Differentially Expressed Genes Shared by Two Distinct Cytoplasmic Male Sterility (CMS) Types of Silene vulgaris Suggest the Importance of Oxidative Stress in Pollen Abortion



Abstract
1. Introduction
2. Materials and Methods
2.1. Trimming and Quality Control of Paired-End Reads
2.2. De Novo Assembly
Evaluation of Assemblies
2.3. Estimation of Transcript Quantification and Differential Gene Expression
2.4. Reciprocal Blast Search
2.5. Estimation of Gene Expression
2.5.1. Plant Material
2.5.2. RNA Extraction, Reverse Transcription and qRT PCR
2.5.3. Statistical Evaluation
3. Results and Discussion
3.1. Transcriptome Assembly, Reduction of Duplicates
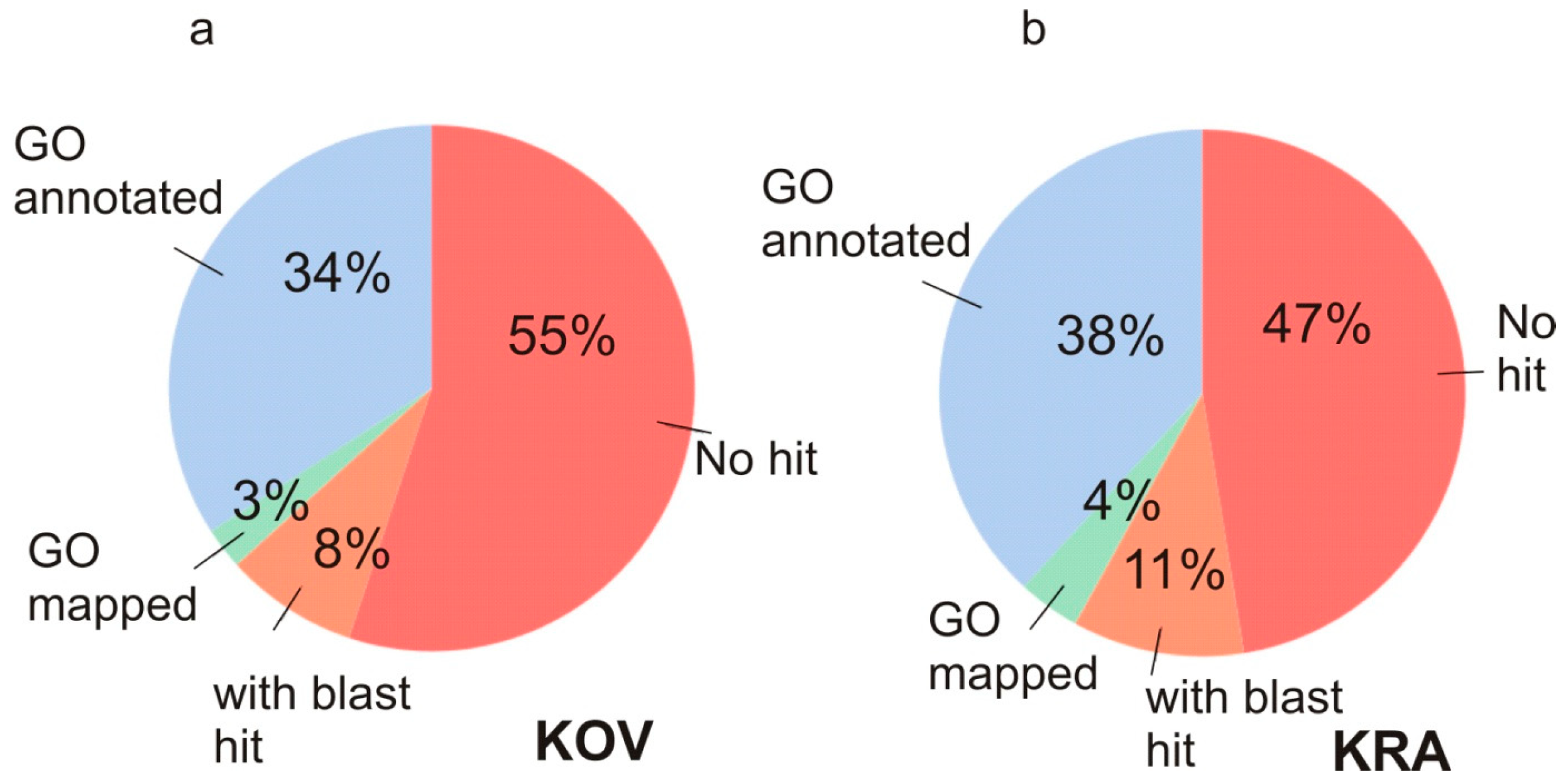
3.2. Functional Annotation
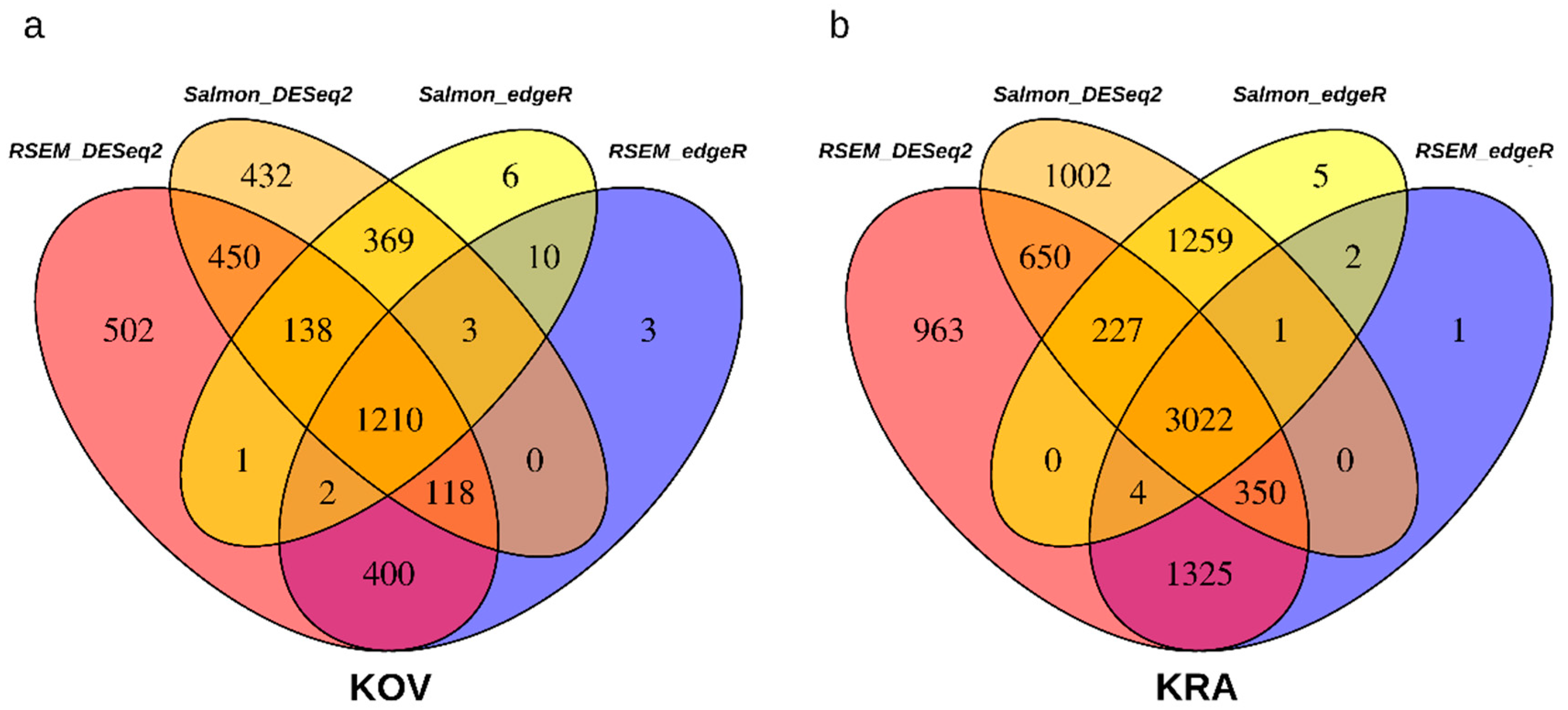
3.3. Estimation of Differentially Expressed (DE) Genes
3.4. Enrichment of GO Categories
3.5. DE Genes Shared by KRA and KOV Plants
3.6. Genes Involved in Male Functions Were Downregulated in S. vulgaris Female Flower Buds
3.7. Pollen Abortion Is Associated with Oxidative Stress
3.8. AOX1 Gene Was Downregulated in Female Flower Buds in S. vulgaris
3.9. Differences between Nuclear-Encoded Transcriptomes of F and H Flower Buds of S. vulgaris Contrast with the Uniformity of Their Organellar Transcriptomes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Horn, R.; Gupta, K.J.; Colombo, N. Mitochondrion role in molecular basis of cytoplasmic male sterility. Mitochondrion 2014, 19, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Touzet, P.; Meyer, E.H. Cytoplasmic male sterility and mitochondrial metabolism in plants. Mitochondrion 2014, 19, 166–171. [Google Scholar] [CrossRef] [PubMed]
- Hanson, M.R. Interactions of Mitochondrial and Nuclear Genes That Affect Male Gametophyte Development. Plant Cell 2004, 16, S154–S169. [Google Scholar] [CrossRef] [PubMed]
- Olson, M.S.; McCauley, D.E. Mitochondrial DNA diversity, population structure, and gender association in the gynodioecious plant Silene vulgaris. Evolution 2002, 56, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Dufay, M.; Champelovier, P.; Käfer, J.; Henry, J.P.; Mousset, S.; Ab Marais, G. An angiosperm-wide analysis of the gynodioecy–dioecy pathway. Ann. Bot. 2014, 114, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Renner, S.S. The relative and absolute frequencies of angiosperm sexual systems: Dioecy, monoecy, gynodioecy, and an updated online database. Am. J. Bot. 2014, 101, 1588–1596. [Google Scholar] [CrossRef]
- Bernasconi, G.; Antonovics, J.; Biere, A.L.; Charlesworth, D.; Delph, L.F.; Filatov, D.A.; Giraud, T.; Hood, M.E.; Marais, G.A.B.; McCauley, D.W.; et al. Silene as a model system in ecology and evolution. Heredity 2009, 103, 5–14. [Google Scholar] [CrossRef]
- Štorchová, H.; Olson, M.S. Comparison between mitochondrial and chloroplast DNA variation in the native range of Silene vulgaris. Mol. Ecol. 2004, 13, 2909–2919. [Google Scholar] [CrossRef]
- Barr, C.M.; Keller, S.R.; Ingvarsson, P.; Sloan, D.B.; Taylor, U.R. Variation in Mutation Rate and Polymorphism Among Mitochondrial Genes of Silene vulgaris. Mol. Biol. Evol. 2007, 24, 1783–1791. [Google Scholar] [CrossRef] [PubMed]
- Sebasky, M.E.; Keller, S.R.; Taylor, D.R. Investigating past range dynamics for a weed of cultivation, Silene vulgaris. Ecol. Evol. 2016, 6, 4800–4811. [Google Scholar] [CrossRef]
- Štorchová, H.; Müller, K.; Lau, S.; Olson, M.S. Mosaic Origins of a Complex Chimeric Mitochondrial Gene in Silene vulgaris. PLoS ONE 2012, 7, e30401. [Google Scholar] [CrossRef] [PubMed]
- Štorchová, H.; Stone, J.D.; Sloan, D.B.; Abeyawardana, O.A.J.; Müller, K.; Walterová, J.; Pažoutová, M. Homologous recombination changes the context of Cytochrome b transcription in the mitochondrial genome of Silene vulgaris KRA. BMC Genom. 2018, 19, 874. [Google Scholar] [CrossRef] [PubMed]
- Sloan, D.B.; Müller, K.; McCauley, D.E.; Taylor, D.R.; Štorchová, H. Intraspecific variation in mitochondrial genome sequence, structure, and gene content inSilene vulgaris, an angiosperm with pervasive cytoplasmic male sterility. New Phytol. 2012, 196, 1228–1239. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Zhang, W.; Wang, Y.; Xu, L.; Zhu, X.; Muleke, E.M.; Liu, L. Comprehensive transcriptome-based characterization of differentially expressed genes involved in microsporogenesis of radish CMS line and its maintainer. Funct. Integr. Genom. 2016, 16, 529–543. [Google Scholar] [CrossRef]
