. 2018, 162, 202–212. [Google Scholar] [CrossRef]
- Saleh, T.; Tyutyunyk-Massey, L.; Gewirtz, D.A. Tumor Cell Escape from Therapy-Induced Senescence as a Model of Disease Recurrence after Dormancy. Cancer Res. 2019, 79, 1044–1046. [Google Scholar] [CrossRef]
- Ohuchida, K.; Mizumoto, K.; Murakami, M.; Qian, L.-W.; Sato, N.; Nagai, E.; Matsumoto, K.; Nakamura, T.; Tanaka, M. Radiation to stromal fibroblasts increases invasiveness of pancreatic cancer cells through tumor-stromal interactions. Cancer Res. 2004, 64, 3215–3222. [Google Scholar] [CrossRef]
- Coppé, J.-P.; Kauser, K.; Campisi, J.; Beauséjour, C.M. Secretion of vascular endothelial growth factor by primary human fibroblasts at senescence. J. Biol. Chem. 2006, 281, 29568–29574. [Google Scholar] [CrossRef]
- Krtolica, A.; Parrinello, S.; Lockett, S.; Desprez, P.-Y.; Campisi, J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: A link between cancer and aging. Proc. Natl. Acad. Sci. USA 2001, 98, 12072–12077. [Google Scholar] [CrossRef]
- Guan, X.; LaPak, K.M.; Hennessey, R.C.; Yu, C.Y.; Shakya, R.; Zhang, J.; Burd, C.E. Stromal Senescence By Prolonged CDK4/6 Inhibition Potentiates Tumor Growth. Mol. Cancer Res. 2017, 15, 237–249. [Google Scholar] [CrossRef]
- Bavik, C.; Coleman, I.; Dean, J.P.; Knudsen, B.; Plymate, S.; Nelson, P.S. The Gene Expression Program of Prostate Fibroblast Senescence Modulates Neoplastic Epithelial Cell Proliferation through Paracrine Mechanisms. Cancer Res. 2006, 66, 794–802. [Google Scholar] [CrossRef]
- Wang, C.; Vegna, S.; Jin, H.; Benedict, B.; Lieftink, C.; Ramirez, C.; de Oliveira, R.L.; Morris, B.; Gadiot, J.; Wang, W.; et al. Inducing and exploiting vulnerabilities for the treatment of liver cancer. Nature 2019, 574, 268–272. [Google Scholar] [CrossRef]
- Tato-Costa, J.; Casimiro, S.; Pacheco, T.; Pires, R.; Fernandes, A.; Alho, I.; Pereira, P.; Costa, P.; Castelo, H.B.; Ferreira, J.; et al. Therapy-Induced Cellular Senescence Induces Epithelial-to-Mesenchymal Transition and Increases Invasiveness in Rectal Cancer. Clin. Colorectal Cancer 2016, 15, 170.e3–178.e3. [Google Scholar] [CrossRef]
- Hassona, Y.; Cirillo, N.; Heesom, K.; Parkinson, E.K.; Prime, S.S. Senescent cancer-associated fibroblasts secrete active MMP-2 that promotes keratinocyte dis-cohesion and invasion. Br. J. Cancer 2014, 111, 1230–1237. [Google Scholar] [CrossRef]
- Kaur, A.; Ecker, B.L.; Douglass, S.M.; Kugel, C.H.; Webster, M.R.; Almeida, F.V.; Somasundaram, R.; Hayden, J.; Ban, E.; Ahmadzadeh, H.; et al. Remodeling of the Collagen Matrix in Aging Skin Promotes Melanoma Metastasis and Affects Immune Cell Motility. Cancer Discov. 2018, 9, 64–81. [Google Scholar] [CrossRef]
- Ksiazek, K.; Jörres, A.; Witowski, J. Senescence Induces a Proangiogenic Switch in Human Peritoneal Mesothelial Cells. Rejuvenation Res. 2008, 11, 681–683. [Google Scholar] [CrossRef]
- Oubaha, M.; Miloudi, K.; Dejda, A.; Guber, V.; Mawambo, G.; Germain, M.-A.; Bourdel, G.; Popovic, N.; Rezende, F.A.; Kaufman, R.J.; et al. Senescence-associated secretory phenotype contributes to pathological angiogenesis in retinopathy. Sci. Transl. Med. 2016, 8, 362ra144. [Google Scholar] [CrossRef]
- Kim, Y.H.; Choi, Y.W.; Lee, J.; Soh, E.Y.; Kim, J.-H.; Park, T.J. Senescent tumor cells lead the collective invasion in thyroid cancer. Nat. Commun. 2017, 8, 15208. [Google Scholar] [CrossRef]
- Webster, M.R.; Xu, M.; Kinzler, K.A.; Kaur, A.; Appleton, J.; O’Connell, M.P.; Marchbank, K.; Valiga, A.; Dang, V.M.; Perego, M.; et al. Wnt5A promotes an adaptive, senescent-like stress response, while continuing to drive invasion in melanoma cells. Pigment Cell Melanoma Res. 2015, 28, 184–195. [Google Scholar] [CrossRef]
- Lee, H.-G.; Kim, J.H.; Sun, W.; Chi, S.-G.; Choi, W.; Lee, K.J. Senescent tumor cells building three-dimensional tumor clusters. Sci. Rep. 2018, 8, 10503. [Google Scholar] [CrossRef]
- Luo, X.; Fu, Y.; Loza, A.J.; Murali, B.; Leahy, K.M.; Ruhland, M.K.; Gang, M.; Su, X.; Zamani, A.; Shi, Y.; et al. Stromal-Initiated Changes in the Bone Promote Metastatic Niche Development. Cell Rep. 2016, 14, 82–92. [Google Scholar] [CrossRef]
- Sosa, M.S.; Bragado, P.; Aguirre-Ghiso, J.A. Mechanisms of disseminated cancer cell dormancy: An awakening field. Nat. Rev. Cancer 2014, 14, 611–622. [Google Scholar] [CrossRef]
- Dalai, S.K.; Khoruzhenko, S.; Drake, C.G.; Jie, C.C.; Sadegh-Nasseri, S. Resolution of infection promotes a state of dormancy and long survival of CD4 memory T cells. Immunol. Cell Biol. 2011, 89, 870–881. [Google Scholar] [CrossRef]
- Manjili, M.H. Tumor Dormancy and Relapse: From a Natural Byproduct of Evolution to a Disease State. Cancer Res. 2017, 77, 2564–2569. [Google Scholar] [CrossRef]
- Aguirre-Ghiso, J.A. Models, mechanisms and clinical evidence for cancer dormancy. Nat. Rev. Cancer 2007, 7, 834–846. [Google Scholar] [CrossRef]
- Kang, Y.; Siegel, P.M.; Shu, W.; Drobnjak, M.; Kakonen, S.M.; Cordón-Cardo, C.; Guise, T.A.; Massagué, J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 2003, 3, 537–549. [Google Scholar] [CrossRef]
