Alterations of the Endoplasmic Reticulum (ER) Calcium Signaling Molecular Components in Alzheimer’s Disease



{kind=link}
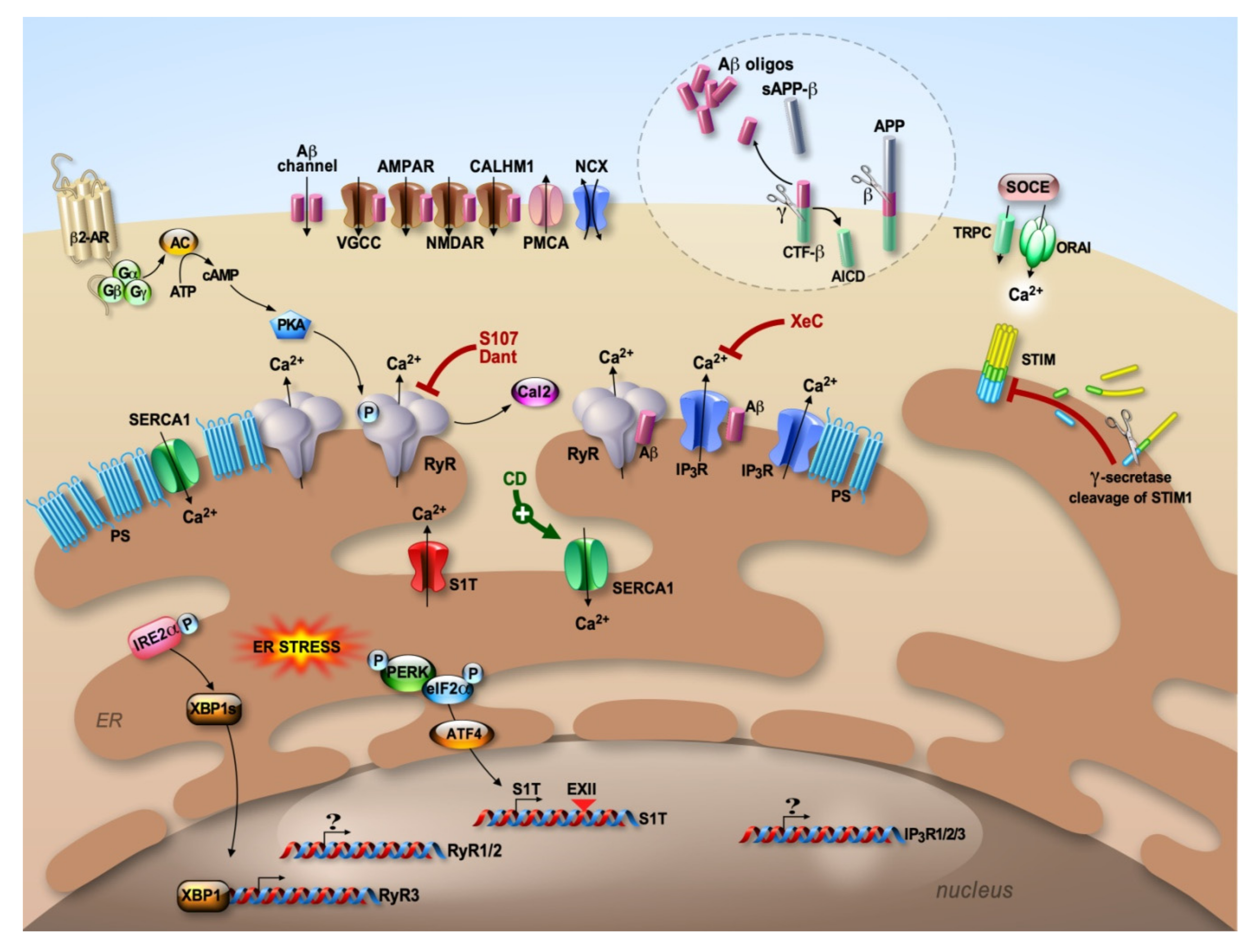{kind=link}
Abstract
1. Introduction
1.1. Ca2+ Signaling
1.2. Alzheimer’s Disease
1.3. Physiology of ER Calcium Handling in Neurons
1.4. Calcium Deregulation in AD
2. The Ryanodine Receptors: RyRs
3. The Inositol 1,4,5-Trisphosphate Receptors: IP3Rs
4. Presenilins 1 and 2: PS1/2
5. The Sarco-Endoplasmic Reticulum (SR/ER) Ca2+-ATPase and Its Truncated Isoform: SERCA and S1T
6. The Molecular Bridge between ER Ca2+ Depletion and Plasma Membrane Ca2+ Entry: STIM/ORAI
7. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [PubMed]
- Bootman, M.D.; Collins, T.J.; Peppiatt, C.M.; Prothero, L.S.; MacKenzie, L.; De Smet, P.; Travers, M.; Tovey, S.C.; Seo, J.T.; Berridge, M.J.; et al. Calcium signalling—An overview. Semin. Cell Dev. Biol. 2001, 12, 3–10. [Google Scholar] [PubMed]
- Cahalan, M.D. STIMulating store-operated Ca(2+) entry. Nat. Cell Biol. 2009, 11, 669–677. [Google Scholar] [PubMed]
- Putney, J.W., Jr. New molecular players in capacitative Ca2+ entry. J. Cell Sci. 2007, 120, 1959–1965. [Google Scholar]
- Smyth, J.T.; Hwang, S.Y.; Tomita, T.; DeHaven, W.I.; Mercer, J.C.; Putney, J.W. Activation and regulation of store-operated calcium entry. J. Cell Mol. Med. 2010, 14, 2337–2349. [Google Scholar]
- Gulisano, W.; Maugeri, D.; Baltrons, M.A.; Fa, M.; Amato, A.; Palmeri, A.; D’Adamio, L.; Grassi, C.; Devanand, D.P.; Honig, L.S.; et al. Role of Amyloid-beta and Tau Proteins in Alzheimer’s Disease: Confuting the Amyloid Cascade. J. Alzheimer’s Dis 2018, 64, S611–S631. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Jones, D.T.; Graff-Radford, J.; Lowe, V.J.; Wiste, H.J.; Gunter, J.L.; Senjem, M.L.; Botha, H.; Kantarci, K.; Boeve, B.F.; Knopman, D.S.; et al. Tau, amyloid, and cascading network failure across the Alzheimer’s disease spectrum. Cortex 2017, 97, 143–159. [Google Scholar] [CrossRef]
- Checler, F. Processing of the beta-amyloid precursor protein and its regulation in Alzheimer’s disease. J. Neurochem. 1995, 65, 1431–1444. [Google Scholar]
- Zhang, Y.W.; Thompson, R.; Zhang, H.; Xu, H. APP processing in Alzheimer’s disease. Mol. Brain 2011, 4, 3. [Google Scholar] [CrossRef]
- Edbauer, D.; Winkler, E.; Regula, J.T.; Pesold, B.; Steiner, H.; Haass, C. Reconstitution of gamma-secretase activity. Nat. Cell Biol. 2003, 5, 486–488. [Google Scholar] [CrossRef] [PubMed]
- Kimberly, W.T.; LaVoie, M.J.; Ostaszewski, B.L.; Ye, W.; Wolfe, M.S.; Selkoe, D.J. Gamma-secretase is a membrane protein complex comprised of presenilin, nicastrin, Aph-1, and Pen-2. Proc. Natl. Acad. Sci. USA 2003, 100, 6382–6387. [Google Scholar] [CrossRef] [PubMed]
- Tanzi, R.E.; Bertram, L. Twenty years of the Alzheimer’s disease amyloid hypothesis: A genetic perspective. Cell 2005, 120, 545–555. [Google Scholar] [PubMed]
- Van Cauwenberghe, C.; Van Broeckhoven, C.; Sleegers, K. The genetic landscape of Alzheimer disease: Clinical implications and perspectives. Genet. Med. 2015, 18, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Golde, T.E.; Cai, X.D.; Shoji, M.; Younkin, S.G. Production of amyloid beta protein from normal amyloid beta-protein precursor (beta APP) and the mutated beta APPS linked to familial Alzheimer’s disease. Ann. N.Y. Acad. Sci. 1993, 695, 103–108. [Google Scholar] [PubMed]
- Wolfe, M.S. When loss is gain: Reduced presenilin proteolytic function leads to increased Abeta42/Abeta40. Talking Point on the role of presenilin mutations in Alzheimer disease. EMBO Rep. 2007, 8, 136–140. [Google Scholar]
- Billings, L.M.; Oddo, S.; Green, K.N.; McGaugh, J.L.; LaFerla, F.M. Intraneuronal Abeta causes the onset of early Alzheimer’s disease-related cognitive deficits in transgenic mice. Neuron 2005, 45, 675–688. [Google Scholar]
- Del Prete, D.; Suski, J.M.; Oules, B.; Debayle, D.; Gay, A.S.; Lacas-Gervais, S.; Bussiere, R.; Bauer, C.; Pinton, P.; Paterlini-Brechot, P.; et al. Localization and Processing of the Amyloid-beta Protein Precursor in Mitochondria-Associated Membranes. J. Alzheimer’s Dis. 2017, 55, 1549–1570. [Google Scholar] [CrossRef]
- Pardossi-Piquard, R.; Petit, A.; Kawarai, T.; Sunyach, C.; Alves da Costa, C.; Vincent, B.; Ring, S.; D’Adamio, L.; Shen, J.; Muller, U.; et al. Presenilin-dependent transcriptional control of the Abeta-degrading enzyme neprilysin by intracellular domains of betaAPP and APLP. Neuron 2005, 46, 541–554. [Google Scholar] [CrossRef]
- Nhan, H.S.; Chiang, K.; Koo, E.H. The multifaceted nature of amyloid precursor protein and its proteolytic fragments: Friends and foes. Acta Neuropathol. 2015, 129, 1–19. [Google Scholar] [CrossRef]
- Baranger, K.; Marchalant, Y.; Bonnet, A.E.; Crouzin, N.; Carrete, A.; Paumier, J.M.; Py, N.A.; Bernard, A.; Bauer, C.; Charrat, E.; et al. MT5-MMP is a new pro-amyloidogenic proteinase that promotes amyloid pathology and cognitive decline in a transgenic mouse model of Alzheimer’s disease. Cell Mol. Life Sci. 2016, 73, 217–236. [Google Scholar] [CrossRef] [PubMed]
- Willem, M.; Tahirovic, S.; Busche, M.A.; Ovsepian, S.V.; Chafai, M.; Kootar, S.; Hornburg, D.; Evans, L.D.; Moore, S.; Daria, A.; et al. eta-Secretase processing of APP inhibits neuronal activity in the hippocampus. Nature 2015, 526, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Lauritzen, I.; Pardossi-Piquard, R.; Bauer, C.; Brigham, E.; Abraham, J.D.; Ranaldi, S.; Fraser, P.; St-George-Hyslop, P.; Le Thuc, O.; Espin, V.; et al. The beta-Secretase-Derived C-Terminal Fragment of betaAPP, C99, But Not Abeta, Is a Key Contributor to Early Intraneuronal Lesions in Triple-Transgenic Mouse Hippocampus. J. Neurosci. 2012, 32, 16243–16255. [Google Scholar] [CrossRef] [PubMed]
- Lauritzen, I.; Pardossi-Piquard, R.; Bourgeois, A.; Pagnotta, S.; Biferi, M.G.; Barkats, M.; Lacor, P.; Klein, W.; Bauer, C.; Checler, F. Intraneuronal aggregation of the beta-CTF fragment of APP (C99) induces Abeta-independent lysosomal-autophagic pathology. Acta Neuropathol. 2016, 132, 257–276. [Google Scholar] [CrossRef]
- Vergara, C.; Houben, S.; Suain, V.; Yilmaz, Z.; De Decker, R.; Vanden Dries, V.; Boom, A.; Mansour, S.; Leroy, K.; Ando, K.; et al. Amyloid-beta pathology enhances pathological fibrillary tau seeding induced by Alzheimer PHF in vivo. Acta Neuropathol. 2019, 137, 397–412. [Google Scholar] [CrossRef]
- He, Z.; Guo, J.L.; McBride, J.D.; Narasimhan, S.; Kim, H.; Changolkar, L.; Zhang, B.; Gathagan, R.J.; Yue, C.; Dengler, C.; et al. Amyloid-beta plaques enhance Alzheimer’s brain tau-seeded pathologies by facilitating neuritic plaque tau aggregation. Nat. Med. 2018, 24, 29–38. [Google Scholar] [CrossRef]
- Holbro, N.; Grunditz, A.; Oertner, T.G. Differential distribution of endoplasmic reticulum controls metabotropic signaling and plasticity at hippocampal synapses. Proc. Natl. Acad. Sci. USA 2009, 106, 15055–15060. [Google Scholar] [CrossRef]
- Dailey, M.E.; Bridgman, P.C. Dynamics of the endoplasmic reticulum and other membranous organelles in growth cones of cultured neurons. J. Neurosci. 1989, 9, 1897–1909. [Google Scholar] [CrossRef]