- Li, C.; Zhao, Z.; Liu, Y.; Liang, B.; Guan, S.; Lan, H.; Wang, J.; Lu, Y.; Cao, M. Comparative transcriptome analysis of isonuclear-alloplasmic lines unmask key transcription factor genes and metabolic pathways involved in sterility of maize CMS-C. PeerJ 2017, 5, e3408. [Google Scholar] [CrossRef] [PubMed]
- Pei, X.; Jing, Z.; Tang, Z.; Zhu, Y. Comparative transcriptome analysis provides insight into differentially expressed genes related to cytoplasmic male sterility in broccoli (Brassica oleracea var. italica). Sci. Hortic. 2017, 217, 234–242. [Google Scholar] [CrossRef]
- Chen, G.; Ye, X.; Zhang, S.; Zhu, S.; Yuan, L.; Hou, J.; Wang, C. Comparative Transcriptome Analysis between Fertile and CMS Flower Buds in Wucai (Brassica campestris L.). BMC Genom. 2018, 19, 908. [Google Scholar] [CrossRef]
- Hamid, R.; Tomar, R.S.; Marashi, H.; Shafaroudi, S.M.; Golakiya, B.; Mohsenpour, M. Transcriptome profiling and cataloging differential gene expression in floral buds of fertile and sterile lines of cotton (Gossypium hirsutum L.). Gene 2018, 660, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Ou, C.; Chen, S.; Cao, Q.; Zhao, Z.; Miao, Z.; Kong, X.; Zhuang, F.-Y. Differentially Expressed Genes between Carrot Petaloid Cytoplasmic Male Sterile and Maintainer during Floral Development. Sci. Rep. 2019, 9, 17384. [Google Scholar] [CrossRef] [PubMed]
- Janzen, G.M.; Wang, L.; Hufford, M.B. The extent of adaptive wild introgression in crops. New Phytol. 2018, 221, 1279–1288. [Google Scholar] [CrossRef] [PubMed]
- Haas, M.; Himmelbach, A.; Mascher, M. The contribution of cis- and trans-acting variants to gene regulation in wild and domesticated barley under cold stress and control conditions. J. Exp. Bot. 2020, 71, 2573–2584. [Google Scholar] [CrossRef] [PubMed]
- Case, A.L.; Willis, J.H. Hybrid male sterility in Mimulus (Phrymaceae) is associated with a geographically restricted mitochondrial rearrangeMENT. Evolution 2008, 62, 1026–1039. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.P.; Shinada, H.; Onodera, Y.; Komaki, C.; Mikami, T.; Kubo, T. A male sterility-associated mitochondrial protein in wild beets causes pollen disruption in transgenic plants. Plant J. 2008, 54, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.D.; Koloušková, P.; Sloan, D.B.; Štorchová, H. Non-coding RNA may be associated with cytoplasmic male sterility in Silene vulgaris. J. Exp. Bot. 2017, 68, 1599–1612. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Krüger, M.; Abeyawardana, O.A.J.; Juříček, M.; Krüger, C.; Štorchová, H. Variation in plastid genomes in the gynodioecious species Silene vulgaris. BMC Plant Biol. 2019, 19, 568. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Florea, L. Rcorrector: Efficient and accurate error correction for Illumina RNA-seq reads. GigaScience 2015, 4, 48. [Google Scholar] [CrossRef] [PubMed]
- Kopylova, E.; Noé, L.; Touzet, H. SortMeRNA: Fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 2012, 28, 3211–3217. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Gilbert, D.G. Longest protein, longest transcript or most expression, for accurate gene reconstruction of transcriptomes? bioRxiv 2019, 829184. [Google Scholar] [CrossRef]
- Seppey, M.; Manni, M.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness. In Gene Prediction; Kollmar, M., Ed.; Methods in Molecular Biology; Humana: New York, NY, USA, 2019; Volume 1962, pp. 227–245. [Google Scholar]
- Li, B.; Fillmore, N.; Bai, Y.; Collins, M.; Thomson, J.A.; Stewart, R.; Dewey, C.N. Evaluation of de novo transcriptome assemblies from RNA-Seq data. Genome Biol. 2014, 15, 553. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ruhlman, T.; Mower, J.P.; Jansen, R. Comparative analyses of two Geraniaceae transcriptomes using next-generation sequencing. BMC Plant Biol. 2013, 13, 228. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, M.I.L.R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef]
- Koloušková, P.; Stone, J.D.; Štorchová, H. Evaluation of reference genes for reverse transcription quantitative real-time PCR (RT-qPCR) studies in Silene vulgaris considering the method of cDNA preparation. PLoS ONE 2017, 12, e0183470. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.D.; Štorchová, H. The application of RNA-seq to the comprehensive analysis of plant mitochondrial transcriptomes. Mol. Genet. Genom. 2014, 290, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Sloan, D.B.; Keller, S.R.; Berardi, A.E.; Sanderson, B.J.; Karpovich, J.F.; Taylor, D.R. De novo transcriptome assembly and polymorphism detection in the flowering plant Silene vulgaris (Caryophyllaceae). Mol. Ecol. Resour. 2011, 12, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Gastaldi, V.; Lucero, L.; Ferrero, L.; Ariel, F.; Gonzalez, D.H. Class-I TCP Transcription Factors Activate the SAUR63 Gene Subfamily in Gibberellin-Dependent Stamen Filament Elongation. Plant Physiol. 2020, 182, 2096–2110. [Google Scholar] [CrossRef] [PubMed]
