Prognosis, Biology, and Targeting of TP53 Dysregulation in Multiple Myeloma

Abstract
1. Introduction
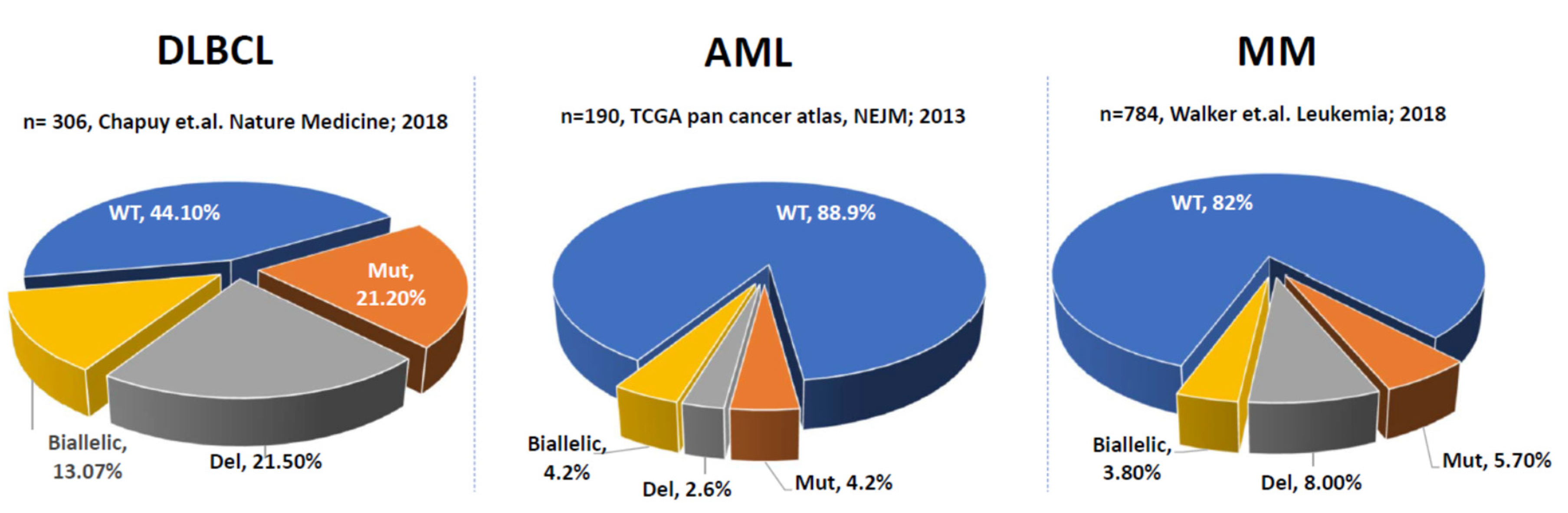
2. P53 Aberrations in Solid Tumors and Hematological Malignancies
3. Prognosis of Del17p/TP53 Inactivation in Multiple Myeloma
3.1. Deletion of 17p in MM
3.2. Biallelic Inactivation of TP53 in MM
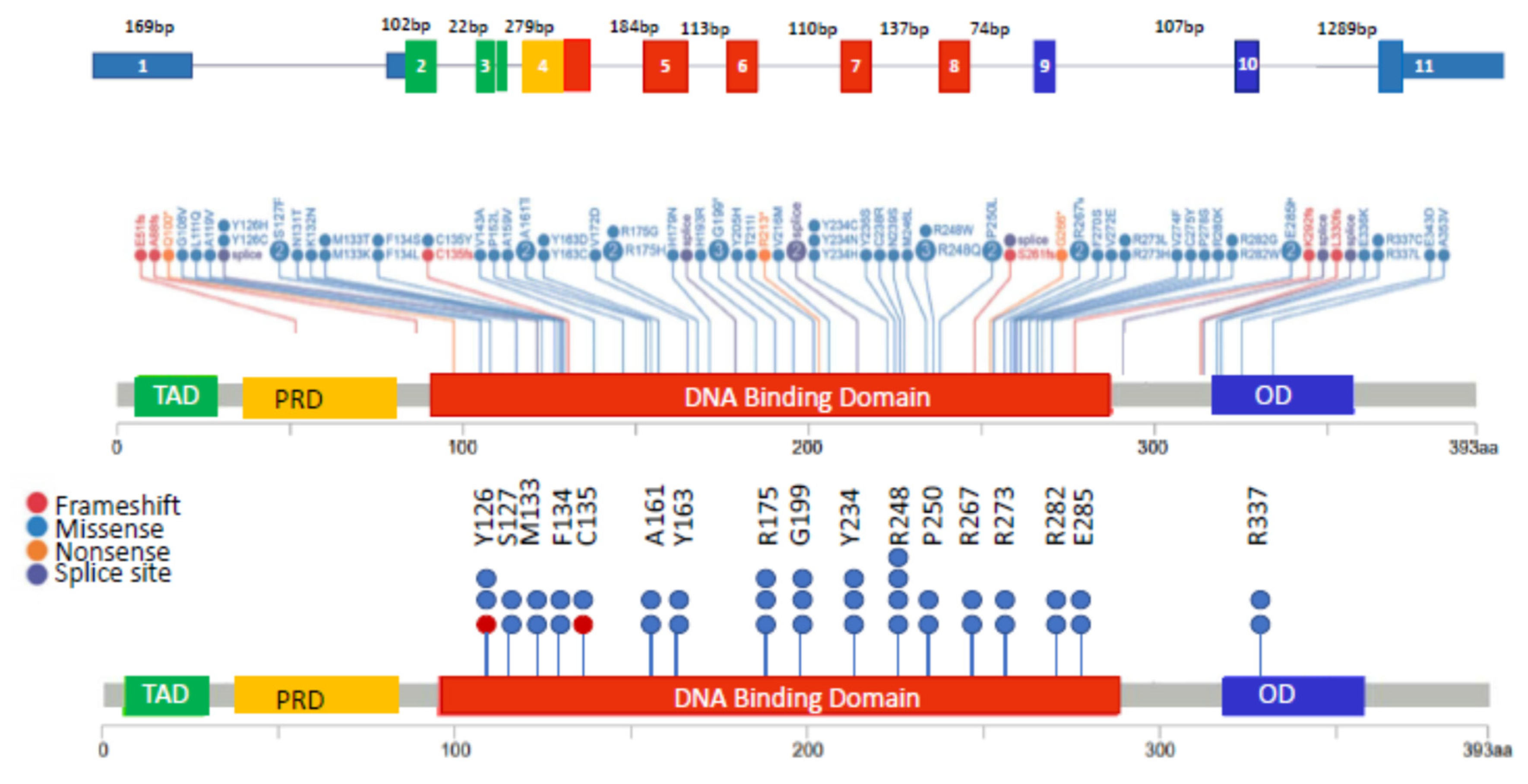
3.3. Monoallelic Mutation of TP53 in MM
4. Biology of Del17p/P53 Inactivation
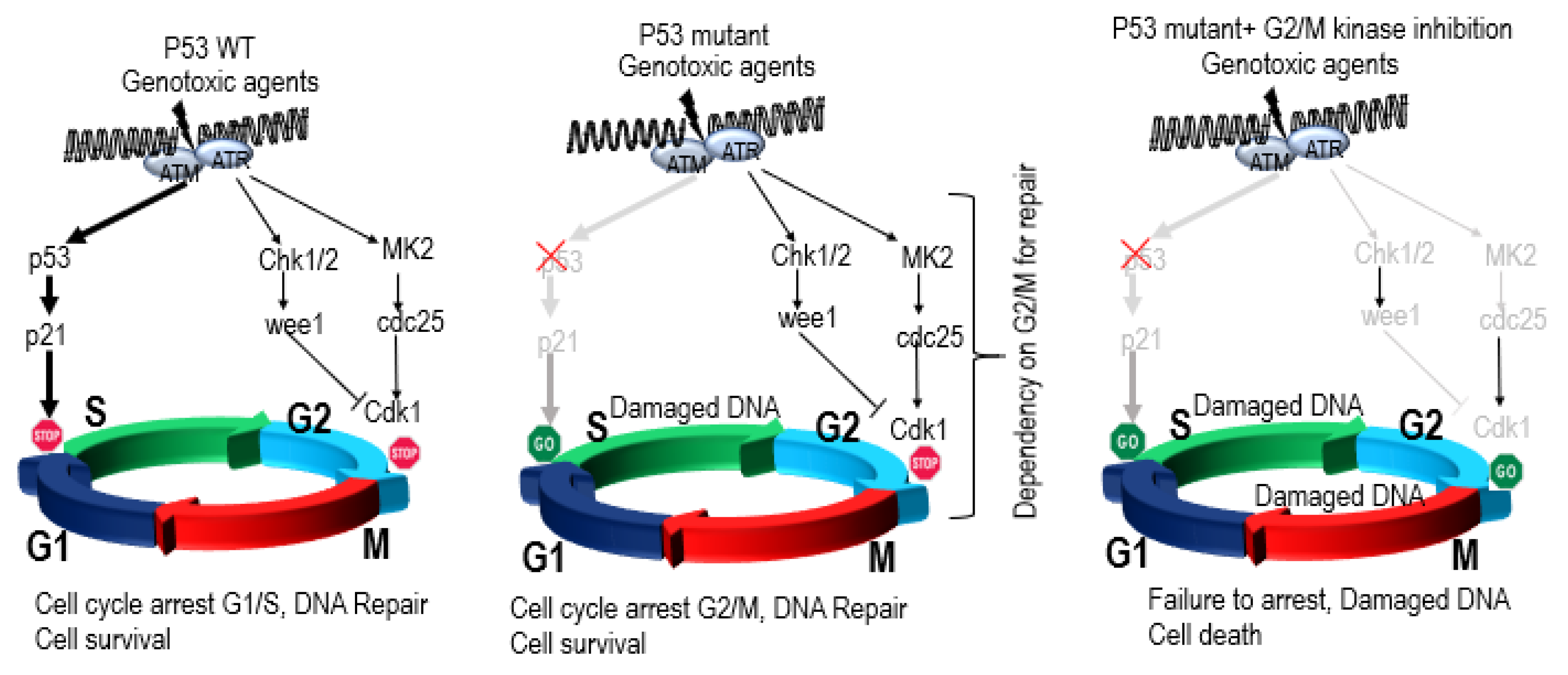
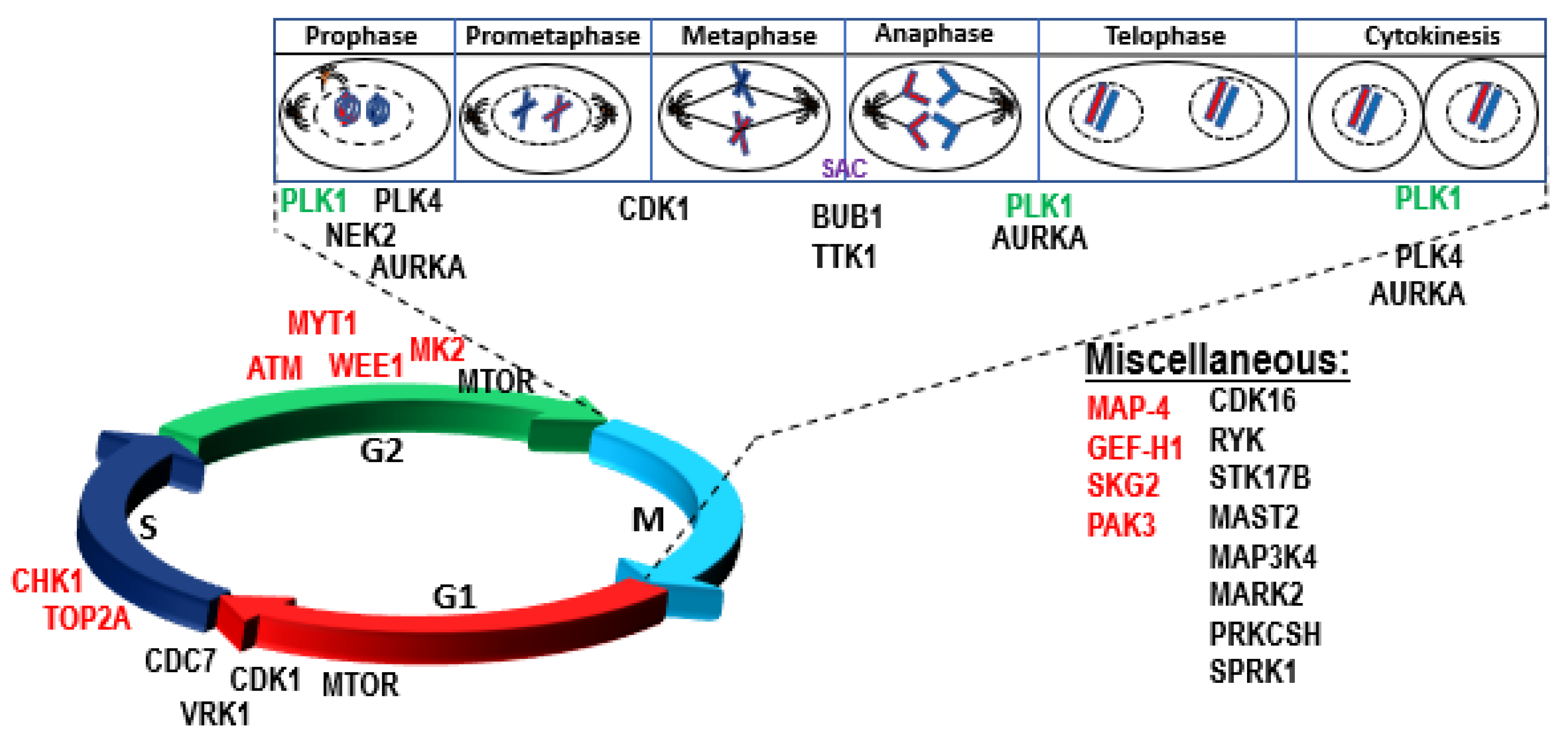
4.1. Role of P53 in Genomic Instability, DNA Repair, Aneuploidy, and Checkpoint Control
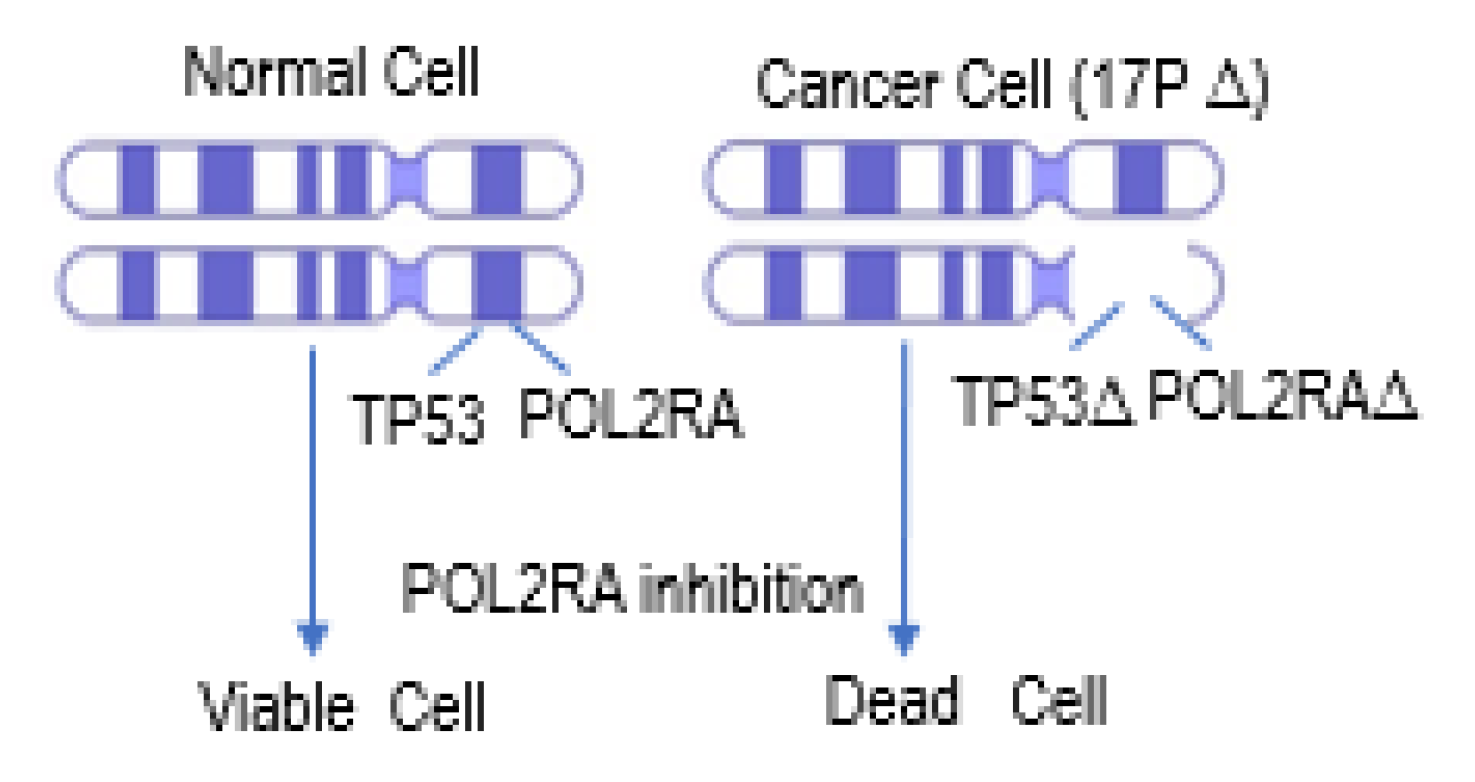
4.2. P53 Synthetic Lethality
P53 Synthetic Lethality in MM
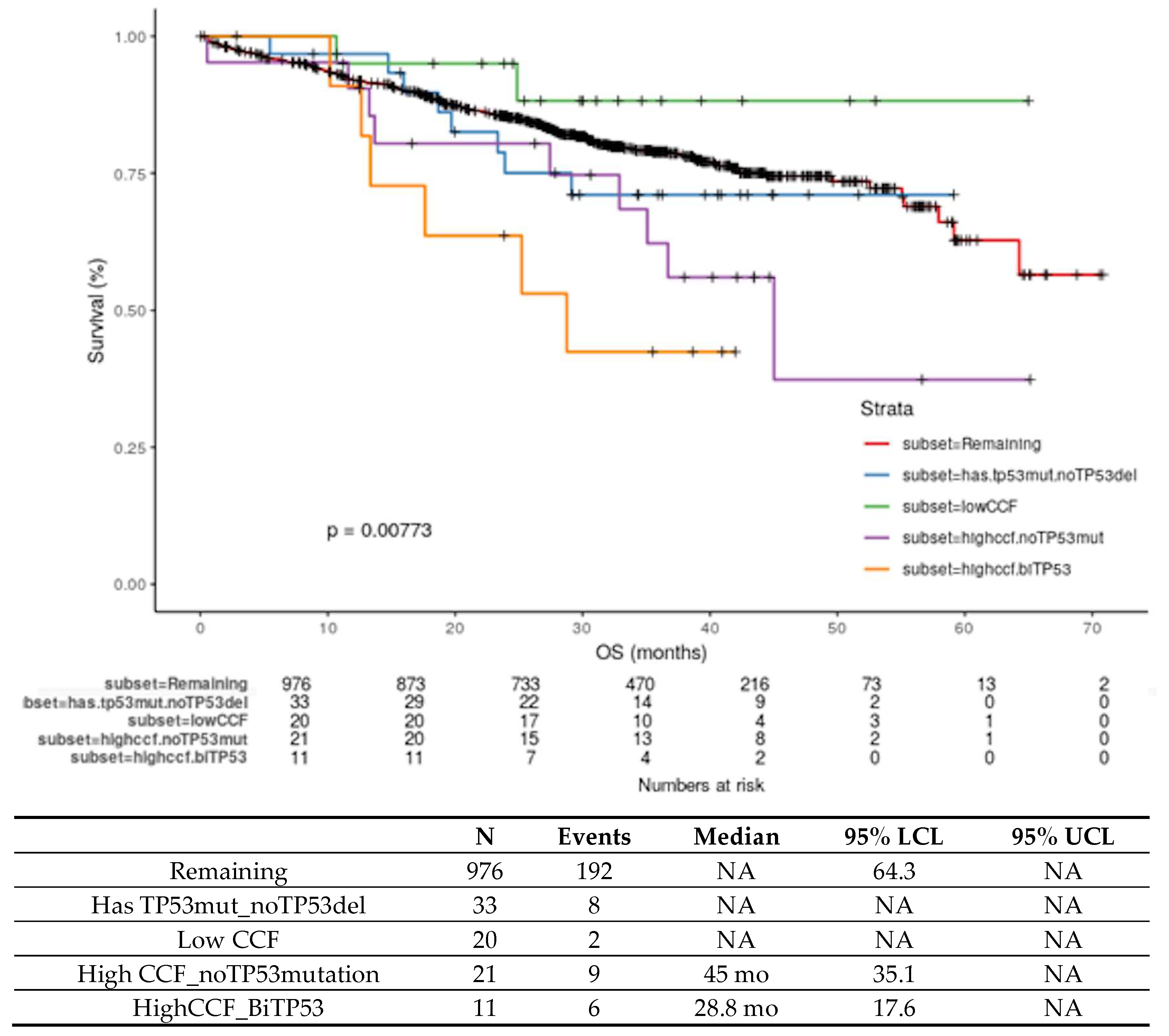
4.3. Biology of High-risk Del17p in MM
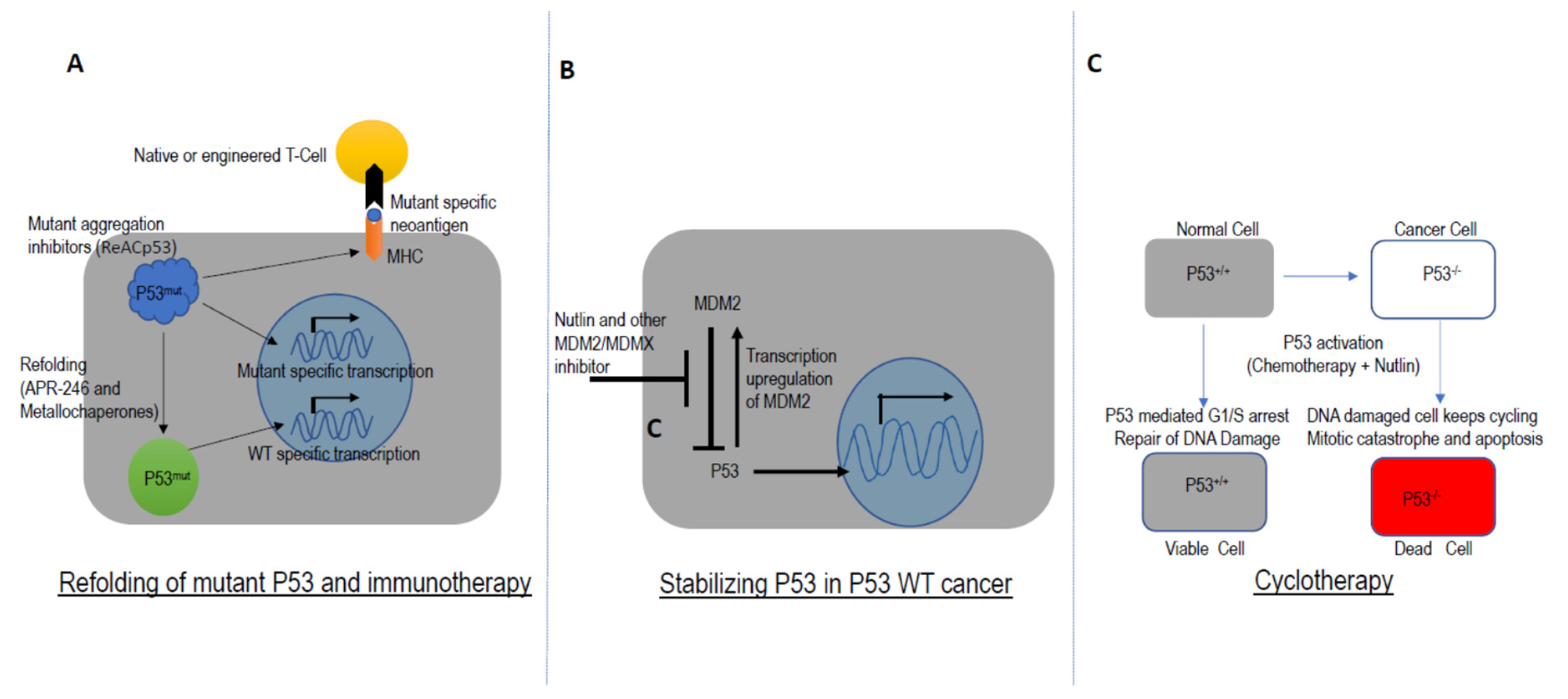
5. Targeting TP53 in Drug Development
6. Conclusion and Future Perspective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- The National Cancer Institute. Cancer Stat Facts: Myeloma. Available online: https://seer.cancer.gov/statfacts/html/mulmy.html (accessed on 26 May 2019).
- Fonseca, R.; Abouzaid, S.; Bonafede, M.; Cai, Q.; Parikh, K.; Cosler, L.; Richardson, P. Trends in overall survival and costs of multiple myeloma, 2000–2014. Leukemia 2016, 31, 1915. [Google Scholar] [CrossRef]
- Kumar, S.K.; Dispenzieri, A.; Lacy, M.Q.; Gertz, M.A.; Buadi, F.K.; Pandey, S.; Kapoor, P.; Dingli, D.; Hayman, S.R.; Leung, N.; et al. Continued improvement in survival in multiple myeloma: Changes in early mortality and outcomes in older patients. Leukemia 2014, 28, 1122–1128. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, A.; Avet-Loiseau, H.; Oliva, S.; Lokhorst, H.M.; Goldschmidt, H.; Rosinol, L.; Richardson, P.; Caltagirone, S.; Lahuerta, J.J.; Facon, T.; et al. Revised International Staging System for Multiple Myeloma: A Report From International Myeloma Working Group. J. Clin. Oncol. 2015, 33, 2863–2869. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.A.; Mavrommatis, K.; Wardell, C.P.; Ashby, T.C.; Bauer, M.; Davies, F.; Rosenthal, A.; Wang, H.; Qu, P.; Hoering, A.; et al. A high-risk, Double-Hit, group of newly diagnosed myeloma identified by genomic analysis. Leukemia 2018. [Google Scholar] [CrossRef] [PubMed]
- Thakurta, A.; Ortiz, M.; Blecua, P.; Towfic, F.; Corre, J.; Serbina, N.V.; Flynt, E.; Yu, Z.; Yang, Z.; Palumbo, A.; et al. High subclonal fraction of 17p deletion is associated with poor prognosis in multiple myeloma. Blood 2019, 133, 1217–1221. [Google Scholar] [CrossRef]
- Lane, D.P.; Crawford, L.V. T antigen is bound to a host protein in SV40-transformed cells. Nature 1979, 278, 261–263. [Google Scholar] [CrossRef]
- Linzer, D.I.; Levine, A.J. Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell 1979, 17, 43–52. [Google Scholar] [CrossRef]
- Kress, M.; May, E.; Cassingena, R.; May, P. Simian virus 40-transformed cells express new species of proteins precipitable by anti-simian virus 40 tumor serum. J. Virol. 1979, 31, 472–483. [Google Scholar] [CrossRef]
- DeLeo, A.B.; Jay, G.; Appella, E.; Dubois, G.C.; Law, L.W.; Old, L.J. Detection of a transformation-related antigen in chemically induced sarcomas and other transformed cells of the mouse. Proc. Natl. Acad. Sci. USA 1979, 76, 2420–2424. [Google Scholar] [CrossRef]
- Finlay, C.A.; Hinds, P.W.; Levine, A.J. The p53 proto-oncogene can act as a suppressor of transformation. Cell 1989, 57, 1083–1093. [Google Scholar] [CrossRef]
- Baker, S.J.; Preisinger, A.C.; Jessup, J.M.; Paraskeva, C.; Markowitz, S.; Willson, J.K.; Hamilton, S.; Vogelstein, B. p53 gene mutations occur in combination with 17p allelic deletions as late events in colorectal tumorigenesis. Cancer Res. 1990, 50, 7717–7722. [Google Scholar] [PubMed]
- Malkin, D.; Li, F.P.; Strong, L.C.; Fraumeni, J.F., Jr.; Nelson, C.E.; Kim, D.H.; Kassel, J.; Gryka, M.A.; Bischoff, F.Z.; Tainsky, M.A.; et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 1990, 250, 1233–1238. [Google Scholar] [CrossRef] [PubMed]
- Ciriello, G.; Miller, M.L.; Aksoy, B.A.; Senbabaoglu, Y.; Schultz, N.; Sander, C. Emerging landscape of oncogenic signatures across human cancers. Nat. Genet. 2013, 45, 1127–1133. [Google Scholar] [CrossRef] [PubMed]
- Hollstein, M.; Sidransky, D.; Vogelstein, B.; Harris, C.C. p53 mutations in human cancers. Science 1991, 253, 49–53. [Google Scholar] [CrossRef]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N.; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Li, V.D.; Li, K.H.; Li, J.T. TP53 mutations as potential prognostic markers for specific cancers: Analysis of data from The Cancer Genome Atlas and the International Agency for Research on Cancer TP53 Database. J. Cancer Res. Clin. Oncol. 2019, 145, 625–636. [Google Scholar] [CrossRef]
- Yachida, S.; White, C.M.; Naito, Y.; Zhong, Y.; Brosnan, J.A.; Macgregor-Das, A.M.; Morgan, R.A.; Saunders, T.; Laheru, D.A.; Herman, J.M.; et al. Clinical significance of the genetic landscape of pancreatic cancer and implications for identification of potential long-term survivors. Clin. Cancer Res. 2012, 18, 6339–6347. [Google Scholar] [CrossRef]
- Ding, L.; Getz, G.; Wheeler, D.A.; Mardis, E.R.; McLellan, M.D.; Cibulskis, K.; Sougnez, C.; Greulich, H.; Muzny, D.M.; Morgan, M.B.; et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008, 455, 1069–1075. [Google Scholar] [CrossRef]