- Emptage, N.J.; Reid, C.A.; Fine, A. Calcium stores in hippocampal synaptic boutons mediate short-term plasticity, store-operated Ca2+ entry, and spontaneous transmitter release. Neuron 2001, 29, 197–208. [Google Scholar] [CrossRef]
- Berridge, M.J.; Bootman, M.D.; Lipp, P. Calcium--a life and death signal. Nature 1998, 395, 645–648. [Google Scholar] [CrossRef]
- Li, W.; Llopis, J.; Whitney, M.; Zlokarnik, G.; Tsien, R.Y. Cell-permeant caged InsP3 ester shows that Ca2+ spike frequency can optimize gene expression. Nature 1998, 392, 936–941. [Google Scholar] [CrossRef] [PubMed]
- Bandtlow, C.E.; Schmidt, M.F.; Hassinger, T.D.; Schwab, M.E.; Kater, S.B. Role of intracellular calcium in NI-35-evoked collapse of neuronal growth cones. Science 1993, 259, 80–83. [Google Scholar] [CrossRef] [PubMed]
- Popugaeva, E.; Bezprozvanny, I. Can the calcium hypothesis explain synaptic loss in Alzheimer’s disease? Neurodegener. Dis. 2014, 13, 139–141. [Google Scholar] [CrossRef] [PubMed]
- Bojarski, L.; Herms, J.; Kuznicki, J. Calcium dysregulation in Alzheimer’s disease. Neurochem. Int. 2008, 52, 621–633. [Google Scholar] [CrossRef] [PubMed]
- Tong, B.C.; Wu, A.J.; Li, M.; Cheung, K.H. Calcium signaling in Alzheimer’s disease & therapies. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1745–1760. [Google Scholar] [CrossRef]
- Del Prete, D.; Checler, F.; Chami, M. Ryanodine receptors: Physiological function and deregulation in Alzheimer disease. Mol. Neurodegener. 2014, 9, 21. [Google Scholar] [CrossRef]
- Cao, L.L.; Guan, P.P.; Liang, Y.Y.; Huang, X.S.; Wang, P. Calcium Ions Stimulate the Hyperphosphorylation of Tau by Activating Microsomal Prostaglandin E Synthase 1. Front. Aging Neurosci. 2019, 11, 108. [Google Scholar] [CrossRef]
- Hayley, M.; Perspicace, S.; Schulthess, T.; Seelig, J. Calcium enhances the proteolytic activity of BACE1: An in vitro biophysical and biochemical characterization of the BACE1-calcium interaction. Biochim. Biophys. Acta 2009, 1788, 1933–1938. [Google Scholar] [CrossRef]
- Ho, M.; Hoke, D.E.; Chua, Y.J.; Li, Q.X.; Culvenor, J.G.; Masters, C.; White, A.R.; Evin, G. Effect of Metal Chelators on gamma-Secretase Indicates That Calcium and Magnesium Ions Facilitate Cleavage of Alzheimer Amyloid Precursor Substrate. Int. J. Alzheimer’s Dis. 2010, 2011, 950932. [Google Scholar] [CrossRef]
- Miller, S.G.; Kennedy, M.B. Regulation of brain type II Ca2+/calmodulin-dependent protein kinase by autophosphorylation: A Ca2+-triggered molecular switch. Cell 1986, 44, 861–870. [Google Scholar] [CrossRef]
- Oka, M.; Fujisaki, N.; Maruko-Otake, A.; Ohtake, Y.; Shimizu, S.; Saito, T.; Hisanaga, S.I.; Iijima, K.M.; Ando, K. Ca2+/calmodulin-dependent protein kinase II promotes neurodegeneration caused by tau phosphorylated at Ser262/356 in a transgenic Drosophila model of tauopathy. J. Biochem. 2017, 162, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Litersky, J.M.; Johnson, G.V.; Jakes, R.; Goedert, M.; Lee, M.; Seubert, P. Tau protein is phosphorylated by cyclic AMP-dependent protein kinase and calcium/calmodulin-dependent protein kinase II within its microtubule-binding domains at Ser-262 and Ser-356. Biochem. J. 1996, 316 (Pt 2), 655–660. [Google Scholar] [CrossRef]
- Chakroborty, S.; Stutzmann, G.E. Early calcium dysregulation in Alzheimer’s disease: Setting the stage for synaptic dysfunction. Sci. China Life Sci. 2011, 54, 752–762. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P. ER calcium and Alzheimer’s disease: In a state of flux. Sci. Signal. 2010, 3, pe10. [Google Scholar] [CrossRef] [PubMed]
- LaFerla, F.M. Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease. Nat. Rev. Neurosci. 2002, 3, 862–872. [Google Scholar] [CrossRef] [PubMed]
- Supnet, C.; Bezprozvanny, I. The dysregulation of intracellular calcium in Alzheimer disease. Cell Calcium 2010, 47, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Bezprozvanny, I.; Mattson, M.P. Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci. 2008, 31, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J. Calcium signalling and Alzheimer’s disease. Neurochem. Res. 2011, 36, 1149–1156. [Google Scholar] [CrossRef]
- Chakroborty, S.; Stutzmann, G.E. Calcium channelopathies and Alzheimer’s disease: Insight into therapeutic success and failures. Eur. J. Pharmacol. 2014, 739, 83–95. [Google Scholar] [CrossRef]
- Arispe, N.; Rojas, E.; Pollard, H.B. Alzheimer disease amyloid beta protein forms calcium channels in bilayer membranes: Blockade by tromethamine and aluminum. Proc. Natl. Acad. Sci. USA 1993, 90, 567–571. [Google Scholar] [CrossRef]
- Alberdi, E.; Sanchez-Gomez, M.V.; Cavaliere, F.; Perez-Samartin, A.; Zugaza, J.L.; Trullas, R.; Domercq, M.; Matute, C. Amyloid beta oligomers induce Ca2+ dysregulation and neuronal death through activation of ionotropic glutamate receptors. Cell Calcium 2010, 47, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Texido, L.; Martin-Satue, M.; Alberdi, E.; Solsona, C.; Matute, C. Amyloid beta peptide oligomers directly activate NMDA receptors. Cell Calcium 2011, 49, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Thibault, O.; Pancani, T.; Landfield, P.W.; Norris, C.M. Reduction in neuronal L-type calcium channel activity in a double knock-in mouse model of Alzheimer’s disease. Biochim. Biophys. Acta 2012, 1822, 546–549. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Siebert, A.P.; Cheung, K.H.; Lee, R.J.; Johnson, B.; Cohen, A.S.; Vingtdeux, V.; Marambaud, P.; Foskett, J.K. Calcium homeostasis modulator 1 (CALHM1) is the pore-forming subunit of an ion channel that mediates extracellular Ca2+ regulation of neuronal excitability. Proc. Natl. Acad. Sci. USA 2012, 109, E1963–E1971. [Google Scholar] [CrossRef]
- Zeiger, W.; Vetrivel, K.S.; Buggia-Prevot, V.; Nguyen, P.D.; Wagner, S.L.; Villereal, M.L.; Thinakaran, G. Ca2+ influx through store-operated Ca2+ channels reduces Alzheimer disease beta-amyloid peptide secretion. J. Biol. Chem. 2013, 288, 26955–26966. [Google Scholar] [CrossRef]
- Yoo, A.S.; Cheng, I.; Chung, S.; Grenfell, T.Z.; Lee, H.; Pack-Chung, E.; Handler, M.; Shen, J.; Xia, W.; Tesco, G.; et al. Presenilin-mediated modulation of capacitative calcium entry. Neuron 2000, 27, 561–572. [Google Scholar] [CrossRef]
- Chernyuk, D.; Zernov, N.; Kabirova, M.; Bezprozvanny, I.; Popugaeva, E. Antagonist of neuronal store-operated calcium entry exerts beneficial effects in neurons expressing PSEN1DeltaE9 mutant linked to familial Alzheimer disease. Neuroscience 2019, 410, 118–127. [Google Scholar] [CrossRef]
- Zhang, H.; Sun, S.; Wu, L.; Pchitskaya, E.; Zakharova, O.; Fon Tacer, K.; Bezprozvanny, I. Store-Operated Calcium Channel Complex in Postsynaptic Spines: A New Therapeutic Target for Alzheimer’s Disease Treatment. J. Neurosci. 2016, 36, 11837–11850. [Google Scholar] [CrossRef]
- Zhang, H.; Wu, L.; Pchitskaya, E.; Zakharova, O.; Saito, T.; Saido, T.; Bezprozvanny, I. Neuronal Store-Operated Calcium Entry and Mushroom Spine Loss in Amyloid Precursor Protein Knock-In Mouse Model of Alzheimer’s Disease. J. Neurosci. 2015, 35, 13275–13286. [Google Scholar] [CrossRef]