- Verma, N. Transcriptional regulation of anther development in Arabidopsis. Gene 2019, 689, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Liang, W.; Yin, C.; Cui, X.; Zong, J.; Wang, X.; Hu, J.; Zhang, D. Rice MADS3 Regulates ROS Homeostasis during Late Anther Development. Plant Cell 2011, 23, 515–533. [Google Scholar] [CrossRef]
- Routray, P.; Li, T.; Yamasaki, A.; Yoshinari, A.; Takano, J.; Choi, W.G.; Sams, C.E.; Roberts, D.M. Nodulin Intrinsic Protein 7;1 Is a Tapetal Boric Acid Channel Involved in Pollen Cell Wall Formation. Plant Physiol. 2018, 178, 1269–1283. [Google Scholar] [CrossRef] [PubMed]
- Cecchetti, V.; Brunetti, P.; Napoli, N.; Fattorini, L.; Altamura, M.M.; Costantino, P.; Cardarelli, M. ABCB1 and ABCB19 auxin transporters have synergistic effects on early and lateArabidopsisanther development. J. Integr. Plant Biol. 2015, 57, 1089–1098. [Google Scholar] [CrossRef]
- Sheng, Y.; Wang, Y.; Jiao, S.; Jin, Y.; Ji, P.; Luan, F. Mapping and Preliminary Analysis of ABORTED MICROSPORES (AMS) as the Candidate Gene Underlying the Male Sterility (MS-5) Mutant in Melon (Cucumis melo L.). Front. Plant Sci. 2017, 8, 902. [Google Scholar] [CrossRef]
- Zluvova, J.; Zak, J.; Janousek, B.; Vyskot, B. Dioecious Silene latifolia plants show sexual dimorphism in the vegetative stage. BMC Plant Biol. 2010, 10, 208. [Google Scholar] [CrossRef]
- Dobritsa, A.A.; Geanconteri, A.; Shrestha, J.; Carlson, A.L.; Kooyers, N.; Coerper, D.; Urbanczyk-Wochniak, E.; Bench, B.J.; Sumner, L.W.; Swanson, R.; et al. A Large-Scale Genetic Screen in Arabidopsis to Identify Genes Involved in Pollen Exine Production. Plant Physiol. 2011, 157, 947–970. [Google Scholar] [CrossRef]
- Jiang, J.; Zhang, Z.; Cao, J. Pollen wall development: The associated enzymes and metabolic pathways. Plant Biol. 2012, 15, 249–263. [Google Scholar] [CrossRef]
- Bosch, M.; Hepler, P.K. Pectin Methylesterases and Pectin Dynamics in Pollen Tubes. Plant Cell 2005, 17, 3219–3226. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.; Song, C.; Gao, L.; Zhang, H.; Yang, C.; Sheng, J.; Ren, J.; Chen, D.; Wang, Y. Transcriptome de novo assembly and analysis of differentially expressed genes related to cytoplasmic male sterility in onion. Plant Physiol. Biochem. 2018, 125, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Mittler, R. Oxidative stress, antioxidants and stress tolerance. Trends Plant Sci. 2002, 7, 405–410. [Google Scholar] [CrossRef]
- Yang, L.; Wu, Y.; Zhang, M.; Zhang, J.; Stewart, J.M.; Xing, C.; Wu, J.; Jin, S. Transcriptome, cytological and biochemical analysis of cytoplasmic male sterility and maintainer line in CMS-D8 cotton. Plant Mol. Biol. 2018, 97, 537–551. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.; Xu, H.; Liu, Z.; Guo, J.; Li, H.; Chen, L.; Fang, C.; Zhang, Q.; Bai, M.; Yao, N.; et al. A detrimental mitochondrial-nuclear interaction causes cytoplasmic male sterility in rice. Nat. Genet. 2013, 45, 573–577. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.; Moon, S.; Lee, Y.-S.; Zhu, L.; Liang, W.; Zhang, D.; Jung, K.-H.; An, G. Defective Tapetum Cell Death 1 (DTC1) Regulates ROS Levels by Binding to Metallothionein during Tapetum Degeneration. Plant Physiol. 2016, 170, 1611–1623. [Google Scholar] [CrossRef] [PubMed]
- Escamez, S.; Stael, S.; Vainonen, J.P.; Willems, P.; Jin, H.; Kimura, S.; Van Breusegem, F.; Gevaert, K.; Wrzaczek, M.; Tuominen, H. Extracellular peptide Kratos restricts cell death during vascular development and stress in Arabidopsis. J. Exp. Bot. 2019, 70, 2199–2210. [Google Scholar] [CrossRef]
- Qin, T.; Tian, Q.; Wang, G.; Xiong, L. LOWER TEMPERATURE 1 Enhances ABA Responses and Plant Drought Tolerance by Modulating the Stability and Localization of C2-Domain ABA-Related Proteins in Arabidopsis. Mol. Plant 2019, 12, 1243–1258. [Google Scholar] [CrossRef]
- Ding, B.; Hao, M.; Mei, D.; Zaman, Q.U.; Sang, S.-F.; Wang, H.; Wang, W.-X.; Fu, L.; Cheng, H.; Hu, Q. Transcriptome and Hormone Comparison of Three Cytoplasmic Male Sterile Systems in Brassica napus. Int. J. Mol. Sci. 2018, 19, 4022. [Google Scholar] [CrossRef]
- Ilyas, M.; Rasheed, A.; Mahmood, T. Functional characterization of germin and germin-like protein genes in various plant species using transgenic approaches. Biotechnol. Lett. 2016, 38, 1405–1421. [Google Scholar] [CrossRef]