- Wood, L.D.; Parsons, D.W.; Jones, S.; Lin, J.; Sjoblom, T.; Leary, R.J.; Shen, D.; Boca, S.M.; Barber, T.; Ptak, J.; et al. The genomic landscapes of human breast and colorectal cancers. Science 2007, 318, 1108–1113. [Google Scholar] [CrossRef]
- Baugh, E.H.; Ke, H.; Levine, A.J.; Bonneau, R.A.; Chan, C.S. Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differ. 2018, 25, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Bouaoun, L.; Sonkin, D.; Ardin, M.; Hollstein, M.; Byrnes, G.; Zavadil, J.; Olivier, M. TP53 Variations in Human Cancers: New Lessons from the IARC TP53 Database and Genomics Data. Hum. Mutat. 2016, 37, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Leroy, B.; Fournier, J.L.; Ishioka, C.; Monti, P.; Inga, A.; Fronza, G.; Soussi, T. The TP53 website: An integrative resource centre for the TP53 mutation database and TP53 mutant analysis. Nucleic Acids Res. 2013, 41, D962–D969. [Google Scholar] [CrossRef] [PubMed]
- Lode, L.; Eveillard, M.; Trichet, V.; Soussi, T.; Wuilleme, S.; Richebourg, S.; Magrangeas, F.; Ifrah, N.; Campion, L.; Traulle, C.; et al. Mutations in TP53 are exclusively associated with del(17p) in multiple myeloma. Haematologica 2010, 95, 1973–1976. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.A.; Leone, P.E.; Chiecchio, L.; Dickens, N.J.; Jenner, M.W.; Boyd, K.D.; Johnson, D.C.; Gonzalez, D.; Dagrada, G.P.; Protheroe, R.K.; et al. A compendium of myeloma-associated chromosomal copy number abnormalities and their prognostic value. Blood 2010, 116, E56–E65. [Google Scholar] [CrossRef]
- Donehower, L.A.; Soussi, T.; Korkut, A.; Liu, Y.; Schultz, A.; Cardenas, M.; Li, X.; Babur, O.; Hsu, T.K.; Lichtarge, O.; et al. Integrated Analysis of TP53 Gene and Pathway Alterations in The Cancer Genome Atlas. Cell Rep. 2019, 28, 3010. [Google Scholar] [CrossRef]
- Teoh, P.J.; Chung, T.H.; Sebastian, S.; Choo, S.N.; Yan, J.; Ng, S.B.; Fonseca, R.; Chng, W.J. p53 haploinsufficiency and functional abnormalities in multiple myeloma. Leukemia 2014, 28, 2066–2074. [Google Scholar] [CrossRef]
- Lohr, J.G.; Stojanov, P.; Carter, S.L.; Cruz-Gordillo, P.; Lawrence, M.S.; Auclair, D.; Sougnez, C.; Knoechel, B.; Gould, J.; Saksena, G.; et al. Widespread genetic heterogeneity in multiple myeloma: Implications for targeted therapy. Cancer Cell 2014, 25, 91–101. [Google Scholar] [CrossRef]
- Brosh, R.; Rotter, V. When mutants gain new powers: News from the mutant p53 field. Nat. Rev. Cancer 2009, 9, 701–713. [Google Scholar] [CrossRef]
- Datta, A.; Ghatak, D.; Das, S.; Banerjee, T.; Paul, A.; Butti, R.; Gorain, M.; Ghuwalewala, S.; Roychowdhury, A.; Alam, S.K.; et al. p53 gain-of-function mutations increase Cdc7-dependent replication initiation. EMBO Rep. 2017, 18, 2030–2050. [Google Scholar] [CrossRef]
- Barta, J.A.; McMahon, S.B. Lung-Enriched Mutations in the p53 Tumor Suppressor: A Paradigm for Tissue-Specific Gain of Oncogenic Function. Mol. Cancer Res. 2019, 17, 3–9. [Google Scholar] [CrossRef]
- Xu, J.; Qian, J.; Hu, Y.; Wang, J.; Zhou, X.; Chen, H.; Fang, J.Y. Heterogeneity of Li-Fraumeni syndrome links to unequal gain-of-function effects of p53 mutations. Sci. Rep. 2014, 4, 4223. [Google Scholar] [CrossRef] [PubMed]
- Van Oijen, M.G.; Slootweg, P.J. Gain-of-function mutations in the tumor suppressor gene p53. Clin. Cancer Res. 2000, 6, 2138–2145. [Google Scholar] [PubMed]
- Dittmer, D.; Pati, S.; Zambetti, G.; Chu, S.; Teresky, A.K.; Moore, M.; Finlay, C.; Levine, A.J. Gain of function mutations in p53. Nat. Genet. 1993, 4, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.J.; Chun, S.M.; Kim, K.R.; Sohn, I.; Sung, C.O. Clinical relevance of gain-of-function mutations of p53 in high-grade serous ovarian carcinoma. PLoS ONE 2013, 8, e72609. [Google Scholar] [CrossRef]
- Olive, K.P.; Tuveson, D.A.; Ruhe, Z.C.; Yin, B.; Willis, N.A.; Bronson, R.T.; Crowley, D.; Jacks, T. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 2004, 119, 847–860. [Google Scholar] [CrossRef] [PubMed]
- De Vries, A.; Flores, E.R.; Miranda, B.; Hsieh, H.M.; van Oostrom, C.T.; Sage, J.; Jacks, T. Targeted point mutations of p53 lead to dominant-negative inhibition of wild-type p53 function. Proc. Natl. Acad. Sci. USA 2002, 99, 2948–2953. [Google Scholar] [CrossRef]
- Hegi, M.E.; Klein, M.A.; Ruedi, D.; Chene, P.; Hamou, M.F.; Aguzzi, A. p53 transdominance but no gain of function in mouse brain tumor model. Cancer Res. 2000, 60, 3019–3024. [Google Scholar]
- Boettcher, S.; Miller, P.G.; Sharma, R.; McConkey, M.; Leventhal, M.; Krivtsov, A.V.; Giacomelli, A.O.; Wong, W.; Kim, J.; Chao, S.; et al. A dominant-negative effect drives selection of TP53 missense mutations in myeloid malignancies. Science 2019, 365, 599–604. [Google Scholar] [CrossRef]
- Lee, M.K.; Teoh, W.W.; Phang, B.H.; Tong, W.M.; Wang, Z.Q.; Sabapathy, K. Cell-type, dose, and mutation-type specificity dictate mutant p53 functions in vivo. Cancer Cell 2012, 22, 751–764. [Google Scholar] [CrossRef]
- Srivastava, S.; Wang, S.; Tong, Y.A.; Hao, Z.M.; Chang, E.H. Dominant negative effect of a germ-line mutant p53: A step fostering tumorigenesis. Cancer Res. 1993, 53, 4452–4455. [Google Scholar] [PubMed]
- Avet-Loiseau, H.; Attal, M.; Moreau, P.; Charbonnel, C.; Garban, F.; Hulin, C.; Leyvraz, S.; Michallet, M.; Yakoub-Agha, I.; Garderet, L.; et al. Genetic abnormalities and survival in multiple myeloma: The experience of the Intergroupe Francophone du Myélome. Blood 2007, 109, 3489–3495. [Google Scholar] [CrossRef]