- Popugaeva, E.; Pchitskaya, E.; Speshilova, A.; Alexandrov, S.; Zhang, H.; Vlasova, O.; Bezprozvanny, I. STIM2 protects hippocampal mushroom spines from amyloid synaptotoxicity. Mol. Neurodegener. 2015, 10, 37. [Google Scholar] [CrossRef]
- Tang, J.; Oliveros, A.; Jang, M.H. Dysfunctional Mitochondrial Bioenergetics and Synaptic Degeneration in Alzheimer Disease. Int. Neurourol. J. 2019, 23, S5–S10. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Escudero, V.; Martin-Maestro, P.; Perry, G.; Avila, J. Deconstructing mitochondrial dysfunction in Alzheimer disease. Oxid. Med. Cell Longev. 2013, 2013, 162152. [Google Scholar] [CrossRef] [PubMed]
- Esteras, N.; Abramov, A.Y. Mitochondrial Calcium Deregulation in the Mechanism of Beta-Amyloid and Tau Pathology. Cells 2020, 9, 2135. [Google Scholar] [CrossRef] [PubMed]
- Perez, M.J.; Ponce, D.P.; Aranguiz, A.; Behrens, M.I.; Quintanilla, R.A. Mitochondrial permeability transition pore contributes to mitochondrial dysfunction in fibroblasts of patients with sporadic Alzheimer’s disease. Redox Biol. 2018, 19, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Jadiya, P.; Kolmetzky, D.W.; Tomar, D.; Di Meco, A.; Lombardi, A.A.; Lambert, J.P.; Luongo, T.S.; Ludtmann, M.H.; Pratico, D.; Elrod, J.W. Impaired mitochondrial calcium efflux contributes to disease progression in models of Alzheimer’s disease. Nat. Commun. 2019, 10, 3885. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, D.M.; Lee, J.H.; Kumar, A.; Lee, S.; Orenstein, S.J.; Nixon, R.A. Autophagy failure in Alzheimer’s disease and the role of defective lysosomal acidification. Eur. J. Neurosci. 2013, 37, 1949–1961. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Yu, W.H.; Kumar, A.; Lee, S.; Mohan, P.S.; Peterhoff, C.M.; Wolfe, D.M.; Martinez-Vicente, M.; Massey, A.C.; Sovak, G.; et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 2010, 141, 1146–1158. [Google Scholar] [CrossRef]
- Lee, J.H.; McBrayer, M.K.; Wolfe, D.M.; Haslett, L.J.; Kumar, A.; Sato, Y.; Lie, P.P.; Mohan, P.; Coffey, E.E.; Kompella, U.; et al. Presenilin 1 Maintains Lysosomal Ca(2+) Homeostasis via TRPML1 by Regulating vATPase-Mediated Lysosome Acidification. Cell Rep. 2015, 12, 1430–1444. [Google Scholar] [CrossRef]
- Tu, H.; Nelson, O.; Bezprozvanny, A.; Wang, Z.; Lee, S.F.; Hao, Y.H.; Serneels, L.; De Strooper, B.; Yu, G.; Bezprozvanny, I. Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer’s disease-linked mutations. Cell 2006, 126, 981–993. [Google Scholar] [CrossRef]
- Nelson, O.; Tu, H.; Lei, T.; Bentahir, M.; de Strooper, B.; Bezprozvanny, I. Familial Alzheimer disease-linked mutations specifically disrupt Ca2+ leak function of presenilin 1. J. Clin. Invest. 2007, 117, 1230–1239. [Google Scholar] [CrossRef]
- Brunello, L.; Zampese, E.; Florean, C.; Pozzan, T.; Pizzo, P.; Fasolato, C. Presenilin-2 dampens intracellular Ca2+ stores by increasing Ca2+ leakage and reducing Ca2+ uptake. J. Cell Mol. Med. 2009, 13, 3358–3369. [Google Scholar] [CrossRef] [PubMed]
- Shilling, D.; Muller, M.; Takano, H.; Mak, D.O.; Abel, T.; Coulter, D.A.; Foskett, J.K. Suppression of InsP3 receptor-mediated Ca2+ signaling alleviates mutant presenilin-linked familial Alzheimer’s disease pathogenesis. J. Neurosci. 2014, 34, 6910–6923. [Google Scholar] [CrossRef] [PubMed]
- Shilling, D.; Mak, D.O.; Kang, D.E.; Foskett, J.K. Lack of evidence for presenilins as endoplasmic reticulum Ca2+ leak channels. J. Biol. Chem. 2012, 287, 10933–10944. [Google Scholar] [CrossRef] [PubMed]
- Green, K.N.; Demuro, A.; Akbari, Y.; Hitt, B.D.; Smith, I.F.; Parker, I.; LaFerla, F.M. SERCA pump activity is physiologically regulated by presenilin and regulates amyloid beta production. J. Cell Biol. 2008, 181, 1107–1116. [Google Scholar] [CrossRef]
- Bussiere, R.; Oules, B.; Mary, A.; Vaillant-Beuchot, L.; Martin, C.; El Manaa, W.; Vallee, D.; Duplan, E.; Paterlini-Brechot, P.; Alves Da Costa, C.; et al. Upregulation of the Sarco-Endoplasmic Reticulum Calcium ATPase 1 Truncated Isoform Plays a Pathogenic Role in Alzheimer’s Disease. Cells 2019, 8, 1539. [Google Scholar] [CrossRef]
- Nelson, O.; Supnet, C.; Liu, H.; Bezprozvanny, I. Familial Alzheimer’s disease mutations in presenilins: Effects on endoplasmic reticulum calcium homeostasis and correlation with clinical phenotypes. J. Alzheimer’s Dis. 2010, 21, 781–793. [Google Scholar] [CrossRef]
- Nelson, O.; Supnet, C.; Tolia, A.; Horre, K.; De Strooper, B.; Bezprozvanny, I. Mutagenesis mapping of the presenilin 1 calcium leak conductance pore. J. Biol. Chem. 2011, 286, 22339–22347. [Google Scholar] [CrossRef]
- Zampese, E.; Fasolato, C.; Kipanyula, M.J.; Bortolozzi, M.; Pozzan, T.; Pizzo, P. Presenilin 2 modulates endoplasmic reticulum (ER)-mitochondria interactions and Ca2+ cross-talk. Proc. Natl. Acad. Sci. USA 2011, 108, 2777–2782. [Google Scholar] [CrossRef]
- Stutzmann, G.E.; Caccamo, A.; LaFerla, F.M.; Parker, I. Dysregulated IP3 signaling in cortical neurons of knock-in mice expressing an Alzheimer’s-linked mutation in presenilin1 results in exaggerated Ca2+ signals and altered membrane excitability. J. Neurosci. 2004, 24, 508–513. [Google Scholar] [CrossRef]
- Stutzmann, G.E.; Smith, I.; Caccamo, A.; Oddo, S.; Parker, I.; Laferla, F. Enhanced ryanodine-mediated calcium release in mutant PS1-expressing Alzheimer’s mouse models. Ann. N.Y. Acad. Sci. 2007, 1097, 265–277. [Google Scholar] [CrossRef]
- Cheung, K.H.; Shineman, D.; Muller, M.; Cardenas, C.; Mei, L.; Yang, J.; Tomita, T.; Iwatsubo, T.; Lee, V.M.; Foskett, J.K. Mechanism of Ca2+ disruption in Alzheimer’s disease by presenilin regulation of InsP(3) receptor channel gating. Neuron 2008, 58, 871–883. [Google Scholar] [CrossRef] [PubMed]
- Querfurth, H.W.; Jiang, J.; Geiger, J.D.; Selkoe, D.J. Caffeine stimulates amyloid beta-peptide release from beta-amyloid precursor protein-transfected HEK293 cells. J. Neurochem. 1997, 69, 1580–1591. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.L.; Mayne, M.; Holden, C.P.; Geiger, J.D.; Mattson, M.P. Presenilin-1 mutations increase levels of ryanodine receptors and calcium release in PC12 cells and cortical neurons. J. Biol. Chem. 2000, 275, 18195–18200. [Google Scholar] [CrossRef] [PubMed]
- Smith, I.F.; Hitt, B.; Green, K.N.; Oddo, S.; LaFerla, F.M. Enhanced caffeine-induced Ca2+ release in the 3xTg-AD mouse model of Alzheimer’s disease. J. Neurochem. 2005, 94, 1711–1718. [Google Scholar] [CrossRef]
- Stutzmann, G.E.; Smith, I.; Caccamo, A.; Oddo, S.; Laferla, F.M.; Parker, I. Enhanced ryanodine receptor recruitment contributes to Ca2+ disruptions in young, adult, and aged Alzheimer’s disease mice. J. Neurosci. 2006, 26, 5180–5189. [Google Scholar] [CrossRef]
- Supnet, C.; Grant, J.; Kong, H.; Westaway, D.; Mayne, M. Amyloid-beta-(1-42) increases ryanodine receptor-3 expression and function in neurons of TgCRND8 mice. J. Biol. Chem. 2006, 281, 38440–38447. [Google Scholar] [CrossRef]
- Chakroborty, S.; Goussakov, I.; Miller, M.B.; Stutzmann, G.E. Deviant ryanodine receptor-mediated calcium release resets synaptic homeostasis in presymptomatic 3xTg-AD mice. J. Neurosci. 2009, 29, 9458–9470. [Google Scholar] [CrossRef]