- Millar, A.H.; Whelan, J.; Soole, K.L.; Day, D.A. Organization and Regulation of Mitochondrial Respiration in Plants. Annu. Rev. Plant Biol. 2011, 62, 79–104. [Google Scholar] [CrossRef] [PubMed]
- Schwarzländer, M.; König, A.-C.; Sweetlove, L.J.; Finkemeier, I. The impact of impaired mitochondrial function on retrograde signalling: A meta-analysis of transcriptomic responses. J. Exp. Bot. 2011, 63, 1735–1750. [Google Scholar] [CrossRef] [PubMed]
- Kühn, K.; Obata, T.; Feher, K.; Bock, R.; Fernie, A.R.; Meyer, E.H. Complete Mitochondrial Complex I Deficiency Induces an Up-Regulation of Respiratory Fluxes That Is Abolished by Traces of Functional Complex I. Plant Physiol. 2015, 168, 1537–1549. [Google Scholar] [CrossRef] [PubMed]
- Merendino, L.; Courtois, F.; Grübler, B.; Bastien, O.; Straetmanns, V.; Chevalier, F.; Lerbs-Mache, S.; Lurin, C.; Pfannschmidt, T. Retrograde signals from mitochondria reprogramme skoto-morphogenesis in Arabidopsis thaliana via alternative oxidase 1a. Philos. Trans. R. Soc. B Biol. Sci. 2020, 375, 20190567. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Rodriguez-Uribe, L.; Xu, J.; Zhang, J. Transcriptome analysis of cytoplasmic male sterility and restoration in CMS-D8 cotton. Plant Cell Rep. 2013, 32, 1531–1542. [Google Scholar] [CrossRef] [PubMed]
- Niazi, A.K.; Delannoy, E.; Iqbal, R.K.; Mileshina, D.; Val, R.; Gabryelska, M.; Wyszko, E.; Taconnat, L.; Szymanski, M.; Barciszewski, J.; et al. Mitochondrial Transcriptome Control and Intercompartment Cross-Talk During Plant Development. Cells 2019, 8, 583. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.-B.; Wu, X.-M.; Ge, X.-X.; Deng, X.-X.; Grosser, J.W.; Guo, W.-W. Comparative Transcript Profiling of a Male Sterile Cybrid Pummelo and Its Fertile Type Revealed Altered Gene Expression Related to Flower Development. PLoS ONE 2012, 7, e43758. [Google Scholar] [CrossRef]
- Hamid, R.; Jacob, F.; Marashi, H.; Rathod, V.; Tomar, R.S. Uncloaking lncRNA-meditated gene expression as a potential regulator of CMS in cotton (Gossypium hirsutum L.). Genomics 2020, 112, 3354–3364. [Google Scholar] [CrossRef]
- Colombatti, F.; Gonzalez, D.H.; Welchen, E. Plant mitochondria under pathogen attack: A sigh of relief or a last breath? Mitochondrion 2014, 19, 238–244. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| KRA | Raw Reads | Final Number of Reads |
|---|---|---|
| Sum of read pairs | 232,291,523 | 96,244,096 |
| Average number of read pairs per sample | 38,715,254 | 16,040,683 |
| KOV | ||
| Sum of read pairs | 199,896,220 | 142,034,627 |
| Average number of read pairs per sample | 33,316,036 | 23,672,438 |
| Assembly | N50 | Percent GC | Total Trinity ‘Transcripts’ | Total Trinity ‘Genes’ | Transcripts’ per ‘Genes’ | Average Contig | Median Contig Length | Total Assembled Bases | Contiguity | Completeness | Detonate Score |
|---|---|---|---|---|---|---|---|---|---|---|---|
| KRA | |||||||||||
| Trinity_146_32 | 1477 | 39.51 | 485,454 | 217,940 | 2.227466275 | 943 | 598 | 457,681,231 | |||
| Trinity_146_32_cdhit | 1486 | 39.4 | 459,631 | 198,260 | 2.318324422 | 960 | 621 | 441,163,523 | 0.827 | 0.905 | −39,167,557,352 |
| Trinity_146_32_evigene | 1387 | 39.71 | 104,013 | 84,004 | 1.238191039 | 951 | 662 | 98,923,714 | 0.783 | 0.85 | −39,482,140,762 |
| KOV | |||||||||||
| Trinity_65 | 1280 | 39.76 | 322,441 | 181,371 | 1.777797994 | 774 | 437 | 249,686,710 | |||
| Trinity_65_cdhit | 1268 | 39.7 | 307,905 | 170,448 | 1.806445368 | 779 | 449 | 239,849,174 | 0.868 | 0.909 | −23,349,399,191 |
| Trinity_65_evigene | 1300 | 40.1 | 72,262 | 64,549 | 1.11949062 | 822 | 470 | 59,426,744 | 0.863 | 0.897 | −28,051,238,486 |
| KOV | KRA | Annotation | Process or Function |
|---|---|---|---|
| DN20956_c0_g1 | DN25290_c0_g1 | aquaporin TIP1-3 | abiotic stress response |
| DN8218_c1_g1 | DN50548_c0_g1 | BURP domain protein RD22 | abiotic stress response |
| DN11001_c0_g2 | DN58656_c0_g2 | SPX domain-containing protein 3 | abiotic stress response |
| DN117378_c0_g1 | DN52115_c3_g1 | ABC transporter G family member 14 | abiotic stress response, transport of cytokinins |
| DN117904_c0_g1 | DN57560_c0_g2 | leucine-rich repeat receptor-like kinase BAM2 | anther and pollen development |
| DN17312_c0_g1 | DN45544_c4_g1 | auxin-responsive protein SAUR64 | anther development |
| DN17927_c0_g2 | DN49199_c0_g1 | aquaporin NIP7-1 | anther development |
| DN19580_c0_g2 | DN42368_c0_g1 | scopoletin glucosyltransferase | biotic stress response |
| DN29923_c0_g1 | DN49507_c0_g1 | glucan endo-1,3-beta-glucosidase 12 | biotic stress response |