- Thanendrarajan, S.; Tian, E.; Qu, P.; Mathur, P.; Schinke, C.; van Rhee, F.; Zangari, M.; Rasche, L.; Weinhold, N.; Alapat, D.; et al. The level of deletion 17p and bi-allelic inactivation of TP53 has a significant impact on clinical outcome in multiple myeloma. Haematologica 2017. [Google Scholar] [CrossRef] [PubMed]
- Pawlyn, C.; Davies, F.E. Towards personalized treatment in multiple myeloma based on molecular characteristics. Blood 2018. [Google Scholar] [CrossRef] [PubMed]
- Chng, W.J.; Price-Troska, T.; Gonzalez-Paz, N.; Van Wier, S.; Jacobus, S.; Blood, E.; Henderson, K.; Oken, M.; Van Ness, B.; Greipp, P.; et al. Clinical significance of TP53 mutation in myeloma. Leukemia 2007, 21, 582–584. [Google Scholar] [CrossRef]
- Shah, V.; Johnson, D.C.; Sherborne, A.L.; Ellis, S.; Aldridge, F.M.; Howard-Reeves, J.; Begum, F.; Price, A.; Kendall, J.; Chiecchio, L.; et al. Sub-clonal TP53 copy number is associated with prognosis in multiple myeloma. Blood 2018, 132, 2465–2469. [Google Scholar] [CrossRef]
- Shah, V.; Sherborne, A.L.; Walker, B.A.; Johnson, D.C.; Boyle, E.M.; Ellis, S.; Begum, D.B.; Proszek, P.Z.; Jones, J.R.; Pawlyn, C.; et al. Prediction of outcome in newly diagnosed myeloma: A meta-analysis of the molecular profiles of 1905 trial patients. Leukemia 2018, 32, 102–110. [Google Scholar] [CrossRef]
- An, G.; Li, Z.; Tai, Y.-T.; Acharya, C.; Li, Q.; Qin, X.; Yi, S.; Xu, Y.; Feng, X.; Li, C.; et al. The Impact of Clone Size on the Prognostic Value of Chromosome Aberrations by Fluorescence In Situ Hybridization in Multiple Myeloma. Clin. Cancer Res. 2015, 21, 2148–2156. [Google Scholar] [CrossRef]
- Boyd, K.D.; Ross, F.M.; Tapper, W.J.; Chiecchio, L.; Dagrada, G.; Konn, Z.J.; Gonzalez, D.; Walker, B.A.; Hockley, S.L.; Wardell, C.P.; et al. The clinical impact and molecular biology of del(17p) in multiple myeloma treated with conventional or thalidomide-based therapy. Genes Chromosomes Cancer 2011, 50, 765–774. [Google Scholar] [CrossRef]
- Neben, K.; Jauch, A.; Bertsch, U.; Heiss, C.; Hielscher, T.; Seckinger, A.; Mors, T.; Müller, N.Z.; Hillengass, J.; Raab, M.S.; et al. Combining chromosomal aberrations t(4;14) and del(17p13) with ISS allows a stratification of myeloma patients undergoing autologous stem cell transplantation. Haematologica 2010, haematol-2009. [Google Scholar]
- Lonial, S.; Dimopoulos, M.; Palumbo, A.; White, D.; Grosicki, S.; Spicka, I.; Walter-Croneck, A.; Moreau, P.; Mateos, M.V.; Magen, H.; et al. Elotuzumab Therapy for Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2015, 373, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Schouten, J.P.; McElgunn, C.J.; Waaijer, R.; Zwijnenburg, D.; Diepvens, F.; Pals, G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002, 30, e57. [Google Scholar] [CrossRef] [PubMed]
- Lakshman, A.; Painuly, U.; Rajkumar, S.V.; Ketterling, R.P.; Kapoor, P.; Greipp, P.T.; Dispenzieri, A.; Gertz, M.A.; Buadi, F.K.; Lacy, M.Q.; et al. Impact of acquired del(17p) in multiple myeloma. Blood Adv. 2019, 3, 1930–1938. [Google Scholar] [CrossRef] [PubMed]
- Avet-Loiseau, H.; Bahlis, N.J.; Chng, W.J.; Masszi, T.; Viterbo, L.; Pour, L.; Ganly, P.; Palumbo, A.; Cavo, M.; Langer, C.; et al. Ixazomib significantly prolongs progression-free survival in high-risk relapsed/refractory myeloma patients. Blood 2017, 130, 2610–2618. [Google Scholar] [CrossRef]
- Chang, H.; Qi, C.; Yi, Q.-L.; Reece, D.; Stewart, A.K. p53 gene deletion detected by fluorescence in situ hybridization is an adverse prognostic factor for patients with multiple myeloma following autologous stem cell transplantation. Blood 2005, 105, 358–360. [Google Scholar] [CrossRef]
- Gaballa, S.; Saliba, R.M.; Srour, S.; Lu, G.; Brammer, J.E.; Shah, N.; Bashir, Q.; Patel, K.; Bock, F.; Parmar, S.; et al. Outcomes in patients with multiple myeloma with TP53 deletion after autologous hematopoietic stem cell transplant. Am. J. Hematol. 2016, 91, E442–E447. [Google Scholar] [CrossRef]
- Lakshman, A.; Painuly, U.; Rajkumar, S.V.; Ketterling, R.P.; Kapoor, P.; Greipp, P.T.; Gertz, M.A.; Buadi, F.K.; Lacy, M.Q.; Dingli, D.; et al. Natural history of multiple myeloma with de novo del(17p). Blood Cancer J. 2019, 9, 32. [Google Scholar] [CrossRef]
- Chin, M.; Sive, J.I.; Allen, C.; Roddie, C.; Chavda, S.J.; Smith, D.; Blombery, P.; Jones, K.; Ryland, G.L.; Popat, R.; et al. Prevalence and timing of TP53 mutations in del(17p) myeloma and effect on survival. Blood Cancer J. 2017, 7, e610. [Google Scholar] [CrossRef]
- Chang, H.; Trieu, Y.; Qi, X.; Jiang, N.N.; Xu, W.; Reece, D. Impact of cytogenetics in patients with relapsed or refractory multiple myeloma treated with bortezomib: Adverse effect of 1q21 gains. Leuk. Res. 2011, 35, 95–98. [Google Scholar] [CrossRef]
- Chen, M.H.; Qi, C.X.; Saha, M.N.; Chang, H. p53 nuclear expression correlates with hemizygous TP53 deletion and predicts an adverse outcome for patients with relapsed/refractory multiple myeloma treated with lenalidomide. Am. J. Clin. Pathol. 2012, 137, 208–212. [Google Scholar] [CrossRef]
- Ashby, C.; Boyle, E.; Tytarenko, R.G.; Wang, H.; Rosenthal, A.; Patel, P.; Wang, Y.; Deshpande, S.; Ortiz, M.; Flynt, E.; et al. Long-Term Follow-up Identifies Double Hit and Key Mutations as Impacting Progression Free and Overall Survival in Multiple Myeloma. Blood 2018, 132, 110. [Google Scholar] [CrossRef]