- Bacac, M.; Stamenkovic, I. Metastatic Cancer Cell. Annu. Rev. Pathol. Mech. Dis. 2008, 3, 221–247. [Google Scholar] [CrossRef]
- Demicheli, R.; Miceli, R.; Moliterni, A.; Zambetti, M.; Hrushesky, W.J.M.; Retsky, M.W.; Valagussa, P.; Bonadonna, G. Breast cancer recurrence dynamics following adjuvant CMF is consistent with tumor dormancy and mastectomy-driven acceleration of the metastatic process. Ann. Oncol. 2005, 16, 1449–1457. [Google Scholar] [CrossRef]
- Aguirre-Ghiso, J.A.; Sosa, M.S. Emerging Topics on Disseminated Cancer Cell Dormancy and the Paradigm of Metastasis. Annu. Rev. Cancer Biol. 2018, 2, 377–393. [Google Scholar] [CrossRef]
- Vera-Ramirez, L.; Vodnala, S.K.; Nini, R.; Hunter, K.W.; Green, J.E. Autophagy promotes the survival of dormant breast cancer cells and metastatic tumour recurrence. Nat. Commun. 2018, 9, 1944. [Google Scholar] [CrossRef]
- Meng, S.; Tripathy, D.; Frenkel, E.P.; Shete, S.; Naftalis, E.Z.; Huth, J.F.; Beitsch, P.D.; Leitch, M.; Hoover, S.; Euhus, D.; et al. Circulating Tumor Cells in Patients with Breast Cancer Dormancy. Clin. Cancer Res. 2004, 10, 8152–8162. [Google Scholar] [CrossRef]
- Borgen, E.; Rypdal, M.C.; Sosa, M.S.; Renolen, A.; Schlichting, E.; Lønning, P.E.; Synnestvedt, M.; Aguirre-Ghiso, J.A.; Naume, B. NR2F1 stratifies dormant disseminated tumor cells in breast cancer patients. Breast Cancer Res. 2018, 20, 120. [Google Scholar] [CrossRef]
- Van ’t Veer, L.J.; Dai, H.; van de Vijver, M.J.; He, Y.D.; Hart, A.A.M.; Mao, M.; Peterse, H.L.; van der Kooy, K.; Marton, M.J.; Witteveen, A.T.; et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature 2002, 415, 530–536. [Google Scholar] [CrossRef]
- Gangnus, R.; Langer, S.; Breit, E.; Pantel, K.; Speicher, M.R. Genomic Profiling of Viable and Proliferative Micrometastatic Cells from Early-Stage Breast Cancer Patients. Clin. Cancer Res. 2004, 10, 3457–3464. [Google Scholar] [CrossRef]
- Caswell, D.R.; Chuang, C.-H.; Yang, D.; Chiou, S.-H.; Cheemalavagu, S.; Kim-Kiselak, C.; Connolly, A.; Winslow, M.M. Obligate progression precedes lung adenocarcinoma dissemination. Cancer Discov. 2014, 4, 781–789. [Google Scholar] [CrossRef]
- Hüsemann, Y.; Geigl, J.B.; Schubert, F.; Musiani, P.; Meyer, M.; Burghart, E.; Forni, G.; Eils, R.; Fehm, T.; Riethmüller, G.; et al. Systemic Spread Is an Early Step in Breast Cancer. Cancer Cell 2008, 13, 58–68. [Google Scholar] [CrossRef]
- Eyles, J.; Puaux, A.-L.; Wang, X.; Toh, B.; Prakash, C.; Hong, M.; Tan, T.G.; Zheng, L.; Ong, L.C.; Jin, Y.; et al. Tumor cells disseminate early, but immunosurveillance limits metastatic outgrowth, in a mouse model of melanoma. J. Clin. Investig. 2010, 120, 2030–2039. [Google Scholar] [CrossRef]
- Nielsen, M.; Thomsen, J.; Primdahl, S.; Dyreborg, U.; Andersen, J. Breast cancer and atypia among young and middle-aged women: A study of 110 medicolegal autopsies. Br. J. Cancer 1987, 56, 814–819. [Google Scholar] [CrossRef]
- Hosseini, H.; Obradović, M.M.S.; Hoffmann, M.; Harper, K.L.; Sosa, M.S.; Werner-Klein, M.; Nanduri, L.K.; Werno, C.; Ehrl, C.; Maneck, M.; et al. Early dissemination seeds metastasis in breast cancer. Nature 2016, 540, 552–558. [Google Scholar] [CrossRef]
- Morris, V.L.; Koop, S.; MacDonald, I.C.; Schmidt, E.E.; Grattan, M.; Percy, D.; Chambers, A.F.; Groom, A.C. Mammary carcinoma cell lines of high and low metastatic potential differ not in extravasation but in subsequent migration and growth. Clin. Exp. Metastasis 1994, 12, 357–367. [Google Scholar] [CrossRef]
- Barkan, D.; Kleinman, H.; Simmons, J.L.; Asmussen, H.; Kamaraju, A.K.; Hoenorhoff, M.J.; Liu, Z.-Y.; Costes, S.V.; Cho, E.H.; Lockett, S.; et al. Inhibition of Metastatic Outgrowth from Single Dormant Tumor Cells by Targeting the Cytoskeleton. Cancer Res. 2008, 68, 6241–6250. [Google Scholar] [CrossRef]
- Barkan, D.; El Touny, L.H.; Michalowski, A.M.; Smith, J.A.; Chu, I.; Davis, A.S.; Webster, J.D.; Hoover, S.; Simpson, R.M.; Gauldie, J.; et al. Metastatic Growth from Dormant Cells Induced by a Col-I-Enriched Fibrotic Environment. Cancer Res. 2010, 70, 5706–5716. [Google Scholar] [CrossRef]
- Rajbhandari, N.; Lin, W.; Wehde, B.L.; Triplett, A.A.; Wagner, K.-U. Autocrine IGF1 Signaling Mediates Pancreatic Tumor Cell Dormancy in the Absence of Oncogenic Drivers. Cell Rep. 2017, 18, 2243–2255. [Google Scholar] [CrossRef]
- Romero, I.; Garrido, F.; Garcia-Lora, A.M. Metastases in Immune-Mediated Dormancy: A New Opportunity for Targeting Cancer. Cancer Res. 2014, 74, 6750–6757. [Google Scholar] [CrossRef]
- Giancotti, F.G. Mechanisms Governing Metastatic Dormancy and Reactivation. Cell 2013, 155, 750–764. [Google Scholar] [CrossRef]
- Lytle, N.K.; Barber, A.G.; Reya, T. Stem cell fate in cancer growth, progression and therapy resistance. Nat. Rev. Cancer 2018, 18, 669–680. [Google Scholar] [CrossRef]