| DN13581_c1_g1 | DN50185_c1_g1 | anthranilate N-benzoyltransferase 2 | biotic stress response |
| DN294_c0_g1 | DN63593_c0_g1 | inositol-pentakisphosphate 2-kinase | biotic stress response |
| DN5945_c0_g1 | DN64094_c0_g1 | ABC transporter G family member 31 | biotic stress response |
| DN26761_c0_g1 | DN39252_c0_g1 | F-box protein CPR30-like | biotic stress response, proteasome |
| DN49774_c0_g1 | DN39675_c0_g1 | F-box/LRR-repeat protein 13-like | biotic stress response, proteasome |
| DN15243_c0_g1 | DN52671_c0_g1 | jasmonate-induced protein homolog | biotic stress response, response to jasmonate |
| DN36891_c0_g2 | DN54344_c0_g1 | protein TIFY 10A-like | biotic stress response, response to jasmonate |
| DN5404_c2_g1 | DN44106_c0_g1 | pectinesterase 11 | cell wall |
| DN3555_c0_g2 | DN48381_c0_g1 | xyloglucan endotransglucosylase/hydrolase | cell wall |
| DN75992_c1_g1 | DN59301_c0_g4 | UDP-arabinopyranose mutase 1 | cell wall |
| DN6141_c0_g1 | DN60757_c0_g1 | cell wall RBR3 | cell wall |
| DN12797_c0_g1 | DN69711_c0_g1 | Pectinesterase 2 | cell wall, pectin modification |
| DN11562_c0_g1 | DN54928_c0_g1 | UDP-glucuronate:xylan alpha-glucuronosyltransferase | cell wall, sugar metabolism |
| DN24940_c0_g2 | DN68925_c2_g1 | microtubule-associated protein 70-2 | cytoskelet |
| DN28090_c0_g1 | DN23353_c0_g1 | LIM domain-containing protein WLIM2b | cytoskelet |
| DN3731_c0_g1 | DN55753_c0_g1 | protein NSP-INTERACTING KINASE 1 | defense against viruses |
| DN5587_c1_g1 | DN52217_c0_g1 | ribosome-inactivating protein lychnin | defense to herbivores |
| DN15662_c0_g1 | DN54291_c1_g1 | benzyl alcohol O-benzoyltransferase | floral scent production |
| DN1200_c0_g1 | DN66313_c0_g2 | benzyl alcohol O-benzoyltransferase | floral scent production |
| DN12658_c0_g1 | DN61319_c0_g1 | ent-copalyl diphosphate synthase, chloroplastic | gibberellin biosynthesis |
| DN10021_c0_g1 | DN67192_c0_g1 | 3-hydroxy-3-methylglutaryl-coenzyme A reductase 3 | isoprenoid biosynthesis |
| DN7797_c0_g3 | DN58587_c0_g1 | caffeoylshikimate esterase-like | lignin biosynthesis |
| DN84_c0_g3 | DN64134_c1_g1 | O-acyltransferase WSD1-like | lipid and wax metabolism, cutin synthesis |
| DN1640_c0_g1 | DN72417_c1_g1 | protein ECERIFERUM 1-like | lipid and wax metabolism, cutin synthesis |
| DN18997_c0_g1 | DN62098_c0_g1 | GDSL esterase/lipase EXL3 | lipid catabolism |
| DN1956_c0_g1 | DN48468_c3_g1 | very-long-chain 3-oxoacyl-CoA reductase 1 | lipid metabolism |
| DN12582_c0_g2 | DN49044_c1_g1 | putative peroxygenase 4 | lipid degradation, ABA signaling |
| DN4033_c0_g1 | DN50049_c3_g1 | delta(12)-fatty-acid desaturase FAD2 | lipid metabolism |
| DN28833_c0_g1 | DN55962_c0_g1 | Acyltransferase-like protein, chloroplastic | lipid and wax metabolism, cutin synthesis |
| DN6801_c0_g1 | DN56028_c0_g2 | acyl-CoA--sterol O-acyltransferase 1 | lipid metabolism |
| DN1868_c0_g1 | DN61281_c1_g1 | 3-oxoacyl-[acyl-carrier-protein] synthase I, chloroplastic | lipid metabolism |
| DN4218_c0_g4 | DN61881_c0_g1 | long chain acyl-CoA synthetase 7, peroxisomal | lipid metabolism |
| DN5733_c0_g3 | DN62382_c1_g1 | NADH--cytochrome b5 reductase 1 | lipid metabolism |
| DN12960_c0_g1 | DN65129_c0_g1 | acyltransferase-like At1g54570, chloroplastic | lipid and wax metabolism, cutin synthesis |
| DN7091_c0_g1 | DN70722_c3_g2 | non-specific lipid-transfer protein A-like | lipid metbolism, cutin formation |
| DN9230_c0_g1 | DN57659_c0_g1 | glycerol-3-phosphate 2-O-acyltransferase 6 | lipid metabolism, cutin synthesis |
| DN8493_c1_g4 | DN58552_c1_g2 | dehydrodolichyl diphosphate synthase subunit NUS1-like | lipid metabolism, glycosylation |
| DN4923_c0_g1 | DN71674_c2_g5 | dehydrodolichyl diphosphate synthase subunit NUS1-like | lipid metabolism, glycosylation |
| DN42303_c0_g1 | DN54377_c1_g1 | 3-ketoacyl-CoA synthase 10 | lipid metabolism, pollen development |
| DN325_c0_g1 | DN69099_c0_g1 | 3-ketoacyl-CoA synthase 7-like | lipid metabolism, very long chain fatty acid |
| DN40822_c0_g1 | DN56377_c0_g1 | Adipose-regulatory protein, putative | lipid storage |
| DN17020_c0_g1 | DN52633_c0_g1 | aluminum-act. malate transporter 2 | malate transport |
| DN140371_c0_g1 | DN62179_c1_g1 | serendip2, partial | male gender-associated gene |
| DN19368_c0_g1 | DN58293_c0_g1 | non-functional NADPH-dependent codeinone reductase 2 | oxidation-reduction processes |
| DN1066_c0_g2 | DN58866_c0_g1 | cytochrome P450 89A2 | oxidation-reduction processes |
| DN8357_c0_g1 | DN63019_c1_g2 | uncharacterized oxidoreductase At1g06690, chloroplastic | oxidation-reduction processes |