- Munawar, U.; Rasche, L.; Muller, N.; Vogt, C.; Da-Via, M.; Haertle, L.; Arampatzi, P.; Dietrich, S.; Roth, M.; Garitano-Trojaola, A.; et al. Hierarchy of mono- and biallelic TP53 alterations in multiple myeloma cell fitness. Blood 2019, 134, 836–840. [Google Scholar] [CrossRef] [PubMed]
- Owen, R.G.; Davis, S.A.; Randerson, J.; Rawstron, A.C.; Davies, F.; Child, J.A.; Jack, A.S.; Morgan, G.J. p53 gene mutations in multiple myeloma. Mol. Pathol. 1997, 50, 18–20. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.A.; Mavrommatis, K.; Wardell, C.P.; Ashby, T.C.; Bauer, M.; Davies, F.E.; Rosenthal, A.; Wang, H.; Qu, P.; Hoering, A.; et al. Identification of novel mutational drivers reveals oncogene dependencies in multiple myeloma. Blood 2018. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, R.; Blood, E.; Rue, M.; Harrington, D.; Oken, M.M.; Kyle, R.A.; Dewald, G.W.; Van Ness, B.; Van Wier, S.A.; Henderson, K.J.; et al. Clinical and biologic implications of recurrent genomic aberrations in myeloma. Blood 2003, 101, 4569–4575. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 7, 20. [Google Scholar] [CrossRef]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 2011, 39, e118. [Google Scholar] [CrossRef]
- Vaser, R.; Adusumalli, S.; Leng, S.N.; Sikic, M.; Ng, P.C. SIFT missense predictions for genomes. Nat. Protoc. 2016, 11, 1–9. [Google Scholar] [CrossRef]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef]
- Eischen, C.M.; Lozano, G. The Mdm network and its regulation of p53 activities: A rheostat of cancer risk. Hum. Mutat. 2014, 35, 728–737. [Google Scholar] [CrossRef]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Filipescu, D.; Naughtin, M.; Podsypanina, K.; Lejour, V.; Wilson, L.; Gurard-Levin, Z.A.; Orsi, G.A.; Simeonova, I.; Toufektchan, E.; Attardi, L.D.; et al. Essential role for centromeric factors following p53 loss and oncogenic transformation. Genes Dev. 2017, 31, 463–480. [Google Scholar] [CrossRef] [PubMed]
- Rausch, T.; Jones, D.T.; Zapatka, M.; Stutz, A.M.; Zichner, T.; Weischenfeldt, J.; Jager, N.; Remke, M.; Shih, D.; Northcott, P.A.; et al. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell 2012, 148, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Duensing, A.; Duensing, S. Guilt by association? p53 and the development of aneuploidy in cancer. Biochem. Biophys. Res. Commun. 2005, 331, 694–700. [Google Scholar] [CrossRef]
- Fukasawa, K.; Choi, T.; Kuriyama, R.; Rulong, S.; Vande Woude, G.F. Abnormal centrosome amplification in the absence of p53. Science 1996, 271, 1744–1747. [Google Scholar] [CrossRef] [PubMed]
- Vitre, B.D.; Cleveland, D.W. Centrosomes, chromosome instability (CIN) and aneuploidy. Curr. Opin. Cell Biol. 2012, 24, 809–815. [Google Scholar] [CrossRef]
- Ha, G.H.; Baek, K.H.; Kim, H.S.; Jeong, S.J.; Kim, C.M.; McKeon, F.; Lee, C.W. p53 activation in response to mitotic spindle damage requires signaling via BubR1-mediated phosphorylation. Cancer Res. 2007, 67, 7155–7164. [Google Scholar] [CrossRef]
- Oikawa, T.; Okuda, M.; Ma, Z.; Goorha, R.; Tsujimoto, H.; Inokuma, H.; Fukasawa, K. Transcriptional control of BubR1 by p53 and suppression of centrosome amplification by BubR1. Mol. Cell Biol. 2005, 25, 4046–4061. [Google Scholar] [CrossRef]
- Vitale, I.; Galluzzi, L.; Vivet, S.; Nanty, L.; Dessen, P.; Senovilla, L.; Olaussen, K.A.; Lazar, V.; Prudhomme, M.; Golsteyn, R.M.; et al. Inhibition of Chk1 kills tetraploid tumor cells through a p53-dependent pathway. PLoS ONE 2007, 2, e1337. [Google Scholar] [CrossRef]
- Wang, X.; Zhou, Y.X.; Qiao, W.; Tominaga, Y.; Ouchi, M.; Ouchi, T.; Deng, C.X. Overexpression of aurora kinase A in mouse mammary epithelium induces genetic instability preceding mammary tumor formation. Oncogene 2006, 25, 7148–7158. [Google Scholar] [CrossRef]
- Choi, B.K.; Dayaram, T.; Parikh, N.; Wilkins, A.D.; Nagarajan, M.; Novikov, I.B.; Bachman, B.J.; Jung, S.Y.; Haas, P.J.; Labrie, J.L.; et al. Literature-based automated discovery of tumor suppressor p53 phosphorylation and inhibition by NEK2. Proc. Natl. Acad. Sci. USA 2018, 115, 10666–10671. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, L.; King, S.; Marcar, L.; Nicol, S.; Dias, S.S.; Schumm, K.; Robertson, P.; Bourdon, J.C.; Perkins, N.; Fuller-Pace, F.; et al. p53-dependent repression of polo-like kinase-1 (PLK1). Cell Cycle 2010, 9, 4200–4212. [Google Scholar] [CrossRef] [PubMed]
- Nabilsi, N.H.; Ryder, D.J.; Peraza-Penton, A.C.; Poudyal, R.; Loose, D.S.; Kladde, M.P. Local depletion of DNA methylation identifies a repressive p53 regulatory region in the NEK2 promoter. J. Biol. Chem. 2013, 288, 35940–35951. [Google Scholar] [CrossRef] [PubMed]
- Ha, G.H.; Breuer, E.K. Mitotic Kinases and p53 Signaling. Biochem. Res. Int. 2012, 2012, 195903. [Google Scholar] [CrossRef]
- Bartek, J.; Lukas, J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 2003, 3, 421–429. [Google Scholar] [CrossRef]
- Sur, S.; Pagliarini, R.; Bunz, F.; Rago, C.; Diaz, L.A., Jr.; Kinzler, K.W.; Vogelstein, B.; Papadopoulos, N. A panel of isogenic human cancer cells suggests a therapeutic approach for cancers with inactivated p53. Proc. Natl. Acad. Sci. USA 2009, 106, 3964–3969. [Google Scholar] [CrossRef] [PubMed]
- Diab, A.; Kao, M.; Kehrli, K.; Kim, H.Y.; Sidorova, J.; Mendez, E. Multiple defects sensitize p53-deficient head and neck cancer cells to the WEE1 kinase inhibition. Mol. Cancer Res. 2019. [Google Scholar] [CrossRef]