- Dembinski, J.L.; Krauss, S. Characterization and functional analysis of a slow cycling stem cell-like subpopulation in pancreas adenocarcinoma. Clin. Exp. Metastasis 2009, 26, 611–623. [Google Scholar] [CrossRef]
- Liu, X.; Johnson, S.; Liu, S.; Kanojia, D.; Yue, W.; Singh, U.P.; Singn, U.; Wang, Q.; Wang, Q.; Nie, Q.; et al. Nonlinear growth kinetics of breast cancer stem cells: Implications for cancer stem cell targeted therapy. Sci. Rep. 2013, 3, 2473. [Google Scholar] [CrossRef]
- Boman, B.M.; Wicha, M.S. Cancer Stem Cells: A Step Toward the Cure. J. Clin. Oncol. 2008, 26, 2795–2799. [Google Scholar] [CrossRef]
- Li, L.; Li, J.-C.; Yang, H.; Zhang, X.; Liu, L.-L.; Li, Y.; Zeng, T.-T.; Zhu, Y.-H.; Li, X.-D.; Li, Y.; et al. Expansion of cancer stem cell pool initiates lung cancer recurrence before angiogenesis. Proc. Natl. Acad. Sci. USA 2018, 115, E8948–E8957. [Google Scholar] [CrossRef]
- Gao, H.; Chakraborty, G.; Lee-Lim, A.P.; Mo, Q.; Decker, M.; Vonica, A.; Shen, R.; Brogi, E.; Brivanlou, A.H.; Giancotti, F.G. The BMP inhibitor Coco reactivates breast cancer cells at lung metastatic sites. Cell 2012, 150, 764–779. [Google Scholar] [CrossRef]
- Bragado, P.; Estrada, Y.; Parikh, F.; Krause, S.; Capobianco, C.; Farina, H.G.; Schewe, D.M.; Aguirre-Ghiso, J.A. TGF-β2 dictates disseminated tumour cell fate in target organs through TGF-β-RIII and p38α/β signalling. Nat. Cell Biol. 2013, 15, 1351–1361. [Google Scholar] [CrossRef]
- Taichman, R.S.; Patel, L.R.; Bedenis, R.; Wang, J.; Weidner, S.; Schumann, T.; Yumoto, K.; Berry, J.E.; Shiozawa, Y.; Pienta, K.J. GAS6 Receptor Status Is Associated with Dormancy and Bone Metastatic Tumor Formation. PLoS ONE 2013, 8, e61873. [Google Scholar] [CrossRef]
- Harper, K.L.; Sosa, M.S.; Entenberg, D.; Hosseini, H.; Cheung, J.F.; Nobre, R.; Avivar-Valderas, A.; Nagi, C.; Girnius, N.; Davis, R.J.; et al. Mechanism of early dissemination and metastasis in Her2+ mammary cancer. Nature 2016, 540, 588–592. [Google Scholar] [CrossRef] [PubMed]
- Sosa, M.S.; Parikh, F.; Maia, A.G.; Estrada, Y.; Bosch, A.; Bragado, P.; Ekpin, E.; George, A.; Zheng, Y.; Lam, H.-M.; et al. NR2F1 controls tumour cell dormancy via SOX9- and RARβ-driven quiescence programmes. Nat. Commun. 2015, 6, 6170. [Google Scholar] [CrossRef] [PubMed]
- Pommier, A.; Anaparthy, N.; Memos, N.; Kelley, Z.L.; Gouronnec, A.; Yan, R.; Auffray, C.; Albrengues, J.; Egeblad, M.; Iacobuzio-Donahue, C.A.; et al. Unresolved endoplasmic reticulum stress engenders immune-resistant, latent pancreatic cancer metastases. Science 2018, 360, eaao4908. [Google Scholar] [CrossRef] [PubMed]
- De Cock, J.M.; Shibue, T.; Dongre, A.; Keckesova, Z.; Reinhardt, F.; Weinberg, R.A. Inflammation Triggers Zeb1-Dependent Escape from Tumor Latency. Cancer Res. 2016, 76, 6778–6784. [Google Scholar] [CrossRef]
- Sosa, M.S.; Avivar-Valderas, A.; Bragado, P.; Wen, H.-C.; Aguirre-Ghiso, J.A. ERK1/2 and p38α/β signaling in tumor cell quiescence: Opportunities to control dormant residual disease. Clin. Cancer Res. 2011, 17, 5850–5857. [Google Scholar] [CrossRef]
- Gao, X.-L.; Zhang, M.; Tang, Y.-L.; Liang, X.-H. Cancer cell dormancy: Mechanisms and implications of cancer recurrence and metastasis. Onco Targets Ther. 2017, 10, 5219–5228. [Google Scholar] [CrossRef]
- Schewe, D.M.; Aguirre-Ghiso, J.A. ATF6-Rheb-mTOR signaling promotes survival of dormant tumor cells in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 10519–10524. [Google Scholar] [CrossRef]
- Hamilton, W.B.; Brickman, J.M. Erk Signaling Suppresses Embryonic Stem Cell Self-Renewal to Specify Endoderm. Cell Rep. 2014, 9, 2056–2070. [Google Scholar] [CrossRef]
- Altshuler, A.; Verbuk, M.; Bhattacharya, S.; Abramovich, I.; Haklai, R.; Hanna, J.H.; Kloog, Y.; Gottlieb, E.; Shalom-Feuerstein, R. RAS Regulates the Transition from Naive to Primed Pluripotent Stem Cells. Stem Cell Rep. 2018, 10, 1088–1101. [Google Scholar] [CrossRef]
- Rybak, A.P.; Tang, D. SOX2 plays a critical role in EGFR-mediated self-renewal of human prostate cancer stem-like cells. Cell. Signal. 2013, 25, 2734–2742. [Google Scholar] [CrossRef]
- Moon, B.-S.; Jeong, W.-J.; Park, J.; Kim, T.I.; Min, D.S.; Choi, K.-Y. Role of Oncogenic K-Ras in Cancer Stem Cell Activation by Aberrant Wnt/β-Catenin Signaling. JNCI J. Natl. Cancer Inst. 2014, 106, djt373. [Google Scholar] [CrossRef] [PubMed]
- Haston, S.; Pozzi, S.; Carreno, G.; Manshaei, S.; Panousopoulos, L.; Gonzalez-Meljem, J.M.; Apps, J.R.; Virasami, A.; Thavaraj, S.; Gutteridge, A.; et al. MAPK pathway control of stem cell proliferation and differentiation in the embryonic pituitary provides insights into the pathogenesis of papillary craniopharyngioma. Development 2017, 144, 2141–2152. [Google Scholar] [CrossRef] [PubMed]
- Ferreirós, A.; Pedrosa, P.; Da Silva-Álvarez, S.; Triana-Martínez, F.; Vilas, J.M.; Picallos-Rabina, P.; González, P.; Gómez, M.; Li, H.; García-Caballero, T.; et al. Context-Dependent Impact of RAS Oncogene Expression on Cellular Reprogramming to Pluripotency. Stem Cell Rep. 2019, 12, 1099–1112. [Google Scholar]