- Paula-Lima, A.C.; Adasme, T.; SanMartin, C.; Sebollela, A.; Hetz, C.; Carrasco, M.A.; Ferreira, S.T.; Hidalgo, C. Amyloid beta-peptide oligomers stimulate RyR-mediated Ca2+ release inducing mitochondrial fragmentation in hippocampal neurons and prevent RyR-mediated dendritic spine remodeling produced by BDNF. Antioxid. Redox Signal. 2011, 14, 1209–1223. [Google Scholar] [CrossRef]
- Ito, E.; Oka, K.; Etcheberrigaray, R.; Nelson, T.J.; McPhie, D.L.; Tofel-Grehl, B.; Gibson, G.E.; Alkon, D.L. Internal Ca2+ mobilization is altered in fibroblasts from patients with Alzheimer disease. Proc. Natl. Acad. Sci. USA 1994, 91, 534–538. [Google Scholar] [CrossRef]
- Goussakov, I.; Miller, M.B.; Stutzmann, G.E. NMDA-mediated Ca(2+) influx drives aberrant ryanodine receptor activation in dendrites of young Alzheimer’s disease mice. J. Neurosci. 2010, 30, 12128–12137. [Google Scholar] [CrossRef]
- Jensen, L.E.; Bultynck, G.; Luyten, T.; Amijee, H.; Bootman, M.D.; Roderick, H.L. Alzheimer’s disease-associated peptide Abeta42 mobilizes ER Ca(2+) via InsP3R-dependent and -independent mechanisms. Front. Mol. Neurosci. 2013, 6, 36. [Google Scholar] [CrossRef] [PubMed]
- Oules, B.; Del Prete, D.; Greco, B.; Zhang, X.; Lauritzen, I.; Sevalle, J.; Moreno, S.; Paterlini-Brechot, P.; Trebak, M.; Checler, F.; et al. Ryanodine receptor blockade reduces amyloid-beta load and memory impairments in Tg2576 mouse model of Alzheimer disease. J. Neurosci. 2012, 32, 11820–11834. [Google Scholar] [CrossRef] [PubMed]
- Chakroborty, S.; Briggs, C.; Miller, M.B.; Goussakov, I.; Schneider, C.; Kim, J.; Wicks, J.; Richardson, J.C.; Conklin, V.; Cameransi, B.G.; et al. Stabilizing ER Ca2+ channel function as an early preventative strategy for Alzheimer’s disease. PLoS ONE 2012, 7, e52056. [Google Scholar] [CrossRef] [PubMed]
- Chakroborty, S.; Kim, J.; Schneider, C.; Jacobson, C.; Molgo, J.; Stutzmann, G.E. Early presynaptic and postsynaptic calcium signaling abnormalities mask underlying synaptic depression in presymptomatic Alzheimer’s disease mice. J. Neurosci. 2012, 32, 8341–8353. [Google Scholar] [CrossRef] [PubMed]
- Santos, S.F.; Pierrot, N.; Morel, N.; Gailly, P.; Sindic, C.; Octave, J.N. Expression of human amyloid precursor protein in rat cortical neurons inhibits calcium oscillations. J. Neurosci. 2009, 29, 4708–4718. [Google Scholar] [CrossRef] [PubMed]
- Gazda, K.; Kuznicki, J.; Wegierski, T. Knockdown of amyloid precursor protein increases calcium levels in the endoplasmic reticulum. Sci. Rep. 2017, 7, 14512. [Google Scholar] [CrossRef]
- Mahoney, R.; Ochoa Thomas, E.; Ramirez, P.; Miller, H.E.; Beckmann, A.; Zuniga, G.; Dobrowolski, R.; Frost, B. Pathogenic Tau Causes a Toxic Depletion of Nuclear Calcium. Cell Rep. 2020, 32, 107900. [Google Scholar] [CrossRef]
- Esteras, N.; Kundel, F.; Amodeo, G.F.; Pavlov, E.V.; Klenerman, D.; Abramov, A.Y. Insoluble tau aggregates induce neuronal death through modification of membrane ion conductance, activation of voltage-gated calcium channels and NADPH oxidase. FEBS J. 2020. [Google Scholar] [CrossRef]
- Britti, E.; Ros, J.; Esteras, N.; Abramov, A.Y. Tau inhibits mitochondrial calcium efflux and makes neurons vulnerable to calcium-induced cell death. Cell Calcium 2020, 86, 102150. [Google Scholar] [CrossRef]
- Furuichi, T.; Furutama, D.; Hakamata, Y.; Nakai, J.; Takeshima, H.; Mikoshiba, K. Multiple types of ryanodine receptor/Ca2+ release channels are differentially expressed in rabbit brain. J. Neurosci. 1994, 14, 4794–4805. [Google Scholar] [CrossRef]
- Lehnart, S.E.; Mongillo, M.; Bellinger, A.; Lindegger, N.; Chen, B.X.; Hsueh, W.; Reiken, S.; Wronska, A.; Drew, L.J.; Ward, C.W.; et al. Leaky Ca2+ release channel/ryanodine receptor 2 causes seizures and sudden cardiac death in mice. J. Clin. Investig. 2008, 118, 2230–2245. [Google Scholar] [CrossRef] [PubMed]
- Meissner, G.; Darling, E.; Eveleth, J. Kinetics of rapid Ca2+ release by sarcoplasmic reticulum. Effects of Ca2+, Mg2+, and adenine nucleotides. Biochemistry 1986, 25, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Meissner, G. The structural basis of ryanodine receptor ion channel function. J. Gen. Physiol. 2017, 149, 1065–1089. [Google Scholar] [CrossRef] [PubMed]
- Laver, D.R.; Honen, B.N. Luminal Mg2+, a key factor controlling RYR2-mediated Ca2+ release: Cytoplasmic and luminal regulation modeled in a tetrameric channel. J. Gen. Physiol. 2008, 132, 429–446. [Google Scholar] [CrossRef] [PubMed]
- Tencerova, B.; Zahradnikova, A.; Gaburjakova, J.; Gaburjakova, M. Luminal Ca2+ controls activation of the cardiac ryanodine receptor by ATP. J. Gen. Physiol. 2012, 140, 93–108. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Bai, X.; Yan, C.; Wu, J.; Li, Z.; Xie, T.; Peng, W.; Yin, C.; Li, X.; Scheres, S.H.W.; et al. Structure of the rabbit ryanodine receptor RyR1 at near-atomic resolution. Nature 2015, 517, 50–55. [Google Scholar] [CrossRef]
- Peng, W.; Shen, H.; Wu, J.; Guo, W.; Pan, X.; Wang, R.; Chen, S.R.; Yan, N. Structural basis for the gating mechanism of the type 2 ryanodine receptor RyR2. Science 2016, 354. [Google Scholar] [CrossRef]
- Denniss, A.; Dulhunty, A.F.; Beard, N.A. Ryanodine receptor Ca(2+) release channel post-translational modification: Central player in cardiac and skeletal muscle disease. Int. J. Biochem. Cell Biol. 2018, 101, 49–53. [Google Scholar] [CrossRef]
- Lanner, J.T.; Georgiou, D.K.; Joshi, A.D.; Hamilton, S.L. Ryanodine receptors: Structure, expression, molecular details, and function in calcium release. Cold Spring Harb. Perspect. Biol. 2010, 2, a003996. [Google Scholar] [CrossRef]
- Balshaw, D.M.; Yamaguchi, N.; Meissner, G. Modulation of intracellular calcium-release channels by calmodulin. J. Membr. Biol. 2002, 185, 1–8. [Google Scholar] [CrossRef]
- MacMillan, D. FK506 binding proteins: Cellular regulators of intracellular Ca2+ signalling. Eur. J. Pharmacol. 2013, 700, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Zalk, R.; Lehnart, S.E.; Marks, A.R. Modulation of the ryanodine receptor and intracellular calcium. Annu. Rev. Biochem. 2007, 76, 367–385. [Google Scholar] [CrossRef] [PubMed]
- Marx, S.O.; Reiken, S.; Hisamatsu, Y.; Jayaraman, T.; Burkhoff, D.; Rosemblit, N.; Marks, A.R. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): Defective regulation in failing hearts. Cell 2000, 101, 365–376. [Google Scholar] [CrossRef]
- Currie, S. Cardiac ryanodine receptor phosphorylation by CaM Kinase II: Keeping the balance right. Front. Biosci. 2009, 14, 5134–5156. [Google Scholar] [CrossRef][Green Version]
- Allen, P.B.; Ouimet, C.C.; Greengard, P. Spinophilin, a novel protein phosphatase 1 binding protein localized to dendritic spines. Proc. Natl. Acad. Sci. USA 1997, 94, 9956–9961. [Google Scholar] [CrossRef]
- Chami, M.; Checler, F. Targeting Post-Translational Remodeling of Ryanodine Receptor: A New Track for Alzheimer’s Disease Therapy? Curr. Alzheimer Res. 2020, 17, 313–323. [Google Scholar] [CrossRef]