- Moser, R.; Xu, C.; Kao, M.; Annis, J.; Lerma, L.A.; Schaupp, C.M.; Gurley, K.E.; Jang, I.S.; Biktasova, A.; Yarbrough, W.G.; et al. Functional kinomics identifies candidate therapeutic targets in head and neck cancer. Clin. Cancer Res. 2014, 20, 4274–4288. [Google Scholar] [CrossRef]
- Pappano, W.N.; Zhang, Q.; Tucker, L.A.; Tse, C.; Wang, J. Genetic inhibition of the atypical kinase Wee1 selectively drives apoptosis of p53 inactive tumor cells. BMC Cancer 2014, 14, 430. [Google Scholar] [CrossRef]
- Webster, P.J.; Littlejohns, A.T.; Gaunt, H.J.; Prasad, K.R.; Beech, D.J.; Burke, D.A. AZD1775 induces toxicity through double-stranded DNA breaks independently of chemotherapeutic agents in p53-mutated colorectal cancer cells. Cell Cycle 2017, 16, 2176–2182. [Google Scholar] [CrossRef]
- Morandell, S.; Reinhardt, H.C.; Cannell, I.G.; Kim, J.S.; Ruf, D.M.; Mitra, T.; Couvillon, A.D.; Jacks, T.; Yaffe, M.B. A reversible gene-targeting strategy identifies synthetic lethal interactions between MK2 and p53 in the DNA damage response in vivo. Cell Rep. 2013, 5, 868–877. [Google Scholar] [CrossRef] [PubMed]
- Gurpinar, E.; Vousden, K.H. Hitting cancers’ weak spots: Vulnerabilities imposed by p53 mutation. Trends Cell Biol. 2015, 25, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, A.; Grueneberg, D.A.; Hellner, K.; Sawyer, J.; Grace, M.; Li, W.; Harlow, E.; Munger, K. Kinase requirements in human cells: V. Synthetic lethal interactions between p53 and the protein kinases SGK2 and PAK3. Proc. Natl. Acad. Sci. USA 2010, 107, 12463–12468. [Google Scholar] [CrossRef]
- Wang, Y.; Decker, S.J.; Sebolt-Leopold, J. Knockdown of Chk1, Wee1 and Myt1 by RNA interference abrogates G2 checkpoint and induces apoptosis. Cancer Biol. Ther. 2004, 3, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Simon, R. Identification of potential synthetic lethal genes to p53 using a computational biology approach. BMC Med. Genom. 2013, 6, 30. [Google Scholar] [CrossRef]
- Wang, X.; Sun, Q. TP53 mutations, expression and interaction networks in human cancers. Oncotarget 2017, 8, 624–643. [Google Scholar] [CrossRef]
- Li, Y.; Liu, Y.; Xu, H.; Jiang, G.; Van der Jeught, K.; Fang, Y.; Zhou, Z.; Zhang, L.; Frieden, M.; Wang, L.; et al. Heterozygous deletion of chromosome 17p renders prostate cancer vulnerable to inhibition of RNA polymerase II. Nat. Commun. 2018, 9, 4394. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, X.; Han, C.; Wan, G.; Huang, X.; Ivan, C.; Jiang, D.; Rodriguez-Aguayo, C.; Lopez-Berestein, G.; Rao, P.H.; et al. TP53 loss creates therapeutic vulnerability in colorectal cancer. Nature 2015, 520, 697–701. [Google Scholar] [CrossRef]
- Morgan, G.J.; Walker, B.A.; Davies, F.E. The genetic architecture of multiple myeloma. Nat. Rev. Cancer 2012, 12, 335–348. [Google Scholar] [CrossRef]
- Herrero, A.B.; San Miguel, J.; Gutierrez, N.C. Deregulation of DNA double-strand break repair in multiple myeloma: Implications for genome stability. PLoS ONE 2015, 10, e0121581. [Google Scholar] [CrossRef]
- Kaelin, W.G., Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer 2005, 5, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Neri, P.; Ren, L.; Gratton, K.; Stebner, E.; Johnson, J.; Klimowicz, A.; Duggan, P.; Tassone, P.; Mansoor, A.; Stewart, D.A.; et al. Bortezomib-induced “BRCAness” sensitizes multiple myeloma cells to PARP inhibitors. Blood 2011, 118, 6368–6379. [Google Scholar] [CrossRef] [PubMed]
- de Bruin, E.C.; McGranahan, N.; Mitter, R.; Salm, M.; Wedge, D.C.; Yates, L.; Jamal-Hanjani, M.; Shafi, S.; Murugaesu, N.; Rowan, A.J.; et al. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science 2014, 346, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Henderson, S.; Chakravarthy, A.; Su, X.; Boshoff, C.; Fenton, T.R. APOBEC-mediated cytosine deamination links PIK3CA helical domain mutations to human papillomavirus-driven tumor development. Cell Rep. 2014, 7, 1833–1841. [Google Scholar] [CrossRef] [PubMed]
- Jamal-Hanjani, M.; Wilson, G.A.; McGranahan, N.; Birkbak, N.J.; Watkins, T.B.K.; Veeriah, S.; Shafi, S.; Johnson, D.H.; Mitter, R.; Rosenthal, R.; et al. Tracking the Evolution of Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 376, 2109–2121. [Google Scholar] [CrossRef]
- Law, E.K.; Sieuwerts, A.M.; LaPara, K.; Leonard, B.; Starrett, G.J.; Molan, A.M.; Temiz, N.A.; Vogel, R.I.; Meijer-van Gelder, M.E.; Sweep, F.C.; et al. The DNA cytosine deaminase APOBEC3B promotes tamoxifen resistance in ER-positive breast cancer. Sci. Adv. 2016, 2, e1601737. [Google Scholar] [CrossRef]
- McGranahan, N.; Favero, F.; de Bruin, E.C.; Birkbak, N.J.; Szallasi, Z.; Swanton, C. Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci. Transl. Med. 2015, 7, 283ra254. [Google Scholar] [CrossRef]
- Sieuwerts, A.M.; Willis, S.; Burns, M.B.; Look, M.P.; Meijer-Van Gelder, M.E.; Schlicker, A.; Heideman, M.R.; Jacobs, H.; Wessels, L.; Leyland-Jones, B.; et al. Elevated APOBEC3B correlates with poor outcomes for estrogen-receptor-positive breast cancers. Horm. Cancer 2014, 5, 405–413. [Google Scholar] [CrossRef]
- Walker, B.A.; Wardell, C.P.; Murison, A.; Boyle, E.M.; Begum, D.B.; Dahir, N.M.; Proszek, P.Z.; Melchor, L.; Pawlyn, C.; Kaiser, M.F.; et al. APOBEC family mutational signatures are associated with poor prognosis translocations in multiple myeloma. Nat. Commun. 2015, 6, 6997. [Google Scholar] [CrossRef]