- Sato, A.; Okada, M.; Shibuya, K.; Watanabe, E.; Seino, S.; Narita, Y.; Shibui, S.; Kayama, T.; Kitanaka, C. Pivotal role for ROS activation of p38 MAPK in the control of differentiation and tumor-initiating capacity of glioma-initiating cells. Stem Cell Res. 2014, 12, 119–131. [Google Scholar] [CrossRef]
- Guinot, A.; Oeztuerk-Winder, F.; Ventura, J.-J. miR-17-92/p38α Dysregulation Enhances Wnt Signaling and Selects Lgr6 + Cancer Stem-like Cells during Lung Adenocarcinoma Progression. Cancer Res. 2016, 76, 4012–4022. [Google Scholar] [CrossRef]
- Soeda, A.; Lathia, J.; Williams, B.J.; Wu, Q.; Gallagher, J.; Androutsellis-Theotokis, A.; Giles, A.J.; Yang, C.; Zhuang, Z.; Gilbert, M.R.; et al. The p38 signaling pathway mediates quiescence of glioma stem cells by regulating epidermal growth factor receptor trafficking. Oncotarget 2017, 8, 33316–33328. [Google Scholar] [CrossRef]
- Lu, H.; Tran, L.; Park, Y.; Chen, I.; Lan, J.; Xie, Y.; Semenza, G.L. Reciprocal Regulation of DUSP9 and DUSP16 Expression by HIF1 Controls ERK and p38 MAP Kinase Activity and Mediates Chemotherapy-Induced Breast Cancer Stem Cell Enrichment. Cancer Res. 2018, 78, 4191–4202. [Google Scholar] [CrossRef]
- Courtois-Cox, S.; Genther Williams, S.M.; Reczek, E.E.; Johnson, B.W.; McGillicuddy, L.T.; Johannessen, C.M.; Hollstein, P.E.; MacCollin, M.; Cichowski, K. A negative feedback signaling network underlies oncogene-induced senescence. Cancer Cell 2006, 10, 459–472. [Google Scholar] [CrossRef]
- Kwong, J.; Hong, L.; Liao, R.; Deng, Q.; Han, J.; Sun, P. p38alpha and p38gamma mediate oncogenic ras-induced senescence through differential mechanisms. J. Biol. Chem. 2009, 284, 11237–11246. [Google Scholar] [CrossRef]
- Xu, Y.; Li, N.; Xiang, R.; Sun, P. Emerging roles of the p38 MAPK and PI3K/AKT/mTOR pathways in oncogene-induced senescence. Trends Biochem. Sci. 2014, 39, 268–276. [Google Scholar] [CrossRef]
- Puig, I.; Tenbaum, S.P.; Chicote, I.; Arqués, O.; Martínez-Quintanilla, J.; Cuesta-Borrás, E.; Ramírez, L.; Gonzalo, P.; Soto, A.; Aguilar, S.; et al. TET2 controls chemoresistant slow-cycling cancer cell survival and tumor recurrence. J. Clin. Investig. 2018, 128, 3887–3905. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Xing, F.; Liu, Y.; Wu, K.; Said, N.; Pochampally, R.; Shiozawa, Y.; Lin, H.-K.; Balaji, K.C.; Watabe, K. Secreted Protein Acidic and Rich in Cysteine (SPARC) Mediates Metastatic Dormancy of Prostate Cancer in Bone. J. Biol. Chem. 2016, 291, 19351–19363. [Google Scholar] [CrossRef] [PubMed]
- Bartosh, T.J.; Ullah, M.; Zeitouni, S.; Beaver, J.; Prockop, D.J. Cancer cells enter dormancy after cannibalizing mesenchymal stem/stromal cells (MSCs). Proc. Natl. Acad. Sci. USA 2016, 113, E6447–E6456. [Google Scholar] [CrossRef] [PubMed]
- Katakura, Y.; Nakata, E.; Miura, T.; Shirahata, S. Transforming Growth Factor β Triggers Two Independent-Senescence Programs in Cancer Cells. Biochem. Biophys. Res. Commun. 1999, 255, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.L.; Birke, K.; Moriniere, J.; Welge-Lüssen, U. TGF-β2 Induces Senescence-Associated Changes in Human Trabecular Meshwork Cells. Investig. Opthalmol. Vis. Sci. 2010, 51, 5718. [Google Scholar] [CrossRef] [PubMed]
- Rapisarda, V.; Borghesan, M.; Miguela, V.; Encheva, V.; Snijders, A.P.; Lujambio, A.; O’Loghlen, A. Integrin Beta 3 Regulates Cellular Senescence by Activating the TGF-β Pathway. Cell Rep. 2017, 18, 2480–2493. [Google Scholar] [CrossRef]
- Senturk, S.; Mumcuoglu, M.; Gursoy-Yuzugullu, O.; Cingoz, B.; Akcali, K.C.; Ozturk, M. Transforming growth factor-beta induces senescence in hepatocellular carcinoma cells and inhibits tumor growth. Hepatology 2010, 52, 966–974. [Google Scholar] [CrossRef]
- Gibaja, A.; Aburto, M.R.; Pulido, S.; Collado, M.; Hurle, J.M.; Varela-Nieto, I.; Magariños, M. TGFβ2-induced senescence during early inner ear development. Sci. Rep. 2019, 9, 5912. [Google Scholar] [CrossRef]
- Yumoto, K.; Eber, M.R.; Wang, J.; Cackowski, F.C.; Decker, A.M.; Lee, E.; Nobre, A.R.; Aguirre-Ghiso, J.A.; Jung, Y.; Taichman, R.S. Axl is required for TGF-β2-induced dormancy of prostate cancer cells in the bone marrow. Sci. Rep. 2016, 6, 36520. [Google Scholar] [CrossRef]
- Linde, N.; Fluegen, G.; Aguirre-Ghiso, J.A. The Relationship Between Dormant Cancer Cells and Their Microenvironment. Adv. Cancer Res. 2016, 132, 45–71. [Google Scholar]
- Bui, A.T.; Laurent, F.; Havard, M.; Dautry, F.; Tchénio, T. SMAD signaling and redox imbalance cooperate to induce prostate cancer cell dormancy. Cell Cycle 2015, 14, 1218–1231. [Google Scholar] [CrossRef][Green Version]
- Brown, J.A.; Yonekubo, Y.; Hanson, N.; Sastre-Perona, A.; Basin, A.; Rytlewski, J.A.; Dolgalev, I.; Meehan, S.; Tsirigos, A.; Beronja, S.; et al. TGF-β-Induced Quiescence Mediates Chemoresistance of Tumor-Propagating Cells in Squamous Cell Carcinoma. Cell Stem Cell 2017, 21, 650.e8–664.e8. [Google Scholar] [CrossRef]