- Lee, S.Y.; Hwang, D.Y.; Kim, Y.K.; Lee, J.W.; Shin, I.C.; Oh, K.W.; Lee, M.K.; Lim, J.S.; Yoon, D.Y.; Hwang, S.J.; et al. PS2 mutation increases neuronal cell vulnerability to neurotoxicants through activation of caspase-3 by enhancing of ryanodine receptor-mediated calcium release. FASEB J. 2006, 20, 151–153. [Google Scholar]
- Guo, Q.; Sopher, B.L.; Furukawa, K.; Pham, D.G.; Robinson, N.; Martin, G.M.; Mattson, M.P. Alzheimer’s presenilin mutation sensitizes neural cells to apoptosis induced by trophic factor withdrawal and amyloid beta-peptide: Involvement of calcium and oxyradicals. J. Neurosci. 1997, 17, 4212–4222. [Google Scholar] [CrossRef]
- Leissring, M.A.; Murphy, M.P.; Mead, T.R.; Akbari, Y.; Sugarman, M.C.; Jannatipour, M.; Anliker, B.; Muller, U.; Saftig, P.; De Strooper, B.; et al. A physiologic signaling role for the gamma -secretase-derived intracellular fragment of APP. Proc. Natl. Acad. Sci. USA 2002, 99, 4697–4702. [Google Scholar] [CrossRef]
- Lopez, J.R.; Lyckman, A.; Oddo, S.; Laferla, F.M.; Querfurth, H.W.; Shtifman, A. Increased intraneuronal resting [Ca(2+)] in adult Alzheimer’s disease mice. J. Neurochem. 2008, 105, 262–271. [Google Scholar] [CrossRef]
- Rojas, G.; Cardenas, A.M.; Fernandez-Olivares, P.; Shimahara, T.; Segura-Aguilar, J.; Caviedes, R.; Caviedes, P. Effect of the knockdown of amyloid precursor protein on intracellular calcium increases in a neuronal cell line derived from the cerebral cortex of a trisomy 16 mouse. Exp. Neurol. 2008, 209, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Su, Z.; Zhao, C.; Song, B.; Zhang, X.; Zhao, N.; Shen, X.; Gong, Y. Effect of amyloid beta on capacitive calcium entry in neural 2a cells. Brain Res. Bull. 2009, 78, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Paula-Lima, A.C.; Hidalgo, C. Amyloid beta-peptide oligomers, ryanodine receptor-mediated Ca(2+) release, and Wnt-5a/Ca(2+) signaling: Opposing roles in neuronal mitochondrial dynamics? Front. Cell Neurosci. 2013, 7, 120. [Google Scholar] [CrossRef] [PubMed]
- Shtifman, A.; Ward, C.W.; Laver, D.R.; Bannister, M.L.; Lopez, J.R.; Kitazawa, M.; LaFerla, F.M.; Ikemoto, N.; Querfurth, H.W. Amyloid-beta protein impairs Ca2+ release and contractility in skeletal muscle. Neurobiol. Aging 2010, 31, 2080–2090. [Google Scholar] [CrossRef] [PubMed]
- Kelliher, M.; Fastbom, J.; Cowburn, R.F.; Bonkale, W.; Ohm, T.G.; Ravid, R.; Sorrentino, V.; O’Neill, C. Alterations in the ryanodine receptor calcium release channel correlate with Alzheimer’s disease neurofibrillary and beta-amyloid pathologies. Neuroscience 1999, 92, 499–513. [Google Scholar] [CrossRef]
- Wu, B.; Yamaguchi, H.; Lai, F.A.; Shen, J. Presenilins regulate calcium homeostasis and presynaptic function via ryanodine receptors in hippocampal neurons. Proc. Natl. Acad. Sci. USA 2013, 110, 15091–15096. [Google Scholar] [CrossRef]
- Kipanyula, M.J.; Contreras, L.; Zampese, E.; Lazzari, C.; Wong, A.K.; Pizzo, P.; Fasolato, C.; Pozzan, T. Ca2+ dysregulation in neurons from transgenic mice expressing mutant presenilin 2. Aging Cell 2012, 11, 885–893. [Google Scholar] [CrossRef]
- Cisse, M.; Duplan, E.; Lorivel, T.; Dunys, J.; Bauer, C.; Meckler, X.; Gerakis, Y.; Lauritzen, I.; Checler, F. The transcription factor XBP1s restores hippocampal synaptic plasticity and memory by control of the Kalirin-7 pathway in Alzheimer model. Mol. Psychiatry 2017, 22, 1562–1575. [Google Scholar] [CrossRef]
- Gerakis, Y.; Dunys, J.; Bauer, C.; Checler, F. Abeta42 oligomers modulate beta-secretase through an XBP-1s-dependent pathway involving HRD1. Sci. Rep. 2016, 6, 37436. [Google Scholar] [CrossRef]
- Casas-Tinto, S.; Zhang, Y.; Sanchez-Garcia, J.; Gomez-Velazquez, M.; Rincon-Limas, D.E.; Fernandez-Funez, P. The ER stress factor XBP1s prevents amyloid-beta neurotoxicity. Hum. Mol. Genet. 2011, 20, 2144–2160. [Google Scholar] [CrossRef]
- Liu, J.; Supnet, C.; Sun, S.; Zhang, H.; Good, L.; Popugaeva, E.; Bezprozvanny, I. The role of ryanodine receptor type 3 in a mouse model of Alzheimer disease. Channels 2014, 8, 230–242. [Google Scholar] [CrossRef] [PubMed]
- Mickelson, J.R.; Gallant, E.M.; Litterer, L.A.; Johnson, K.M.; Rempel, W.E.; Louis, C.F. Abnormal sarcoplasmic reticulum ryanodine receptor in malignant hyperthermia. J. Biol. Chem. 1988, 263, 9310–9315. [Google Scholar] [PubMed]
- Kushnir, A.; Wajsberg, B.; Marks, A.R. Ryanodine receptor dysfunction in human disorders. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1687–1697. [Google Scholar] [CrossRef] [PubMed]
- Kushnir, A.; Betzenhauser, M.J.; Marks, A.R. Ryanodine receptor studies using genetically engineered mice. FEBS Lett. 2010, 584, 1956–1965. [Google Scholar] [CrossRef]
- Takeshima, H.; Iino, M.; Takekura, H.; Nishi, M.; Kuno, J.; Minowa, O.; Takano, H.; Noda, T. Excitation-contraction uncoupling and muscular degeneration in mice lacking functional skeletal muscle ryanodine-receptor gene. Nature 1994, 369, 556–559. [Google Scholar] [CrossRef]
- Takeshima, H.; Ikemoto, T.; Nishi, M.; Nishiyama, N.; Shimuta, M.; Sugitani, Y.; Kuno, J.; Saito, I.; Saito, H.; Endo, M.; et al. Generation and characterization of mutant mice lacking ryanodine receptor type 3. J. Biol. Chem. 1996, 271, 19649–19652. [Google Scholar] [CrossRef]
- Matsuo, N.; Tanda, K.; Nakanishi, K.; Yamasaki, N.; Toyama, K.; Takao, K.; Takeshima, H.; Miyakawa, T. Comprehensive behavioral phenotyping of ryanodine receptor type 3 (RyR3) knockout mice: Decreased social contact duration in two social interaction tests. Front. Behav. Neurosci. 2009, 3, 3. [Google Scholar] [CrossRef]
- Balschun, D.; Wolfer, D.P.; Bertocchini, F.; Barone, V.; Conti, A.; Zuschratter, W.; Missiaen, L.; Lipp, H.P.; Frey, J.U.; Sorrentino, V. Deletion of the ryanodine receptor type 3 (RyR3) impairs forms of synaptic plasticity and spatial learning. EMBO J. 1999, 18, 5264–5273. [Google Scholar] [CrossRef]
- Futatsugi, A.; Kato, K.; Ogura, H.; Li, S.T.; Nagata, E.; Kuwajima, G.; Tanaka, K.; Itohara, S.; Mikoshiba, K. Facilitation of NMDAR-independent LTP and spatial learning in mutant mice lacking ryanodine receptor type 3. Neuron 1999, 24, 701–713. [Google Scholar] [CrossRef]
- Shimuta, M.; Yoshikawa, M.; Fukaya, M.; Watanabe, M.; Takeshima, H.; Manabe, T. Postsynaptic modulation of AMPA receptor-mediated synaptic responses and LTP by the type 3 ryanodine receptor. Mol. Cell Neurosci. 2001, 17, 921–930. [Google Scholar] [CrossRef]
- Gong, S.; Su, B.B.; Tovar, H.; Mao, C.; Gonzalez, V.; Liu, Y.; Lu, Y.; Wang, K.S.; Xu, C. Polymorphisms Within RYR3 Gene Are Associated With Risk and Age at Onset of Hypertension, Diabetes, and Alzheimer’s Disease. Am. J. Hypertens. 2018, 31, 818–826. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Song, F.; Wang, J.; Han, G.; Bai, Z.; Xie, B.; Feng, X.; Jia, J.; Duan, Y.; Lei, H. Hidden risk genes with high-order intragenic epistasis in Alzheimer’s disease. J. Alzheimer’s Dis. 2014, 41, 1039–1056. [Google Scholar] [CrossRef] [PubMed]