- Caval, V.; Suspene, R.; Shapira, M.; Vartanian, J.P.; Wain-Hobson, S. A prevalent cancer susceptibility APOBEC3A hybrid allele bearing APOBEC3B 3’UTR enhances chromosomal DNA damage. Nat. Commun. 2014, 5, 5129. [Google Scholar] [CrossRef]
- Landry, S.; Narvaiza, I.; Linfesty, D.C.; Weitzman, M.D. APOBEC3A can activate the DNA damage response and cause cell-cycle arrest. EMBO Rep. 2011, 12, 444–450. [Google Scholar] [CrossRef]
- Nikkila, J.; Kumar, R.; Campbell, J.; Brandsma, I.; Pemberton, H.N.; Wallberg, F.; Nagy, K.; Scheer, I.; Vertessy, B.G.; Serebrenik, A.A.; et al. Elevated APOBEC3B expression drives a kataegic-like mutation signature and replication stress-related therapeutic vulnerabilities in p53-defective cells. Br. J. Cancer 2017, 117, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, M.; Towfic, F.; Samur, M.K.; Flynt, E.; Jang, I.S.; Wang, K.; Ashby, C.; Walker, B.A.; Trotter, M.; Morgan, G.; et al. A High-Risk Multiple Myeloma Group Identified By Integrative Multi-Omics Segmentation of Newly Diagnosed Patients. Blood 2018, 132, 3165. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, C.; Xu, Z.; Scuoppo, C.; Rillahan, C.D.; Gao, J.; Spitzer, B.; Bosbach, B.; Kastenhuber, E.R.; Baslan, T.; et al. Deletions linked to TP53 loss drive cancer through p53-independent mechanisms. Nature 2016, 531, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Cheok, C.F.; Lane, D.P. Exploiting the p53 Pathway for Therapy. Cold Spring Harb. Perspect. Med. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.; Breunig, C.; Figueroa, V.; Lehners, N.; Baumann, A.; Pálfi, A.; Müller, C.; Lutz, C.; Hechler, T.; Kulke, M.; et al. Preclinical Evaluation of Hdp-101, a Novel Anti-BCMA Antibody-Drug Conjugate, in Multiple Myeloma. Blood 2017, 130, 3070. [Google Scholar] [CrossRef]
- Palfi, A.; Hechler, T.; Mueller, C.; Pahl, A.; Kulke, M. Abstract 2973: CD269 - A promising target for amanitin based ADCs. Cancer Res. 2016, 76, 2973. [Google Scholar]
- Deneberg, S.; Cherif, H.; Lazarevic, V.; Andersson, P.O.; von Euler, M.; Juliusson, G.; Lehmann, S. An open-label phase I dose-finding study of APR-246 in hematological malignancies. Blood Cancer J. 2016, 6, e447. [Google Scholar] [CrossRef]
- Foster, B.A.; Coffey, H.A.; Morin, M.J.; Rastinejad, F. Pharmacological rescue of mutant p53 conformation and function. Science 1999, 286, 2507–2510. [Google Scholar] [CrossRef]
- Lehmann, S.; Bykov, V.J.; Ali, D.; Andren, O.; Cherif, H.; Tidefelt, U.; Uggla, B.; Yachnin, J.; Juliusson, G.; Moshfegh, A.; et al. Targeting p53 in vivo: A first-in-human study with p53-targeting compound APR-246 in refractory hematologic malignancies and prostate cancer. J. Clin. Oncol. 2012, 30, 3633–3639. [Google Scholar] [CrossRef]
- Tal, P.; Eizenberger, S.; Cohen, E.; Goldfinger, N.; Pietrokovski, S.; Oren, M.; Rotter, V. Cancer therapeutic approach based on conformational stabilization of mutant p53 protein by small peptides. Oncotarget 2016, 7, 11817–11837. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Vazquez, A.; Levine, A.J.; Carpizo, D.R. Allele-specific p53 mutant reactivation. Cancer Cell 2012, 21, 614–625. [Google Scholar] [CrossRef] [PubMed]
- Blanden, A.R.; Yu, X.; Loh, S.N.; Levine, A.J.; Carpizo, D.R. Reactivating mutant p53 using small molecules as zinc metallochaperones: Awakening a sleeping giant in cancer. Drug Discov. Today 2015, 20, 1391–1397. [Google Scholar] [CrossRef] [PubMed]
- Soragni, A.; Janzen, D.M.; Johnson, L.M.; Lindgren, A.G.; Thai-Quynh Nguyen, A.; Tiourin, E.; Soriaga, A.B.; Lu, J.; Jiang, L.; Faull, K.F.; et al. A Designed Inhibitor of p53 Aggregation Rescues p53 Tumor Suppression in Ovarian Carcinomas. Cancer Cell 2016, 29, 90–103. [Google Scholar] [CrossRef]
- Momand, J.; Zambetti, G.P.; Olson, D.C.; George, D.; Levine, A.J. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 1992, 69, 1237–1245. [Google Scholar] [CrossRef]
- Chen, J.; Marechal, V.; Levine, A.J. Mapping of the p53 and mdm-2 interaction domains. Mol. Cell Biol. 1993, 13, 4107–4114. [Google Scholar] [CrossRef]
- Tao, W.; Levine, A.J. Nucleocytoplasmic shuttling of oncoprotein Hdm2 is required for Hdm2-mediated degradation of p53. Proc. Natl. Acad. Sci. USA 1999, 96, 3077–3080. [Google Scholar] [CrossRef]
- Honda, R.; Tanaka, H.; Yasuda, H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997, 420, 25–27. [Google Scholar] [CrossRef]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Kubbutat, M.H.; Jones, S.N.; Vousden, K.H. Regulation of p53 stability by Mdm2. Nature 1997, 387, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Bayle, J.H.; Olson, D.; Levine, A.J. The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 1993, 7, 1126–1132. [Google Scholar] [CrossRef] [PubMed]
- Picksley, S.M.; Lane, D.P. The p53-mdm2 autoregulatory feedback loop: A paradigm for the regulation of growth control by p53? Bioessays 1993, 15, 689–690. [Google Scholar] [CrossRef] [PubMed]
- Teoh, G.; Urashima, M.; Ogata, A.; Chauhan, D.; DeCaprio, J.A.; Treon, S.P.; Schlossman, R.L.; Anderson, K.C. MDM2 protein overexpression promotes proliferation and survival of multiple myeloma cells. Blood 1997, 90, 1982–1992. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [PubMed]
- Saha, M.N.; Jiang, H.; Chang, H. Molecular mechanisms of nutlin-induced apoptosis in multiple myeloma: Evidence for p53-transcription-dependent and -independent pathways. Cancer Biol. Ther. 2010, 10, 567–578. [Google Scholar] [CrossRef]