| DN8866_c0_g2 | DN64912_c1_g4 | nudix hydrolase 12, mitochondrial-like | oxidation-reduction processes |
| DN1426_c0_g1 | DN64077_c0_g1 | cell number regulator 13-like | plant growth |
| DN6949_c0_g2 | DN64967_c1_g1 | 2-methyl-6-phytyl-1,4-HQ methyltransferase, chloroplastic | plastoquinon synthesis |
| DN3074_c0_g2 | DN41000_c0_g1 | plant self-incompatibility protein S1 family | pollen development |
| DN5323_c0_g2 | DN43413_c0_g1 | Plant self-incompatibility S1 | pollen development |
| DN881_c0_g1 | DN70592_c0_g1 | polygalacturonase QRT3-like | Pollen development |
| DN10605_c0_g1 | DN62065_c1_g1 | serine/threonine-protein phosphatase PP1 | protein dephosphorylation |
| DN5982_c0_g1 | DN46942_c0_g5 | peptidyl-prolyl cis-trans isomerase | protein folding |
| DN3850_c0_g1 | DN67053_c0_g2 | beta-1,3-galactosyltransferase 8 | protein glycosylation |
| DN24059_c0_g1 | DN43126_c0_g1 | ervatamin-B isoform X2 | proteolysis |
| DN96143_c0_g2 | DN50973_c0_g1 | F-box/kelch-repeat protein At3g23880 | proteolysis |
| DN10994_c0_g2 | DN55415_c0_g4 | probable aspartic protease At2g35615 | proteolysis |
| DN43449_c0_g1 | DN66566_c0_g1 | F-box/kelch-repeat protein At3g23880 | proteolysis |
| DN1779_c0_g1 | DN56281_c3_g1 | HIG_1_N domain-containing protein | respiration in mitochondria |
| DN8887_c0_g1 | DN56446_c0_g1 | alternative oxidase1, mitochondrial | respiration in mitochondria |
| DN6741_c1_g2 | DN67021_c1_g1 | aldehyde dehydrogenase family 2 member C4 | response to biotic stress, redox processes |
| DN15427_c0_g1 | DN70278_c1_g1 | aldehyde dehydrogenase family 3 member F1 | response to osmotic stress |
| DN2886_c0_g2 | DN65942_c2_g2 | oxidation resistance protein 1 | response to oxidative stress |
| DN960_c0_g2 | DN54803_c0_g2 | plastid-lipid-associated protein 3, chloroplastic | response to stress, jasmonate synthesis |
| DN79910_c0_g1 | DN59524_c0_g2 | protein SUPPRESSOR OF MAX2 1 | response to strigolactones |
| DN3860_c0_g1 | DN39636_c0_g2 | retroelement pol polyprotein-like | retroelement |
| DN22296_c0_g2 | DN43632_c0_g1 | RNA-binding protein pno1-like | RNA binding |
| DN100112_c0_g1 | DN53301_c0_g3 | annexin-like protein RJ4 | abiotic stress response |
| DN11360_c0_g1 | DN69045_c0_g1 | cation/H(+) antiporter 20 | abiotic stress response, stomata opening |
| DN25736_c0_g1 | DN48226_c0_g1 | mitogen-activated protein KKK 1 | stress response |
| DN88774_c0_g1 | DN50043_c0_g2 | Bet beta-fructofuranosidase | sugar metabolism |
| DN49169_c0_g2 | DN54840_c0_g2 | rhamnogalacturonan I rhamnosyltransferase | sugar metabolism |
| DN664_c0_g1 | DN73158_c6_g2 | protein TSS | suppression of meristem proliferation |
| DN14694_c0_g1 | DN52676_c1_g1 | zinc finger protein ZAT4 | TF, abiotic stress response |
| DN64000_c0_g1 | DN50222_c0_g3 | transcription factor MYB26 | TF, anther development |
| DN31894_c0_g1 | DN60959_c0_g2 | protein FAR1-RELATED SEQUENCE 5 | TF, development |
| DN26183_c0_g1 | DN53805_c0_g3 | bZIP transcription factor 11 | TF, hypo-osmolarity stress response |
| DN29604_c0_g1 | DN47072_c0_g5 | NAC transcription factor 25 | TF, pollen development |
| DN2940_c0_g1 | DN63243_c0_g1 | transcription factor MYB97 | TF, pollen development |
| DN1229_c0_g1 | DN59645_c1_g1 | transcription factor ABORTED MICROSPORES | TF, pollen development |
| DN2456_c0_g1 | DN62952_c0_g1 | BEL1-like homeodomain protein 11 | TF, positive regulation of plastid development |
| DN14971_c1_g2 | DN52803_c0_g1 | transcription factor MYB41 | TF, salt stress response |
| DN5746_c0_g1 | DN50022_c0_g1 | transcription factor MYB44 | TF, stress response |
| DN11354_c3_g1 | DN56745_c0_g3 | ubiquitin-60S ribosomal protein L40 | translation |
| DN4532_c0_g2 | DN46765_c0_g1 | eukaryotic translation initiation factor 3 subunit G | translation initiation |
| DN8567_c0_g1 | DN58593_c1_g3 | 40S ribosomal protein S27-2 | translation, abiotic stress response |
| DN263_c0_g1 | DN57992_c0_g2 | Ctr copper transporter | transport of copper |
| DN22803_c0_g1 | DN49878_c0_g1 | fe(2+) transport protein 1 | transport of iron |
| DN34096_c0_g1 | DN63070_c0_g1 | metal-nicotianamine transporter YSL7 | transport of metals |
| DN8241_c1_g1 | DN73009_c4_g1 | ABC transporter-like | transport of phytohormones |
| DN6894_c0_g1 | DN63590_c0_g1 | purine permease 5 | transport of purines |
| DN16716_c1_g1 | DN55273_c0_g1 | equilibrative nucleotide transporter 3 | transport of nucleotides, cytokinins |
| DN4674_c0_g1 | DN50701_c1_g1 | homogentisate solanesyltransferase, chloroplastic | ubiquinone synthesis |
| DN10150_c0_g1 | DN63897_c0_g1 | methyltransferase At1g78140, chloroplastic | ubiquinone synthesis |