- Koran, M.E.; Hohman, T.J.; Thornton-Wells, T.A. Genetic interactions found between calcium channel genes modulate amyloid load measured by positron emission tomography. Hum. Genet. 2014, 133, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Wehrens, X.H.; Lehnart, S.E.; Reiken, S.R.; Deng, S.X.; Vest, J.A.; Cervantes, D.; Coromilas, J.; Landry, D.W.; Marks, A.R. Protection from cardiac arrhythmia through ryanodine receptor-stabilizing protein calstabin2. Science 2004, 304, 292–296. [Google Scholar] [CrossRef]
- Andersson, D.C.; Betzenhauser, M.J.; Reiken, S.; Meli, A.C.; Umanskaya, A.; Xie, W.; Shiomi, T.; Zalk, R.; Lacampagne, A.; Marks, A.R. Ryanodine receptor oxidation causes intracellular calcium leak and muscle weakness in aging. Cell Metab. 2011, 14, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Bellinger, A.M.; Reiken, S.; Carlson, C.; Mongillo, M.; Liu, X.; Rothman, L.; Matecki, S.; Lacampagne, A.; Marks, A.R. Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat. Med. 2009, 15, 325–330. [Google Scholar] [CrossRef]
- Bellinger, A.M.; Reiken, S.; Dura, M.; Murphy, P.W.; Deng, S.X.; Landry, D.W.; Nieman, D.; Lehnart, S.E.; Samaru, M.; LaCampagne, A.; et al. Remodeling of ryanodine receptor complex causes “leaky” channels: A molecular mechanism for decreased exercise capacity. Proc. Natl. Acad. Sci. USA 2008, 105, 2198–2202. [Google Scholar] [CrossRef]
- Fauconnier, J.; Meli, A.C.; Thireau, J.; Roberge, S.; Shan, J.; Sassi, Y.; Reiken, S.R.; Rauzier, J.M.; Marchand, A.; Chauvier, D.; et al. Ryanodine receptor leak mediated by caspase-8 activation leads to left ventricular injury after myocardial ischemia-reperfusion. Proc. Natl. Acad. Sci. USA 2011, 108, 13258–13263. [Google Scholar] [CrossRef]
- Liu, X.; Betzenhauser, M.J.; Reiken, S.; Meli, A.C.; Xie, W.; Chen, B.X.; Arancio, O.; Marks, A.R. Role of leaky neuronal ryanodine receptors in stress-induced cognitive dysfunction. Cell 2012, 150, 1055–1067. [Google Scholar] [CrossRef]
- Lacampagne, A.; Liu, X.; Reiken, S.; Bussiere, R.; Meli, A.C.; Lauritzen, I.; Teich, A.F.; Zalk, R.; Saint, N.; Arancio, O.; et al. Post-translational remodeling of ryanodine receptor induces calcium leak leading to Alzheimer’s disease-like pathologies and cognitive deficits. Acta Neuropathol. 2017, 134, 749–767. [Google Scholar] [CrossRef]
- Petrotchenko, E.V.; Yamaguchi, N.; Pasek, D.A.; Borchers, C.H.; Meissner, G. Mass spectrometric analysis and mutagenesis predict involvement of multiple cysteines in redox regulation of the skeletal muscle ryanodine receptor ion channel complex. Res. Rep. Biol. 2011, 2011, 13–21. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sun, J.; Yamaguchi, N.; Xu, L.; Eu, J.P.; Stamler, J.S.; Meissner, G. Regulation of the cardiac muscle ryanodine receptor by O(2) tension and S-nitrosoglutathione. Biochemistry 2008, 47, 13985–13990. [Google Scholar] [CrossRef] [PubMed]
- Bussiere, R.; Lacampagne, A.; Reiken, S.; Liu, X.; Scheuerman, V.; Zalk, R.; Martin, C.; Checler, F.; Marks, A.R.; Chami, M. Amyloid beta production is regulated by beta2-adrenergic signaling-mediated post-translational modifications of the ryanodine receptor. J. Biol. Chem. 2017, 292, 10153–10168. [Google Scholar] [CrossRef]
- Hidalgo, C.; Carrasco, M.A. Redox control of brain calcium in health and disease. Antioxid. Redox Signal. 2011, 14, 1203–1207. [Google Scholar] [CrossRef] [PubMed]
- Von Bernhardi, R.; Eugenin, J. Alzheimer’s disease: Redox dysregulation as a common denominator for diverse pathogenic mechanisms. Antioxid. Redox Signal. 2012, 16, 974–1031. [Google Scholar] [CrossRef]
- Echeverria, V.; Ducatenzeiler, A.; Chen, C.H.; Cuello, A.C. Endogenous beta-amyloid peptide synthesis modulates cAMP response element-regulated gene expression in PC12 cells. Neuroscience 2005, 135, 1193–1202. [Google Scholar] [CrossRef] [PubMed]
- Igbavboa, U.; Johnson-Anuna, L.N.; Rossello, X.; Butterick, T.A.; Sun, G.Y.; Wood, W.G. Amyloid beta-protein1-42 increases cAMP and apolipoprotein E levels which are inhibited by beta1 and beta2-adrenergic receptor antagonists in mouse primary astrocytes. Neuroscience 2006, 142, 655–660. [Google Scholar] [CrossRef] [PubMed]
- Prapong, T.; Uemura, E.; Hsu, W.H. G protein and cAMP-dependent protein kinase mediate amyloid beta-peptide inhibition of neuronal glucose uptake. Exp. Neurol. 2001, 167, 59–64. [Google Scholar] [CrossRef]
- Palavicini, J.P.; Wang, H.; Bianchi, E.; Xu, S.; Rao, J.S.; Kang, D.E.; Lakshmana, M.K. RanBP9 aggravates synaptic damage in the mouse brain and is inversely correlated to spinophilin levels in Alzheimer’s brain synaptosomes. Cell Death Dis. 2013, 4, e667. [Google Scholar] [CrossRef]
- Marambaud, P.; Ancolio, K.; Alves da Costa, C.; Checler, F. Effect of protein kinase A inhibitors on the production of Abeta40 and Abeta42 by human cells expressing normal and Alzheimer’s disease-linked mutated betaAPP and presenilin 1. Br. J. Pharmacol. 1999, 126, 1186–1190. [Google Scholar] [CrossRef]
- Bekris, L.M.; Yu, C.E.; Bird, T.D.; Tsuang, D.W. Genetics of Alzheimer disease. J. Geriatr. Psychiatry Neurol. 2010, 23, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Branca, C.; Wisely, E.V.; Hartman, L.K.; Caccamo, A.; Oddo, S. Administration of a selective beta2 adrenergic receptor antagonist exacerbates neuropathology and cognitive deficits in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2014, 35, 2726–2735. [Google Scholar] [CrossRef] [PubMed]
- Dobarro, M.; Gerenu, G.; Ramirez, M.J. Propranolol reduces cognitive deficits, amyloid and tau pathology in Alzheimer’s transgenic mice. Int. J. Neuropsychopharmacol. 2013, 16, 2245–2257. [Google Scholar] [CrossRef] [PubMed]
- Dang, V.; Medina, B.; Das, D.; Moghadam, S.; Martin, K.J.; Lin, B.; Naik, P.; Patel, D.; Nosheny, R.; Wesson Ashford, J.; et al. Formoterol, a long-acting beta2 adrenergic agonist, improves cognitive function and promotes dendritic complexity in a mouse model of Down syndrome. Biol. Psychiatry 2014, 75, 179–188. [Google Scholar] [CrossRef]
- Peng, J.; Liang, G.; Inan, S.; Wu, Z.; Joseph, D.J.; Meng, Q.; Peng, Y.; Eckenhoff, M.F.; Wei, H. Dantrolene ameliorates cognitive decline and neuropathology in Alzheimer triple transgenic mice. Neurosci. Lett. 2012, 516, 274–279. [Google Scholar] [CrossRef]
- Mikoshiba, K. IP3 receptor/Ca2+ channel: From discovery to new signaling concepts. J. Neurochem. 2007, 102, 1426–1446. [Google Scholar] [CrossRef]
- Ross, C.A.; Meldolesi, J.; Milner, T.A.; Satoh, T.; Supattapone, S.; Snyder, S.H. Inositol 1,4,5-trisphosphate receptor localized to endoplasmic reticulum in cerebellar Purkinje neurons. Nature 1989, 339, 468–470. [Google Scholar] [CrossRef]
- Furuichi, T.; Yoshikawa, S.; Miyawaki, A.; Wada, K.; Maeda, N.; Mikoshiba, K. Primary structure and functional expression of the inositol 1,4,5-trisphosphate-binding protein P400. Nature 1989, 342, 32–38. [Google Scholar] [CrossRef]
- Mignery, G.A.; Sudhof, T.C.; Takei, K.; De Camilli, P. Putative receptor for inositol 1,4,5-trisphosphate similar to ryanodine receptor. Nature 1989, 342, 192–195. [Google Scholar] [CrossRef]
- Foskett, J.K.; White, C.; Cheung, K.H.; Mak, D.O. Inositol trisphosphate receptor Ca2+ release channels. Physiol. Rev. 2007, 87, 593–658. [Google Scholar] [CrossRef]