- Scandura, J.M.; Boccuni, P.; Massague, J.; Nimer, S.D. Transforming growth factor-induced cell cycle arrest of human hematopoietic cells requires p57KIP2 up-regulation. Proc. Natl. Acad. Sci. USA 2004, 101, 15231–15236. [Google Scholar] [CrossRef]
- Cassar, L.; Nicholls, C.; Pinto, A.R.; Chen, R.; Wang, L.; Li, H.; Liu, J.-P. TGF-beta receptor mediated telomerase inhibition, telomere shortening and breast cancer cell senescence. Protein Cell 2017, 8, 39–54. [Google Scholar] [CrossRef]
- Lu, W.-J.; Chua, M.-S.; So, S.K. Suppressing N-Myc downstream regulated gene 1 reactivates senescence signaling and inhibits tumor growth in hepatocellular carcinoma. Carcinogenesis 2014, 35, 915–922. [Google Scholar] [CrossRef]
- Su, D.; Zhu, S.; Han, X.; Feng, Y.; Huang, H.; Ren, G.; Pan, L.; Zhang, Y.; Lu, J.; Huang, B. BMP4-Smad signaling pathway mediates adriamycin-induced premature senescence in lung cancer cells. J. Biol. Chem. 2009, 284, 12153–12164. [Google Scholar] [CrossRef]
- Zhu, D.; Wu, J.; Spee, C.; Ryan, S.J.; Hinton, D.R. BMP4 mediates oxidative stress-induced retinal pigment epithelial cell senescence and is overexpressed in age-related macular degeneration. J. Biol. Chem. 2009, 284, 9529–9539. [Google Scholar] [CrossRef]
- Buckley, S.; Shi, W.; Driscoll, B.; Ferrario, A.; Anderson, K.; Warburton, D. BMP4 signaling induces senescence and modulates the oncogenic phenotype of A549 lung adenocarcinoma cells. Am. J. Physiol. Cell. Mol. Physiol. 2004, 286, L81–L86. [Google Scholar] [CrossRef]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.-W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef]
- Bird, T.G.; Müller, M.; Boulter, L.; Vincent, D.F.; Ridgway, R.A.; Lopez-Guadamillas, E.; Lu, W.-Y.; Jamieson, T.; Govaere, O.; Campbell, A.D.; et al. TGFβ inhibition restores a regenerative response in acute liver injury by suppressing paracrine senescence. Sci. Transl. Med. 2018, 10, eaan1230. [Google Scholar] [CrossRef]
- Schafer, M.J.; White, T.A.; Iijima, K.; Haak, A.J.; Ligresti, G.; Atkinson, E.J.; Oberg, A.L.; Birch, J.; Salmonowicz, H.; Zhu, Y.; et al. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun. 2017, 8, 14532. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Niu, J.; Li, X.; Wang, X.; Guo, Z.; Zhang, F. TGF-β1 induces senescence of bone marrow mesenchymal stem cells via increase of mitochondrial ROS production. BMC Dev. Biol. 2014, 14, 21. [Google Scholar] [CrossRef] [PubMed]
- Marches, R.; Hsueh, R.; Uhr, J.W. Cancer dormancy and cell signaling: Induction of p21(waf1) initiated by membrane IgM engagement increases survival of B lymphoma cells. Proc. Natl. Acad. Sci. USA 1999, 96, 8711–8715. [Google Scholar] [CrossRef] [PubMed]
- Noda, A.; Ning, Y.; Venable, S.F.; Pereira-Smith, O.M.; Smith, J.R. Cloning of Senescent Cell-Derived Inhibitors of DNA Synthesis Using an Expression Screen. Exp. Cell Res. 1994, 211, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Young, A.P.; Schlisio, S.; Minamishima, Y.A.; Zhang, Q.; Li, L.; Grisanzio, C.; Signoretti, S.; Kaelin, W.G. VHL loss actuates a HIF-independent senescence programme mediated by Rb and p400. Nat. Cell Biol. 2008, 10, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Liggett, W.H.; Sidransky, D. Role of the p16 tumor suppressor gene in cancer. J. Clin. Oncol. 1998, 16, 1197–1206. [Google Scholar] [CrossRef]
- Yoshida, A.; Lee, E.K.; Diehl, J.A. Molecular and Cellular Pathobiology Induction of Therapeutic Senescence in Vemurafenib-Resistant Melanoma by Extended Inhibition of CDK4/6. Cancer Res. 2016, 76, 2990–3002. [Google Scholar] [CrossRef]
- Torres-Guzmán, R.; Calsina, B.; Hermoso, A.; Baquero, C.; Alvarez, B.; Amat, J.; McNulty, A.M.; Gong, X.; Boehnke, K.; Du, J.; et al. Preclinical characterization of abemaciclib in hormone receptor positive breast cancer. Oncotarget 2017, 8, 69493–69507. [Google Scholar] [CrossRef]
- Kovatcheva, M.; Liao, W.; Klein, M.E.; Robine, N.; Geiger, H.; Crago, A.M.; Dickson, M.A.; Tap, W.D.; Singer, S.; Koff, A. ATRX is a regulator of therapy induced senescence in human cells. Nat. Commun. 2017, 8, 386. [Google Scholar] [CrossRef]
- Miettinen, T.P.; Peltier, J.; Härtlova, A.; Gierliński, M.; Jansen, V.M.; Trost, M.; Björklund, M. Thermal proteome profiling of breast cancer cells reveals proteasomal activation by CDK4/6 inhibitor palbociclib. EMBO J. 2018, 37, e98359. [Google Scholar] [CrossRef]
- Vijayaraghavan, S.; Karakas, C.; Doostan, I.; Chen, X.; Bui, T.; Yi, M.; Raghavendra, A.S.; Zhao, Y.; Bashour, S.I.; Ibrahim, N.K.; et al. CDK4/6 and autophagy inhibitors synergistically induce senescence in Rb positive cytoplasmic cyclin E negative cancers. Nat. Commun. 2017, 8, 15916. [Google Scholar] [CrossRef] [PubMed]
- Georgakilas, A.G.; Martin, O.A.; Bonner, W.M. p21: A Two-Faced Genome Guardian. Trends Mol. Med. 2017, 23, 310–319. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Wu, P.C.; Dong, D.Z.; Ivanova, I.; Chu, E.; Zeliadt, S.; Vesselle, H.; Wu, D.Y. Polyploidy road to therapy-induced cellular senescence and escape. Int. J. Cancer 2013, 132, 1505–1515. [Google Scholar] [CrossRef] [PubMed]