| KOV | KRA | Annotation | Process or Function |
|---|---|---|---|
| KOV_DN16097_c0_g1 | KRA_DN47101_c0_g1 | caffeine synthase 1-like | alkaloid metabolism |
| KOV_DN17890_c0_g1 | KRA_DN67656_c0_g2 | CBL-interacting serine/threonine-protein kinase 20 | ABA response |
| KOV_DN2739_c0_g1 | KRA_DN70411_c3_g1 | ABC transporter G family member 25 | ABA transport |
| KOV_DN93524_c0_g1 | KRA_DN48556_c0_g2 | protein C2-DOMAIN ABA-RELATED 4-like | ABA signaling |
| KOV_DN84273_c0_g1 | KRA_DN67096_c1_g1 | UPF0496 protein At1g20180-like | abiotic stress response |
| KOV_DN1869_c0_g1 | KRA_DN49644_c0_g1 | homocysteine S-methyltransferase 3 | amino acid metabolism |
| KOV_DN30024_c0_g1 | KRA_DN52586_c0_g1 | cystinosin homolog | amino acid transport |
| KOV_DN17245_c0_g1 | KRA_DN59599_c0_g1 | amino acid permease 6-like | amino acid transport, nutrition |
| KOV_DN2748_c0_g1 | KRA_DN70324_c1_g1 | gamma aminobutyrate transaminase 1, mitochondrial | amino-acid metabolism |
| KOV_DN1890_c0_g4 | KRA_DN56949_c0_g1 | branched-chain-amino-acid aminotransferase 2, chloroplastic | amino-acid synthesis |
| KOV_DN60253_c0_g1 | KRA_DN60612_c1_g1 | protein PIN-LIKES 7-like | auxin response |
| KOV_DN51736_c2_g4 | KRA_DN41168_c0_g1 | NDR1/HIN1-like protein 1 | biotic stress response |
| KOV_DN4107_c0_g1 | KRA_DN47018_c0_g1 | isoflavone 3′-hydroxylase-like | biotic stress response |
| KOV_DN3773_c0_g1 | KRA_DN52943_c0_g3 | UPF0496 protein At4g34320-like | biotic stress response |
| KOV_DN1836_c0_g3 | KRA_DN55452_c0_g3 | acidic mammalian chitinase-like | biotic stress response |
| KOV_DN4184_c0_g2 | KRA_DN59528_c0_g4 | protein ENHANCED PSEUDOMONAS SUSCEPTIBILTY 1 | biotic stress response |
| KOV_DN1416_c0_g1 | KRA_DN64933_c1_g2 | probable serine/threonine-protein kinase PBL19 | biotic stress response |
| KOV_DN4164_c0_g1 | KRA_DN54390_c0_g2 | protein NRT1/PTR FAMILY 5.2-like | biotic stress response, peptide transport |
| KOV_DN14790_c0_g1 | KRA_DN69798_c2_g1 | cyclic nucleotide-gated ion channel 2-like | biotic stress response, programmed cell death |
| KOV_DN9654_c0_g1 | KRA_DN55792_c0_g1 | formate dehydrogenase, mitochondrial | cell death, biotic stress response |
| KOV_DN6043_c0_g1 | KRA_DN35718_c0_g2 | metacaspase 9-like | cell death |
| KOV_DN18472_c0_g1 | KRA_DN61497_c0_g1 | protein NEN1 | cell death, nuclease |
| KOV_DN16884_c0_g1 | KRA_DN59879_c0_g1 | endonuclease 1 | cell death, senescence |
| KOV_DN1688_c0_g1 | KRA_DN51302_c1_g2 | vignain-like | programmed cell death, proteolysis |
| KOV_DN3555_c0_g1 | KRA_DN51950_c0_g1 | probable xyloglucan endotransglucosylase/hydrolase30 | cell wall |
| KOV_DN490_c0_g1 | KRA_DN48287_c2_g1 | 1-aminocyclopropane-1-carboxylate oxidase-like | ethylene production, senescence |
| KOV_DN1335_c0_g1 | KRA_DN44770_c0_g1 | gibberellin 2-beta-dioxygenase 8-like | GA metabolism |
| KOV_DN174_c0_g1 | KRA_DN60929_c0_g1 | serine/threonine-protein kinase SAPK1-like | hyperosmotic stress response |
| KOV_DN5953_c0_g1 | KRA_DN57332_c1_g2 | GDSL esterase/lipase 2-like | lipid metabolism |
| KOV_DN16253_c0_g2 | KRA_DN63290_c0_g1 | Acetyl-coenzyme A synthetase, mitochondrial | lipid metabolism |
| KOV_DN33595_c1_g1 | KRA_DN54044_c1_g1 | caleosin 1 | lipid metabolism, stress response |
| KOV_DN58204_c0_g1 | KRA_DN61032_c0_g1 | lipase-like PAD4 | lipid metabolism, stress response |
| KOV_DN19808_c0_g2 | KRA_DN65272_c0_g1 | glutelin type-A 3-like | nutrition |
| KOV_DN772_c0_g1 | KRA_DN58790_c0_g1 | protein NRT1/PTR FAMILY 2.9 | nutrition, nitrate transport |
| KOV_DN17288_c1_g1 | KRA_DN38866_c0_g1 | peroxidase 27-like | oxidative stress response |
| KOV_DN28823_c0_g1 | KRA_DN48528_c1_g2 | peroxidase 5-like | oxidative stress response |
| KOV_DN23154_c0_g1 | KRA_DN56053_c0_g3 | peroxidase 5-like | oxidative stress response |
| KOV_DN13196_c0_g1 | KRA_DN63168_c0_g1 | aldehyde dehydrogenase family 3 member H1-like | oxidative stress response |
| KOV_DN6075_c0_g1 | KRA_DN52711_c1_g1 | flavonoid 3′-monooxygenase-like | oxidative stress response, antioxidant synth. |
| KOV_DN11776_c0_g1 | KRA_DN52985_c1_g1 | heavy metal-associated isoprenylated plant protein 6 | oxidative stress response, heavy metal ass. |
| KOV_DN3256_c0_g1 | KRA_DN64806_c0_g1 | protein SRG1-like | oxidative stress response, senescence |
| KOV_DN8862_c1_g1 | KRA_DN71589_c1_g2 | protein NRT1/PTR FAMILY 1.2-like | peptide transport |
| KOV_DN3658_c0_g1 | KRA_DN57822_c0_g1 | aspartic proteinase PCS1 | proteolysis |
| KOV_DN12803_c0_g1 | KRA_DN41740_c0_g1 | basic 7S globulin-like | proteolysis, nutrition |
| KOV_DN16367_c0_g2 | KRA_DN47392_c0_g2 | Basic 7S globulin | proteolysis, nutrition |