- Inoue, T.; Kato, K.; Kohda, K.; Mikoshiba, K. Type 1 inositol 1,4,5-trisphosphate receptor is required for induction of long-term depression in cerebellar Purkinje neurons. J. Neurosci. 1998, 18, 5366–5373. [Google Scholar] [CrossRef] [PubMed]
- Baker, K.D.; Edwards, T.M.; Rickard, N.S. The role of intracellular calcium stores in synaptic plasticity and memory consolidation. Neurosci. Biobehav. Rev. 2013, 37, 1211–1239. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, T.; Hisatsune, C.; Le, T.D.; Hashikawa, T.; Hirono, M.; Hattori, M.; Nagao, S.; Mikoshiba, K. Type 1 inositol trisphosphate receptor regulates cerebellar circuits by maintaining the spine morphology of purkinje cells in adult mice. J. Neurosci. 2013, 33, 12186–12196. [Google Scholar] [CrossRef] [PubMed]
- Egorova, P.A.; Bezprozvanny, I.B. Inositol 1,4,5-trisphosphate receptors and neurodegenerative disorders. FEBS J. 2018, 285, 3547–3565. [Google Scholar] [CrossRef]
- Leissring, M.A.; Paul, B.A.; Parker, I.; Cotman, C.W.; LaFerla, F.M. Alzheimer’s presenilin-1 mutation potentiates inositol 1,4,5-trisphosphate-mediated calcium signaling in Xenopus oocytes. J. Neurochem. 1999, 72, 1061–1068. [Google Scholar] [CrossRef]
- Cedazo-Minguez, A.; Popescu, B.O.; Ankarcrona, M.; Nishimura, T.; Cowburn, R.F. The presenilin 1 deltaE9 mutation gives enhanced basal phospholipase C activity and a resultant increase in intracellular calcium concentrations. J. Biol. Chem. 2002, 277, 36646–36655. [Google Scholar] [CrossRef]
- Bezprozvanny, I.; Supnet, C.; Sun, S.; Zhang, H.; De Strooper, B. Response to Shilling et al. (10.1074/jbc.M111.300491). J. Biol. Chem. 2012, 287, 20469. [Google Scholar] [CrossRef]
- Mak, D.O.; Cheung, K.H.; Toglia, P.; Foskett, J.K.; Ullah, G. Analyzing and Quantifying the Gain-of-Function Enhancement of IP3 Receptor Gating by Familial Alzheimer’s Disease-Causing Mutants in Presenilins. PLoS Comput. Biol. 2015, 11, e1004529. [Google Scholar] [CrossRef]
- Toglia, P.; Ullah, G. The gain-of-function enhancement of IP3-receptor channel gating by familial Alzheimer’s disease-linked presenilin mutants increases the open probability of mitochondrial permeability transition pore. Cell Calcium 2016, 60, 13–24. [Google Scholar] [CrossRef]
- Wang, Z.J.; Zhao, F.; Wang, C.F.; Zhang, X.M.; Xiao, Y.; Zhou, F.; Wu, M.N.; Zhang, J.; Qi, J.S.; Yang, W. Xestospongin C, a Reversible IP3 Receptor Antagonist, Alleviates the Cognitive and Pathological Impairments in APP/PS1 Mice of Alzheimer’s Disease. J. Alzheimer’s Dis. 2019, 72, 1217–1231. [Google Scholar] [CrossRef]
- Ronco, V.; Grolla, A.A.; Glasnov, T.N.; Canonico, P.L.; Verkhratsky, A.; Genazzani, A.A.; Lim, D. Differential deregulation of astrocytic calcium signalling by amyloid-beta, TNFalpha, IL-1beta and LPS. Cell Calcium 2014, 55, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Grolla, A.A.; Sim, J.A.; Lim, D.; Rodriguez, J.J.; Genazzani, A.A.; Verkhratsky, A. Amyloid-beta and Alzheimer’s disease type pathology differentially affects the calcium signalling toolkit in astrocytes from different brain regions. Cell Death Dis. 2013, 4, e623. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B.; Beullens, M.; Contreras, B.; Levesque, L.; Craessaerts, K.; Cordell, B.; Moechars, D.; Bollen, M.; Fraser, P.; George-Hyslop, P.S.; et al. Phosphorylation, subcellular localization, and membrane orientation of the Alzheimer’s disease-associated presenilins. J. Biol. Chem. 1997, 272, 3590–3598. [Google Scholar] [CrossRef] [PubMed]
- Sherrington, R.; Rogaev, E.I.; Liang, Y.; Rogaeva, E.A.; Levesque, G.; Ikeda, M.; Chi, H.; Lin, C.; Li, G.; Holman, K.; et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 1995, 375, 754–760. [Google Scholar] [CrossRef]
- Rogaev, E.I.; Sherrington, R.; Rogaeva, E.A.; Levesque, G.; Ikeda, M.; Liang, Y.; Chi, H.; Lin, C.; Holman, K.; Tsuda, T.; et al. Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature 1995, 376, 775–778. [Google Scholar] [CrossRef]
- Levy-Lahad, E.; Wasco, W.; Poorkaj, P.; Romano, D.M.; Oshima, J.; Pettingell, W.H.; Yu, C.E.; Jondro, P.D.; Schmidt, S.D.; Wang, K.; et al. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science 1995, 269, 973–977. [Google Scholar] [CrossRef]
- Lu, P.; Bai, X.C.; Ma, D.; Xie, T.; Yan, C.; Sun, L.; Yang, G.; Zhao, Y.; Zhou, R.; Scheres, S.H.W.; et al. Three-dimensional structure of human gamma-secretase. Nature 2014, 512, 166–170. [Google Scholar] [CrossRef]
- Bai, X.C.; Yan, C.; Yang, G.; Lu, P.; Ma, D.; Sun, L.; Zhou, R.; Scheres, S.H.W.; Shi, Y. An atomic structure of human gamma-secretase. Nature 2015, 525, 212–217. [Google Scholar] [CrossRef]
- Yang, G.; Zhou, R.; Shi, Y. Cryo-EM structures of human gamma-secretase. Curr. Opin. Struct. Biol. 2017, 46, 55–64. [Google Scholar] [CrossRef]
- Lee, M.K.; Slunt, H.H.; Martin, L.J.; Thinakaran, G.; Kim, G.; Gandy, S.E.; Seeger, M.; Koo, E.; Price, D.L.; Sisodia, S.S. Expression of presenilin 1 and 2 (PS1 and PS2) in human and murine tissues. J. Neurosci. 1996, 16, 7513–7525. [Google Scholar] [CrossRef]
- Wong, P.C.; Zheng, H.; Chen, H.; Becher, M.W.; Sirinathsinghji, D.J.; Trumbauer, M.E.; Chen, H.Y.; Price, D.L.; Van der Ploeg, L.H.; Sisodia, S.S. Presenilin 1 is required for Notch1 and DII1 expression in the paraxial mesoderm. Nature 1997, 387, 288–292. [Google Scholar] [CrossRef] [PubMed]
- Donoviel, D.B.; Hadjantonakis, A.K.; Ikeda, M.; Zheng, H.; Hyslop, P.S.; Bernstein, A. Mice lacking both presenilin genes exhibit early embryonic patterning defects. Genes Dev. 1999, 13, 2801–2810. [Google Scholar] [CrossRef] [PubMed]
- Beglopoulos, V.; Sun, X.; Saura, C.A.; Lemere, C.A.; Kim, R.D.; Shen, J. Reduced beta-amyloid production and increased inflammatory responses in presenilin conditional knock-out mice. J. Biol. Chem. 2004, 279, 46907–46914. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wu, B.; Beglopoulos, V.; Wines-Samuelson, M.; Zhang, D.; Dragatsis, I.; Sudhof, T.C.; Shen, J. Presenilins are essential for regulating neurotransmitter release. Nature 2009, 460, 632–636. [Google Scholar] [CrossRef]
- Wolfe, M.S.; Xia, W.; Ostaszewski, B.L.; Diehl, T.S.; Kimberly, W.T.; Selkoe, D.J. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature 1999, 398, 513–517. [Google Scholar] [CrossRef]
- Borchelt, D.R.; Thinakaran, G.; Eckman, C.B.; Lee, M.K.; Davenport, F.; Ratovitsky, T.; Prada, C.M.; Kim, G.; Seekins, S.; Yager, D.; et al. Familial Alzheimer’s disease-linked presenilin 1 variants elevate Abeta1-42/1-40 ratio in vitro and in vivo. Neuron 1996, 17, 1005–1013. [Google Scholar] [CrossRef]
- Tomita, T.; Maruyama, K.; Saido, T.C.; Kume, H.; Shinozaki, K.; Tokuhiro, S.; Capell, A.; Walter, J.; Grunberg, J.; Haass, C.; et al. The presenilin 2 mutation (N141I) linked to familial Alzheimer disease (Volga German families) increases the secretion of amyloid beta protein ending at the 42nd (or 43rd) residue. Proc. Natl. Acad. Sci. USA 1997, 94, 2025–2030. [Google Scholar] [CrossRef]
- Scheuner, D.; Eckman, C.; Jensen, M.; Song, X.; Citron, M.; Suzuki, N.; Bird, T.D.; Hardy, J.; Hutton, M.; Kukull, W.; et al. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat. Med. 1996, 2, 864–870. [Google Scholar] [CrossRef]