- Chu, I.M.; Hengst, L.; Slingerland, J.M. The Cdk inhibitor p27 in human cancer: Prognostic potential and relevance to anticancer therapy. Nat. Rev. Cancer 2008, 8, 253–267. [Google Scholar] [CrossRef]
- Alimonti, A.; Nardella, C.; Chen, Z.; Clohessy, J.G.; Carracedo, A.; Trotman, L.C.; Cheng, K.; Varmeh, S.; Kozma, S.C.; Thomas, G.; et al. A novel type of cellular senescence that can be enhanced in mouse models and human tumor xenografts to suppress prostate tumorigenesis. J. Clin. Investig. 2010, 120, 681–693. [Google Scholar] [CrossRef]
- Litovchick, L.; Florens, L.A.; Swanson, S.K.; Washburn, M.P.; DeCaprio, J.A. DYRK1A protein kinase promotes quiescence and senescence through DREAM complex assembly. Genes Dev. 2011, 25, 801–813. [Google Scholar] [CrossRef]
- Litovchick, L.; Sadasivam, S.; Florens, L.; Zhu, X.; Swanson, S.K.; Velmurugan, S.; Chen, R.; Washburn, M.P.; Liu, X.S.; DeCaprio, J.A. Evolutionarily Conserved Multisubunit RBL2/p130 and E2F4 Protein Complex Represses Human Cell Cycle-Dependent Genes in Quiescence. Mol. Cell 2007, 26, 539–551. [Google Scholar] [CrossRef]
- Deng, X.; Ewton, D.Z.; Friedman, E. Mirk/Dyrk1B maintains the viability of quiescent pancreatic cancer cells by reducing levels of reactive oxygen species. Cancer Res. 2009, 69, 3317–3324. [Google Scholar] [CrossRef]
- Boichuk, S.; Parry, J.A.; Makielski, K.R.; Litovchick, L.; Baron, J.L.; Zewe, J.P.; Wozniak, A.; Mehalek, K.R.; Korzeniewski, N.; Seneviratne, D.S.; et al. The DREAM Complex Mediates GIST Cell Quiescence and Is a Novel Therapeutic Target to Enhance Imatinib-Induced Apoptosis. Cancer Res. 2013, 73, 5120–5129. [Google Scholar] [CrossRef]
- MacDonald, J.; Ramos-Valdes, Y.; Perampalam, P.; Litovchick, L.; DiMattia, G.E.; Dick, F.A. A Systematic Analysis of Negative Growth Control Implicates the DREAM Complex in Cancer Cell Dormancy. Mol. Cancer Res. 2017, 15, 371–381. [Google Scholar] [CrossRef]
- Kovatcheva, M.; Klein, M.E.; Tap, W.D.; Koff, A. Mechanistic understanding of the role of ATRX in senescence provides new insight for combinatorial therapies with CDK4 inhibitors. Mol. Cell. Oncol. 2018, 5, e1384882. [Google Scholar] [CrossRef] [PubMed]
- Buczacki, S.J.A.; Popova, S.; Biggs, E.; Koukorava, C.; Buzzelli, J.; Vermeulen, L.; Hazelwood, L.; Francies, H.; Garnett, M.J.; Winton, D.J. Itraconazole targets cell cycle heterogeneity in colorectal cancer. J. Exp. Med. 2018, 215, 1891–1912. [Google Scholar] [CrossRef] [PubMed]
- Ghajar, C.M. Metastasis prevention by targeting the dormant niche. Nat. Rev. Cancer 2015, 15, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Kfoury, Y.; Scadden, D.T. Mesenchymal cell contributions to the stem cell niche. Cell Stem Cell 2015, 16, 239–253. [Google Scholar] [CrossRef]
- Gnecchi, M.; He, H.; Liang, O.D.; Melo, L.G.; Morello, F.; Mu, H.; Noiseux, N.; Zhang, L.; Pratt, R.E.; Ingwall, J.S.; et al. Paracrine action accounts for marked protection of ischemic heart by Akt-modified mesenchymal stem cells. Nat. Med. 2005, 11, 367–368. [Google Scholar] [CrossRef]
- Madrigal, M.; Rao, K.S.; Riordan, N.H. A review of therapeutic effects of mesenchymal stem cell secretions and induction of secretory modification by different culture methods. J. Transl. Med. 2014, 12, 260. [Google Scholar] [CrossRef]
- Ryan, J.M.; Barry, F.; Murphy, J.M.; Mahon, B.P. Interferon-γ does not break, but promotes the immunosuppressive capacity of adult human mesenchymal stem cells. Clin. Exp. Immunol. 2007, 149, 353–363. [Google Scholar] [CrossRef]
- Bartosh, T.J.; Ylostalo, J.H.; Mohammadipoor, A.; Bazhanov, N.; Coble, K.; Claypool, K.; Lee, R.H.; Choi, H.; Prockop, D.J. Aggregation of human mesenchymal stromal cells (MSCs) into 3D spheroids enhances their antiinflammatory properties. Proc. Natl. Acad. Sci. USA 2010, 107, 13724–13729. [Google Scholar] [CrossRef]
- Shiozawa, Y.; Pedersen, E.A.; Havens, A.M.; Jung, Y.; Mishra, A.; Joseph, J.; Kim, J.K.; Patel, L.R.; Ying, C.; Ziegler, A.M.; et al. Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. J. Clin. Investig. 2011, 121, 1298–1312. [Google Scholar] [CrossRef]
- Axelrod, H.D.; Valkenburg, K.C.; Amend, S.R.; Hicks, J.L.; Parsana, P.; Torga, G.; DeMarzo, A.M.; Pienta, K.J. AXL Is a Putative Tumor Suppressor and Dormancy Regulator in Prostate Cancer. Mol. Cancer Res. 2019, 17, 356–369. [Google Scholar] [CrossRef]
- Jin, C.; Wang, H.; Chen, Y.; Tang, M.; Fan, G.; Wang, Z.; Li, L.; Zhang, Y.; Zhang, W.; Zhong, M. Gas6 Delays Senescence in Vascular Smooth Muscle Cells through the PI3K/ Akt/FoxO Signaling Pathway. Cell. Physiol. Biochem. 2015, 35, 1151–1166. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhao, J.; Jin, C.; Li, Y.; Tang, M.; Wang, Z.; Zhang, W.; Zhang, Y.; Li, L.; Zhong, M. Testosterone delays vascular smooth muscle cell senescence and inhibits collagen synthesis via the Gas6/Axl signaling pathway. Age (Omaha) 2016, 38, 60. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed]