| KOV_DN1246_c0_g1 | KRA_DN45140_c0_g1 | F-box protein SKP2A-like | proteolysis, ubiquitination |
| KOV_DN817_c0_g1 | KRA_DN73170_c8_g1 | putative ABC transporter C family memb. 15 | response to oxidative stress, metabolism of glutathion |
| KOV_DN3628_c0_g1 | KRA_DN72682_c4_g2 | alpha-aminoadipic semialdehyde synthase | lysine degradation, stress response |
| KOV_DN8089_c0_g2 | KRA_DN72242_c0_g1 | DEAD-box ATP-dependent RNA helicase 14 | rRNA processing, ribosome biogenesis |
| KOV_DN11614_c0_g1 | KRA_DN48220_c0_g3 | Senescence regulator | senescence |
| KOV_DN9969_c0_g3 | KRA_DN65248_c0_g2 | protein DMP3 | senescence |
| KOV_DN15831_c0_g2 | KRA_DN53939_c3_g1 | cytochrome P450 71A4-like | senescence, flavor production |
| KOV_DN25654_c0_g1 | KRA_DN40946_c0_g1 | ribonuclease 1-like | senescence, PI starvation |
| KOV_DN4090_c0_g1 | KRA_DN58419_c0_g3 | 60 kDa jasmonate-induced protein | stress response |
| KOV_DN9074_c0_g1 | KRA_DN68068_c0_g1 | pumilio homolog 5 | stress response |
| KOV_DN20724_c0_g1 | KRA_DN59779_c0_g3 | bifunctional epoxide hydrolase 2 | stress response, cutin biosynthesis |
| KOV_DN418_c0_g1 | KRA_DN46150_c0_g1 | auxin-binding protein ABP19a-like | stress response, germin |
| KOV_DN2633_c0_g2 | KRA_DN69004_c2_g1 | auxin-binding protein ABP19a-like | stress response, germin |
| KOV_DN429_c0_g1 | KRA_DN35498_c0_g1 | alpha-amylase-like | sugar metabolism |
| KOV_DN1973_c0_g1 | KRA_DN44419_c0_g2 | cell wall/vacuolar inhibitor of fructosidase 1 | sugar metabolism |
| KOV_DN3558_c0_g1 | KRA_DN66372_c0_g1 | putative beta-galactosidase | sugar metabolism |
| KOV_DN2574_c0_g1 | KRA_DN59605_c4_g1 | beta-D-xylosidase 1 | sugar metabolism, cell wall |
| KOV_DN6104_c0_g1 | KRA_DN52927_c0_g4 | probable trehalose-phosphate phosphatase D | sugar metabolism, drought stress response |
| KOV_DN5263_c0_g1 | KRA_DN43951_c0_g1 | NAC domain-containing protein 92- | TF, abotic stress response |
| KOV_DN6211_c0_g3 | KRA_DN58757_c2_g1 | ethylene-responsive transcription factor ABR1 | TF, cold stress response |
| KOV_DN40310_c0_g1 | KRA_DN60425_c0_g1 | ethylene-responsive transcription factor ERF073 | TF, cold stress response |
| KOV_DN88395_c0_g2 | KRA_DN62269_c0_g1 | ethylene-responsive transcription factor 2 | TF, cold stress response |
| KOV_DN45283_c0_g1 | KRA_DN57848_c2_g2 | homeobox-leucine zipper protein ATHB-6 | TF, drought stress response |
| KOV_DN650_c0_g1 | KRA_DN58037_c6_g1 | zinc finger protein ZAT10-like | TF, drought stress response |
| KOV_DN2019_c0_g1 | KRA_DN59737_c0_g1 | homeobox-leucine zipper protein ATHB-12 | TF, drought stress response |
| KOV_DN883_c0_g1 | KRA_DN52066_c0_g1 | NAC domain-containing protein 72-like | TF, oxidative stress response |
| KOV_DN3909_c0_g1 | KRA_DN53835_c0_g2 | B3 domain-containing protein At2g36080-like | TF, repressor |
| KOV_DN1263_c0_g1 | KRA_DN54658_c0_g1 | homeobox-leucine zipper protein HAT5 | TF, salt stress response |
| KOV_DN4889_c0_g2 | KRA_DN44027_c0_g1 | protein CUP-SHAPED COTYLEDON 3 | TF, SAM development, organ separation |
| KOV_DN6423_c0_g2 | KRA_DN47072_c0_g3 | NAC transcription factor 29-like | TF, senescence |
| KOV_DN3487_c0_g1 | KRA_DN57553_c3_g2 | 30S ribosomal protein | translation |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krüger, M.; Abeyawardana, O.A.J.; Krüger, C.; Juříček, M.; Štorchová, H. Differentially Expressed Genes Shared by Two Distinct Cytoplasmic Male Sterility (CMS) Types of Silene vulgaris Suggest the Importance of Oxidative Stress in Pollen Abortion. Cells 2020, 9, 2700. https://doi.org/10.3390/cells9122700
Krüger M, Abeyawardana OAJ, Krüger C, Juříček M, Štorchová H. Differentially Expressed Genes Shared by Two Distinct Cytoplasmic Male Sterility (CMS) Types of Silene vulgaris Suggest the Importance of Oxidative Stress in Pollen Abortion. Cells. 2020; 9(12):2700. https://doi.org/10.3390/cells9122700
Chicago/Turabian StyleKrüger, Manuela, Oushadee A. J. Abeyawardana, Claudia Krüger, Miloslav Juříček, and Helena Štorchová. 2020. "Differentially Expressed Genes Shared by Two Distinct Cytoplasmic Male Sterility (CMS) Types of Silene vulgaris Suggest the Importance of Oxidative Stress in Pollen Abortion" Cells 9, no. 12: 2700. https://doi.org/10.3390/cells9122700
APA StyleKrüger, M., Abeyawardana, O. A. J., Krüger, C., Juříček, M., & Štorchová, H. (2020). Differentially Expressed Genes Shared by Two Distinct Cytoplasmic Male Sterility (CMS) Types of Silene vulgaris Suggest the Importance of Oxidative Stress in Pollen Abortion. Cells, 9(12), 2700. https://doi.org/10.3390/cells9122700