- Saha, M.N.; Jiang, H.; Jayakar, J.; Reece, D.; Branch, D.R.; Chang, H. MDM2 antagonist nutlin plus proteasome inhibitor velcade combination displays a synergistic anti-myeloma activity. Cancer Biol. Ther. 2010, 9, 936–944. [Google Scholar] [CrossRef]
- Andreeff, M.; Kelly, K.R.; Yee, K.; Assouline, S.; Strair, R.; Popplewell, L.; Bowen, D.; Martinelli, G.; Drummond, M.W.; Vyas, P.; et al. Results of the Phase I Trial of RG7112, a Small-Molecule MDM2 Antagonist in Leukemia. Clin. Cancer Res. 2016, 22, 868–876. [Google Scholar] [CrossRef]
- Rao, B.; Lain, S.; Thompson, A.M. p53-Based cyclotherapy: Exploiting the ‘guardian of the genome’ to protect normal cells from cytotoxic therapy. Br. J. Cancer 2013, 109, 2954–2958. [Google Scholar] [CrossRef]
- Choong, M.L.; Yang, H.; Lee, M.A.; Lane, D.P. Specific activation of the p53 pathway by low dose actinomycin D: A new route to p53 based cyclotherapy. Cell Cycle 2009, 8, 2810–2818. [Google Scholar] [CrossRef]
- Van Leeuwen, I.M. Cyclotherapy: Opening a therapeutic window in cancer treatment. Oncotarget 2012, 3, 596–600. [Google Scholar] [CrossRef]
- Campisi, J. Cellular senescence as a tumor-suppressor mechanism. Trends Cell Biol. 2001, 11, S27–S31. [Google Scholar] [CrossRef]
- Beausejour, C.M.; Krtolica, A.; Galimi, F.; Narita, M.; Lowe, S.W.; Yaswen, P.; Campisi, J. Reversal of human cellular senescence: Roles of the p53 and p16 pathways. EMBO J. 2003, 22, 4212–4222. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Trotman, L.C.; Shaffer, D.; Lin, H.K.; Dotan, Z.A.; Niki, M.; Koutcher, J.A.; Scher, H.I.; Ludwig, T.; Gerald, W.; et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005, 436, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, H.; Yamada, Y.; Iha, H.; Tsukasaki, K.; Nagai, K.; Atogami, S.; Sugahara, K.; Tsuruda, K.; Ishizaki, A.; Kamihira, S. Activation of p53 by Nutlin-3a, an antagonist of MDM2, induces apoptosis and cellular senescence in adult T-cell leukemia cells. Leukemia 2009, 23, 2090–2101. [Google Scholar] [CrossRef] [PubMed]
- Ablain, J.; Rice, K.; Soilihi, H.; de Reynies, A.; Minucci, S.; de The, H. Activation of a promyelocytic leukemia-tumor protein 53 axis underlies acute promyelocytic leukemia cure. Nat. Med. 2014, 20, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Wiley, C.D.; Schaum, N.; Alimirah, F.; Lopez-Dominguez, J.A.; Orjalo, A.V.; Scott, G.; Desprez, P.Y.; Benz, C.; Davalos, A.R.; Campisi, J. Small-molecule MDM2 antagonists attenuate the senescence-associated secretory phenotype. Sci. Rep. 2018, 8, 2410. [Google Scholar] [CrossRef]
- Tonnessen-Murray, C.A.; Frey, W.D.; Rao, S.G.; Shahbandi, A.; Ungerleider, N.A.; Olayiwola, J.O.; Murray, L.B.; Vinson, B.T.; Chrisey, D.B.; Lord, C.J.; et al. Chemotherapy-induced senescent cancer cells engulf other cells to enhance their survival. J. Cell Biol. 2019, 218, 3827–3844. [Google Scholar] [CrossRef]
- Zeimet, A.G.; Marth, C. Why did p53 gene therapy fail in ovarian cancer? Lancet Oncol 2003, 4, 415–422. [Google Scholar] [CrossRef]
- Fedoseyeva, E.V.; Boisgerault, F.; Anosova, N.G.; Wollish, W.S.; Arlotta, P.; Jensen, P.E.; Ono, S.J.; Benichou, G. CD4+ T cell responses to self- and mutated p53 determinants during tumorigenesis in mice. J. Immunol. 2000, 164, 5641–5651. [Google Scholar] [CrossRef]
- Roth, J.; Dittmer, D.; Rea, D.; Tartaglia, J.; Paoletti, E.; Levine, A.J. p53 as a target for cancer vaccines: Recombinant canarypox virus vectors expressing p53 protect mice against lethal tumor cell challenge. Proc. Natl. Acad. Sci. USA 1996, 93, 4781–4786. [Google Scholar] [CrossRef]
- McArdle, S.E.; Rees, R.C.; Mulcahy, K.A.; Saba, J.; McIntyre, C.A.; Murray, A.K. Induction of human cytotoxic T lymphocytes that preferentially recognise tumour cells bearing a conformational p53 mutant. Cancer Immunol. Immunother. 2000, 49, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Offringa, R.; Vierboom, M.P.; van der Burg, S.H.; Erdile, L.; Melief, C.J. p53: A potential target antigen for immunotherapy of cancer. Ann. N. Y. Acad. Sci. 2000, 910, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Kuball, J.; Wen, S.F.; Leissner, J.; Atkins, D.; Meinhardt, P.; Quijano, E.; Engler, H.; Hutchins, B.; Maneval, D.C.; Grace, M.J.; et al. Successful adenovirus-mediated wild-type p53 gene transfer in patients with bladder cancer by intravesical vector instillation. J. Clin. Oncol. 2002, 20, 957–965. [Google Scholar] [CrossRef] [PubMed]
- Bertholet, S.; Iggo, R.; Corradin, G. Cytotoxic T lymphocyte responses to wild-type and mutant mouse p53 peptides. Eur. J. Immunol. 1997, 27, 798–801. [Google Scholar] [CrossRef]
- Vermeij, R.; Leffers, N.; van der Burg, S.H.; Melief, C.J.; Daemen, T.; Nijman, H.W. Immunological and clinical effects of vaccines targeting p53-overexpressing malignancies. J. Biomed. Biotechnol. 2011, 2011, 702146. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Author | N | Prevalence Del17p in Full Dataset | Method | Range (Median) of % Positive Cells | Threshold/CCF for High-Risk | mPFS | mOS |
|---|---|---|---|---|---|---|---|
| Chang [56] | 105 | 9.5% (n = 10) | FISH | 18–95% (Median 53%) | None | Median 7.9 mo | Median 14.7 mo |
| Avet-Loiseau [43] | 532 | 11% (n = 58) | FISH | 32–94% (Median 75%) | ≥60% PCs | mEFS 14.6 mo | Median 22.4 mo |
| Neben [51] | 289 | 10% (n = 29) | FISH | NR | 60–70% PCs | 3-yr: 27% | 3-yr: 50% |
| Lode [25] | 92 | 57% n = 54) | FISH | NR | ≥60% PCs | NR | NR |
| Boyd [50] | 85 | 100% (n = 85) (selected population of del17p patients) | FISH | NR | None | 14.7 mo | 26.6 mo |