- Capparelli, C.; Guido, C.; Whitaker-Menezes, D.; Bonuccelli, G.; Balliet, R.; Pestell, T.G.; Goldberg, A.F.; Pestell, R.G.; Howell, A.; Sneddon, S.; et al. Autophagy and senescence in cancer-associated fibroblasts metabolically supports tumor growth and metastasis, via glycolysis and ketone production. Cell Cycle 2012, 11, 2285–2302. [Google Scholar] [CrossRef] [PubMed]
- Tonnessen-Murray, C.A.; Frey, W.D.; Rao, S.G.; Shahbandi, A.; Ungerleider, N.A.; Olayiwola, J.O.; Murray, L.B.; Vinson, B.T.; Chrisey, D.B.; Lord, C.J.; et al. Chemotherapy-induced senescent cancer cells engulf other cells to enhance their survival. J. Cell Biol. 2019, 218, 3827–3844. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Furness, A.J.S.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef]
- Huang, A.C.; Orlowski, R.J.; Xu, X.; Mick, R.; George, S.M.; Yan, P.K.; Manne, S.; Kraya, A.A.; Wubbenhorst, B.; Dorfman, L.; et al. A single dose of neoadjuvant PD-1 blockade predicts clinical outcomes in resectable melanoma. Nat. Med. 2019, 25, 454–461. [Google Scholar] [CrossRef]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef]
- Przybyla, A.; Zhang, T.; Li, R.; Roen, D.R.; Mackiewicz, A.; Lehmann, P.V. Natural T cell autoreactivity to melanoma antigens: Clonally expanded melanoma-antigen specific CD8 + memory T cells can be detected in healthy humans. Cancer Immunol. Immunother. 2019, 68, 709–720. [Google Scholar] [CrossRef]
- Wang, H.-F.; Wang, S.-S.; Huang, M.-C.; Liang, X.-H.; Tang, Y.-J.; Tang, Y.-L. Targeting Immune-Mediated Dormancy: A Promising Treatment of Cancer. Front. Oncol. 2019, 9, 498. [Google Scholar] [CrossRef]
- Baxevanis, C.N.; Perez, S.A. Cancer Dormancy: A Regulatory Role for Endogenous Immunity in Establishing and Maintaining the Tumor Dormant State. Vaccines 2015, 3, 597–619. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.W.; Yevsa, T.; Woller, N.; Hoenicke, L.; Wuestefeld, T.; Dauch, D.; Hohmeyer, A.; Gereke, M.; Rudalska, R.; Potapova, A.; et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011, 479, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Eggert, T.; Wolter, K.; Ji, J.; Ma, C.; Yevsa, T.; Klotz, S.; Medina-Echeverz, J.; Longerich, T.; Forgues, M.; Reisinger, F.; et al. Distinct Functions of Senescence-Associated Immune Responses in Liver Tumor Surveillance and Tumor Progression. Cancer Cell 2016, 30, 533–547. [Google Scholar] [CrossRef]
- Pereira, B.I.; Devine, O.P.; Vukmanovic-Stejic, M.; Chambers, E.S.; Subramanian, P.; Patel, N.; Virasami, A.; Sebire, N.J.; Kinsler, V.; Valdovinos, A.; et al. Senescent cells evade immune clearance via HLA-E-mediated NK and CD8+ T cell inhibition. Nat. Commun. 2019, 10, 2387. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, D.P.; Yannone, S.M.; Daemen, A.; Sun, Y.; Vakar-Lopez, F.; Kawahara, M.; Freund, A.M.; Rodier, F.; Wu, J.D.; Desprez, P.Y.; et al. Targetable mechanisms driving immunoevasion of persistent senescent cells link chemotherapy-resistant cancer to aging. JCI Insight 2019, 5, 124716. [Google Scholar] [CrossRef] [PubMed]
- Maciel-Barón, L.A.; Morales-Rosales, S.L.; Aquino-Cruz, A.A.; Triana-Martínez, F.; Galván-Arzate, S.; Luna-López, A.; González-Puertos, V.Y.; López-Díazguerrero, N.E.; Torres, C.; Königsberg, M. Senescence associated secretory phenotype profile from primary lung mice fibroblasts depends on the senescence induction stimuli. Age (Omaha) 2016, 38, 26. [Google Scholar]
- Basisty, N.; Kale, A.; Jeon, O.H.; Kuehnemann, C.; Payne, T.; Rao, C.; Holtz, A.; Shah, S.; Sharma, V.; Ferrucci, L.; et al. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol. 2020, 18, e3000599. [Google Scholar] [CrossRef]
- Goel, S.; Decristo, M.J.; Watt, A.C.; Brinjones, H.; Sceneay, J.; Li, B.B.; Khan, N.; Ubellacker, J.M.; Xie, S.; Metzger-Filho, O.; et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature 2017, 548, 471–475. [Google Scholar] [CrossRef]
- Teo, Z.L.; Versaci, S.; Dushyanthen, S.; Caramia, F.; Savas, P.; Mintoff, C.P.; Zethoven, M.; Virassamy, B.; Luen, S.J.; McArthur, G.A.; et al. Combined CDK4/6 and PI3Kα inhibition is synergistic and immunogenic in triple-negative breast cancer. Cancer Res. 2017, 77, 6340–6352. [Google Scholar] [CrossRef]
- Schaer, D.A.; Beckmann, R.P.; Dempsey, J.A.; Huber, L.; Forest, A.; Amaladas, N.; Li, Y.; Wang, Y.C.; Rasmussen, E.R.; Chin, D.; et al. The CDK4/6 Inhibitor Abemaciclib Induces a T Cell Inflamed Tumor Microenvironment and Enhances the Efficacy of PD-L1 Checkpoint Blockade. Cell Rep. 2018, 22, 2978–2994. [Google Scholar] [CrossRef]
- Tolaney, S.M.; Kabos, P.; Dickler, M.N.; Gianni, L.; Jansen, V.; Lu, Y.; Young, S.; Rugo, H.S. Updated efficacy, safety, & PD-L1 status of patients with HR+, HER2- metastatic breast cancer administered abemaciclib plus pembrolizumab. J. Clin. Oncol. 2018, 36, 1059. [Google Scholar]
- Albrengues, J.; Shields, M.A.; Ng, D.; Park, C.G.; Ambrico, A.; Poindexter, M.E.; Upadhyay, P.; Uyeminami, D.L.; Pommier, A.; Küttner, V.; et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 2018, 361, eaao4227. [Google Scholar] [CrossRef] [PubMed]
- Tivari, S.; Lu, H.; Dasgupta, T.; De Lorenzo, M.S.; Wieder, R. Reawakening of dormant estrogen-dependent human breast cancer cells by bone marrow stroma secretory senescence. Cell Commun. Signal. 2018, 16, 48. [Google Scholar] [CrossRef] [PubMed]
- Roberson, R.S.; Kussick, S.J.; Vallieres, E.; Chen, S.-Y.J.; Wu, D.Y. Escape from Therapy-Induced Accelerated Cellular Senescence in p53-Null Lung Cancer Cells and in Human Lung Cancers. Cancer Res. 2005, 65, 2795–2803. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune–metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16Ink4a-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef]
- Baar, M.P.; Brandt, R.M.C.; Putavet, D.A.; Klein, J.D.D.; Derks, K.W.J.; Bourgeois, B.R.M.; Stryeck, S.; Rijksen, Y.; van Willigenburg, H.; Feijtel, D.A.; et al. Targeted Apoptosis of Senescent Cells Restores Tissue Homeostasis in Response to Chemotoxicity and Aging. Cell 2017, 169, 132.e16–147.e16. [Google Scholar] [CrossRef]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.-M.; Demaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 2016, 22, 78–83. [Google Scholar] [CrossRef]
- Jeon, O.H.; Kim, C.; Laberge, R.-M.; Demaria, M.; Rathod, S.; Vasserot, A.P.; Chung, J.W.; Kim, D.H.; Poon, Y.; David, N.; et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat. Med. 2017, 23, 775–781. [Google Scholar] [CrossRef] [PubMed]
- Dörr, J.R.; Yu, Y.; Milanovic, M.; Beuster, G.; Zasada, C.; Däbritz, J.H.M.; Lisec, J.; Lenze, D.; Gerhardt, A.; Schleicher, K.; et al. Synthetic lethal metabolic targeting of cellular senescence in cancer therapy. Nature 2013, 501, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Leite de Oliveira, R.; Wang, C.; Fernandes Neto, J.M.; Mainardi, S.; Evers, B.; Lieftink, C.; Morris, B.; Jochems, F.; Willemsen, L.; et al. High-Throughput Functional Genetic and Compound Screens Identify Targets for Senescence Induction in Cancer. Cell Rep. 2017, 21, 773–783. [Google Scholar] [CrossRef] [PubMed]
- Samaraweera, L.; Adomako, A.; Rodriguez-Gabin, A.; McDaid, H.M. A Novel Indication for Panobinostat as a Senolytic Drug in NSCLC and HNSCC. Sci. Rep. 2017, 7, 1900. [Google Scholar] [CrossRef] [PubMed]
- Sieben, C.J.; Sturmlechner, I.; van de Sluis, B.; van Deursen, J.M. Two-Step Senescence-Focused Cancer Therapies. Trends Cell Biol. 2018, 28, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Myrianthopoulos, V.; Evangelou, K.; Vasileiou, P.V.S.; Cooks, T.; Vassilakopoulos, T.P.; Pangalis, G.A.; Kouloukoussa, M.; Kittas, C.; Georgakilas, A.G.; Gorgoulis, V.G. Senescence and senotherapeutics: A new field in cancer therapy. Pharmacol. Ther. 2019, 193, 31–49. [Google Scholar] [CrossRef]
- Short, S.; Fielder, E.; Miwa, S.; von Zglinicki, T. Senolytics and senostatics as adjuvant tumour therapy. EBioMedicine 2019, 41, 683–692. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Rovira, M.; Galiana, I.; Giménez, C.; Lozano-Torres, B.; Paez-Ribes, M.; Llanos, S.; Chaib, S.; Muñoz-Martín, M.; Ucero, A.C.; et al. A versatile drug delivery system targeting senescent cells. EMBO Mol. Med. 2018, 10, e9355. [Google Scholar] [CrossRef]
- Fleury, H.; Malaquin, N.; Tu, V.; Gilbert, S.; Martinez, A.; Olivier, M.-A.; Sauriol, A.; Communal, L.; Leclerc-Desaulniers, K.; Carmona, E.; et al. Exploiting interconnected synthetic lethal interactions between PARP inhibition and cancer cell reversible senescence. Nat. Commun. 2019, 10, 2556. [Google Scholar] [CrossRef]
- Triana-Martínez, F.; Picallos-Rabina, P.; Da Silva-Álvarez, S.; Pietrocola, F.; Llanos, S.; Rodilla, V.; Soprano, E.; Pedrosa, P.; Ferreirós, A.; Barradas, M.; et al. Identification and characterization of Cardiac Glycosides as senolytic compounds. Nat. Commun. 2019, 10, 4731. [Google Scholar] [CrossRef]
- Guerrero, A.; Herranz, N.; Sun, B.; Wagner, V.; Gallage, S.; Guiho, R.; Wolter, K.; Pombo, J.; Irvine, E.E.; Innes, A.J.; et al. Cardiac glycosides are broad-spectrum senolytics. Nat. Metab. 2019, 1, 1074–1088. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Fang, J.; Chen, J. Tumor cell senescence response produces aggressive variants. Cell Death Discov. 2017, 3, 17049. [Google Scholar] [CrossRef] [PubMed]
- Justice, J.N.; Nambiar, A.M.; Tchkonia, T.; LeBrasseur, N.K.; Pascual, R.; Hashmi, S.K.; Prata, L.; Masternak, M.M.; Kritchevsky, S.B.; Musi, N.; et al. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. EBioMedicine 2019, 40, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Hickson, L.J.; Langhi Prata, L.G.P.; Bobart, S.A.; Evans, T.K.; Giorgadze, N.; Hashmi, S.K.; Herrmann, S.M.; Jensen, M.D.; Jia, Q.; Jordan, K.L.; et al. Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine 2019, 47, 446–456. [Google Scholar] [CrossRef]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).