| An [49] | 333 | 6.6% (n = 22) | FISH | 25–100% (Median 65%) | >50% PCs | 4 mo | 16 mo |
| Lonial [52] | 646 | 32% (n = 206) | FISH | NR | ≥1 cell | NR | NR |
| Thanendrarajan [44] | 779 | 10% (>20% cutoff; n = 76) 8% (>40% cutoff; n = 62) 7% (>60%; cutoff n = 51) 4% (>80% cutoff; n = 34) | FISH | Investigated >20, >40, >60, and >80% cutoffs | ≥20% PCs | 3 yr: 61% | 3 yr: 67% |
| Shah [48] | 1905 | 9% (n = 175) | MLPA | NR | NR | NR; HR: 1.57 | NR; HR: 2.10 |
| Shah [47] | 1777 | 10.8% (MLPA < 0.8) (n = 192) | MLPA | Investigated > 0.8, 0.7–0.79, 0.55–0.69, and <0.55 cutoffs | MLPA value >0.8 (>20% PCs) | NR | ≥0.7 to <0.8: HR = 1.8 ≥0.55 to <0.7: HR = 3.1 <0.5: HR = 2.2 |
| Gaballa [57] | 145 | 23.4% (n = 34) | FISH | NR | NR | 8 mo | 21 mo |
| Lakshman [58] | 310 | 100% (n = 310) (selected population of del17p patients) | FISH | 8–100% (Median 69.5%) | Investigated ≥ 20 vs. <20% ≥30 vs. <30% ≥40 vs. <40% ≥50 vs. <50% ≥60 vs. <60% | 19.2 vs. 32.5 18.8 vs. 30.8 18.3 vs. 30.8 17.8 vs. 30.3 16.8 vs. 28.3 | 45.3 vs. NR 45.2 vs. 89.6 45.2 vs. 89.6 44.8 vs. 58.3 38.1 vs. 58.3 |
| Thakurta [6] | 605 | 100% (n = 605) (selected population of del17p patients) | FISH (discovery) | Investigated CCF range 0.3 to 0.8 | >0.55 CCF | 14.3 mo | 36.1 mo |
| 235 | 100% (n = 235) (selected population of del17p patients) | FISH (replication) | >0.55 CCF | 17 mo | 32 mo | ||
| 108 | 100% (selected population of del17p patients [n = 108] from n = 1273 MGP) | NGS | >0.55 CCF | 26 mo | 36 mo |
| Author | Total N | Prevalence Del17p in Full Dataset | Method | Range (Median) of % Positive Cells | Threshold/CCF for High-Risk | mPFS | mOS |
|---|---|---|---|---|---|---|---|
| Lakshman [39] | 228 (152 control + 76 acquired del17p) | 33% (n = 65) | FISH | 9–100% (Median 89%) | None | 23.0 mo (from diagnosis) 5.4 mo (after detection of del17p) | 68.2 mo (from diagnosis) 18.1 mo (after detection of del17p) |
| Chin [59] | 188 | 22.3% (n = 42) | FISH | NR | None | NR; Mixed NDMM and RRMM | NR |
| Chang [60] | 85 | 22% (n = 17) | FISH | NR | >10% | del17p+ vs. neg. 5.4 vs. 5.0 ns p = 0.60 | 11.5 vs. 15 ns P = 0.41 |
| Chen [61] | 88 | 15% (n = 13) 13% (n = 11) | FISH IHC | NR 10–90% (40%) | >10% >10% | P53/del17p+ vs. neg 2 3.4 vs. 11 mo 3.4 vs. 11 mo | 12.1 vs. 28.8 mo 7.2 vs. 28.8 mo |
| Avet-Loiseau [55] | 552 | 10% (n = 69) Ixazomib Rd n = 36 placebo-Rd n = 33 | FISH | Investigated >5% >20% >60% | None | IRd vs. Rd 21.4 vs. 9.7 21.4 vs. 6.7 15.7 vs. 5.1 | NR |
| Mutation | Score and Predicted Impact | |||
|---|---|---|---|---|
| Mutation Assessor | SIFT | PolyPhen2 | ||
| 1 | Y126C | 3.25, Medium | 0.00, Deleterious | 1.00, Probably damaging |
| 2 | Y126H | 3.25, Medium | 0.00, Deleterious | 1.00, Probably damaging |
| 3 | Y126splice | NA | NA | NA |
| 4 | S127F | 3.29, Medium | 0.00, Deleterious | 1.00, Probably damaging |
| 5 | M133K | 0.00, Neutral | 0.00, Deleterious | 0.12, Benign |
| 6 | M133T | 0.00, Neutral | 0.00, Deleterious | 0.09, Benign |
| 7 | F134S | 2.00, Medium | 0.00, Deleterious | 1.00, Probably damaging |
| 8 | F134L | 2.64, Medium | 0.00, Deleterious | 1.00, Probably damaging |
| 9 | C135Y | 3.08, Medium | 0.00, Deleterious | 1.00, Probably damaging |
| 10 | C135FS | NA | NA | NA |
| 11 | A161T | 2.99, Medium | 0.00, Deleterious | 1.00, Probably damaging |
| 12 | Y163D | 3.17, Medium | 0.00, Deleterious | 1.00, Probably damaging |
| 13 | Y163C | 3.17, Medium | 0.00, Deleterious | 1.00, Probably damaging |
| 14 | R175G | 3.28, Medium | 0.00, Deleterious | 1.00, Probably damaging |
| 15 | R175H | 2.58, Medium | 0.11, Tolerated | 0.31, Benign |
| 16 | G199V | 3.11, Medium | 0.00, Deleterious | 1.00, Probably damaging |
| 17 | Y234C | 2.99, Medium | 0.00, Deleterious | 0.99, Probably damaging |
| 18 | R248W | 3.28, Medium | 0.00, Deleterious | 1.00, Probably damaging |
| 19 | R248Q | 2.94, Medium | 0.00, Deleterious | 1.00, Probably damaging |
| 20 | P250L | 3.27, Medium | 0.00, Deleterious | 1.00, Probably damaging |
| 21 | R267W | 3.22, Medium | 0.05, Tolerated | 0.73, Probably damaging |
| 22 | R273L | 3.18, Medium | 0.00, Deleterious | 0.99, Probably damaging |
| 23 | R273H | 2.08, Medium | 0.13, Tolerated | 0.63, Probably damaging |
| 24 | R282G | 2.46, Medium | 0.03, Deleterious | 0.28, Benign |
| 25 | E285K | 3.04, Medium | 0.13, Tolerated | 0.98, Probably damaging |
| 26 | R337C | 1.56, Low | 0.09, Tolerated | 0.34, Benign |
| 27 | R337L | 2.95, Medium | 0.01, Deleterious | 0.91, Probably damaging |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flynt, E.; Bisht, K.; Sridharan, V.; Ortiz, M.; Towfic, F.; Thakurta, A. Prognosis, Biology, and Targeting of TP53 Dysregulation in Multiple Myeloma. Cells 2020, 9, 287. https://doi.org/10.3390/cells9020287
Flynt E, Bisht K, Sridharan V, Ortiz M, Towfic F, Thakurta A. Prognosis, Biology, and Targeting of TP53 Dysregulation in Multiple Myeloma. Cells. 2020; 9(2):287. https://doi.org/10.3390/cells9020287
Chicago/Turabian StyleFlynt, Erin, Kamlesh Bisht, Vinidhra Sridharan, María Ortiz, Fadi Towfic, and Anjan Thakurta. 2020. "Prognosis, Biology, and Targeting of TP53 Dysregulation in Multiple Myeloma" Cells 9, no. 2: 287. https://doi.org/10.3390/cells9020287
APA StyleFlynt, E., Bisht, K., Sridharan, V., Ortiz, M., Towfic, F., & Thakurta, A. (2020). Prognosis, Biology, and Targeting of TP53 Dysregulation in Multiple Myeloma. Cells, 9(2), 287. https://doi.org/10.3390/cells9020287