- Mehra, R.; Kepp, K.P. Identification of Structural Calcium Binding Sites in Membrane-Bound Presenilin 1 and 2. J. Phys. Chem. B 2020, 124, 4697–4711. [Google Scholar] [CrossRef]
- Thinakaran, G.; Borchelt, D.R.; Lee, M.K.; Slunt, H.H.; Spitzer, L.; Kim, G.; Ratovitsky, T.; Davenport, F.; Nordstedt, C.; Seeger, M.; et al. Endoproteolysis of presenilin 1 and accumulation of processed derivatives in vivo. Neuron 1996, 17, 181–190. [Google Scholar] [CrossRef]
- Takeda, T.; Asahi, M.; Yamaguchi, O.; Hikoso, S.; Nakayama, H.; Kusakari, Y.; Kawai, M.; Hongo, K.; Higuchi, Y.; Kashiwase, K.; et al. Presenilin 2 regulates the systolic function of heart by modulating Ca2+ signaling. FASEB J. 2005, 19, 2069–2071. [Google Scholar] [CrossRef] [PubMed]
- Pack-Chung, E.; Meyers, M.B.; Pettingell, W.P.; Moir, R.D.; Brownawell, A.M.; Cheng, I.; Tanzi, R.E.; Kim, T.W. Presenilin 2 interacts with sorcin, a modulator of the ryanodine receptor. J. Biol. Chem. 2000, 275, 14440–14445. [Google Scholar] [CrossRef] [PubMed]
- Hayrapetyan, V.; Rybalchenko, V.; Rybalchenko, N.; Koulen, P. The N-terminus of presenilin-2 increases single channel activity of brain ryanodine receptors through direct protein-protein interaction. Cell Calcium 2008, 44, 507–518. [Google Scholar] [CrossRef] [PubMed]
- Rybalchenko, V.; Hwang, S.Y.; Rybalchenko, N.; Koulen, P. The cytosolic N-terminus of presenilin-1 potentiates mouse ryanodine receptor single channel activity. Int. J. Biochem. Cell Biol. 2008, 40, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Greotti, E.; Capitanio, P.; Wong, A.; Pozzan, T.; Pizzo, P.; Pendin, D. Familial Alzheimer’s disease-linked presenilin mutants and intracellular Ca(2+) handling: A single-organelle, FRET-based analysis. Cell Calcium 2019, 79, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Baba-Aissa, F.; Raeymaekers, L.; Wuytack, F.; Dode, L.; Casteels, R. Distribution and isoform diversity of the organellar Ca2+ pumps in the brain. Mol. Chem. Neuropathol. 1998, 33, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Britzolaki, A.; Saurine, J.; Klocke, B.; Pitychoutis, P.M. A Role for SERCA Pumps in the Neurobiology of Neuropsychiatric and Neurodegenerative Disorders. Adv. Exp. Med. Biol. 2020, 1131, 131–161. [Google Scholar] [CrossRef]
- Jin, H.; Sanjo, N.; Uchihara, T.; Watabe, K.; St George-Hyslop, P.; Fraser, P.E.; Mizusawa, H. Presenilin-1 holoprotein is an interacting partner of sarco endoplasmic reticulum calcium-ATPase and confers resistance to endoplasmic reticulum stress. J. Alzheimer’s Dis. 2010, 20, 261–273. [Google Scholar] [CrossRef]
- Krajnak, K.; Dahl, R. A new target for Alzheimer’s disease: A small molecule SERCA activator is neuroprotective in vitro and improves memory and cognition in APP/PS1 mice. Bioorg. Med. Chem. Lett. 2018, 28, 1591–1594. [Google Scholar] [CrossRef]
- Zhao, L.; Ackerman, S.L. Endoplasmic reticulum stress in health and disease. Curr. Opin. Cell Biol. 2006, 18, 444–452. [Google Scholar] [CrossRef]
- Hetz, C.; Papa, F.R. The Unfolded Protein Response and Cell Fate Control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Hoozemans, J.J.; Veerhuis, R.; Van Haastert, E.S.; Rozemuller, J.M.; Baas, F.; Eikelenboom, P.; Scheper, W. The unfolded protein response is activated in Alzheimer’s disease. Acta Neuropathol. 2005, 110, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Hoozemans, J.J.; van Haastert, E.S.; Nijholt, D.A.; Rozemuller, A.J.; Eikelenboom, P.; Scheper, W. The unfolded protein response is activated in pretangle neurons in Alzheimer’s disease hippocampus. Am. J. Pathol. 2009, 174, 1241–1251. [Google Scholar] [CrossRef] [PubMed]
- Chang, R.C.; Suen, K.C.; Ma, C.H.; Elyaman, W.; Ng, H.K.; Hugon, J. Involvement of double-stranded RNA-dependent protein kinase and phosphorylation of eukaryotic initiation factor-2alpha in neuronal degeneration. J. Neurochem. 2002, 83, 1215–1225. [Google Scholar] [CrossRef]
- Page, G.; Rioux Bilan, A.; Ingrand, S.; Lafay-Chebassier, C.; Pain, S.; Perault Pochat, M.C.; Bouras, C.; Bayer, T.; Hugon, J. Activated double-stranded RNA-dependent protein kinase and neuronal death in models of Alzheimer’s disease. Neuroscience 2006, 139, 1343–1354. [Google Scholar] [CrossRef]
- Kim, H.S.; Choi, Y.; Shin, K.Y.; Joo, Y.; Lee, Y.K.; Jung, S.Y.; Suh, Y.H.; Kim, J.H. Swedish amyloid precursor protein mutation increases phosphorylation of eIF2alpha in vitro and in vivo. J. Neurosci. Res. 2007, 85, 1528–1537. [Google Scholar] [CrossRef]
- Chami, M.; Gozuacik, D.; Lagorce, D.; Brini, M.; Falson, P.; Peaucellier, G.; Pinton, P.; Lecoeur, H.; Gougeon, M.L.; le Maire, M.; et al. SERCA1 truncated proteins unable to pump calcium reduce the endoplasmic reticulum calcium concentration and induce apoptosis. J. Cell Biol. 2001, 153, 1301–1314. [Google Scholar] [CrossRef]
- Chami, M.; Oules, B.; Szabadkai, G.; Tacine, R.; Rizzuto, R.; Paterlini-Brechot, P. Role of SERCA1 truncated isoform in the proapoptotic calcium transfer from ER to mitochondria during ER stress. Mol. Cell 2008, 32, 641–651. [Google Scholar] [CrossRef]
- Nishitsuji, K.; Tomiyama, T.; Ishibashi, K.; Ito, K.; Teraoka, R.; Lambert, M.P.; Klein, W.L.; Mori, H. The E693Delta mutation in amyloid precursor protein increases intracellular accumulation of amyloid beta oligomers and causes endoplasmic reticulum stress-induced apoptosis in cultured cells. Am. J. Pathol. 2009, 174, 957–969. [Google Scholar] [CrossRef]
- O’Connor, T.; Sadleir, K.R.; Maus, E.; Velliquette, R.A.; Zhao, J.; Cole, S.L.; Eimer, W.A.; Hitt, B.; Bembinster, L.A.; Lammich, S.; et al. Phosphorylation of the translation initiation factor eIF2alpha increases BACE1 levels and promotes amyloidogenesis. Neuron 2008, 60, 988–1009. [Google Scholar] [CrossRef]
- Devi, L.; Ohno, M. PERK mediates eIF2alpha phosphorylation responsible for BACE1 elevation, CREB dysfunction and neurodegeneration in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2014, 35, 2272–2281. [Google Scholar] [CrossRef] [PubMed]
- Chami, L.; Checler, F. BACE1 is at the crossroad of a toxic vicious cycle involving cellular stress and beta-amyloid production in Alzheimer’s disease. Mol. Neurodegener. 2012, 7, 52. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kauppinen, A.; Suuronen, T.; Kaarniranta, K.; Ojala, J. ER stress in Alzheimer’s disease: A novel neuronal trigger for inflammation and Alzheimer’s pathology. J. Neuroinflamm. 2009, 6, 41. [Google Scholar] [CrossRef] [PubMed]
- Popugaeva, E.; Bezprozvanny, I. STIM proteins as regulators of neuronal store-operated calcium influx. Neurodegener. Dis. Manag. 2018, 8, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Secondo, A.; Bagetta, G.; Amantea, D. On the Role of Store-Operated Calcium Entry in Acute and Chronic Neurodegenerative Diseases. Front. Mol. Neurosci. 2018, 11, 87. [Google Scholar] [CrossRef]
- Kraft, R. STIM and ORAI proteins in the nervous system. Channels 2015, 9, 245–252. [Google Scholar] [CrossRef]
- Luik, R.M.; Wu, M.M.; Buchanan, J.; Lewis, R.S. The elementary unit of store-operated Ca2+ entry: Local activation of CRAC channels by STIM1 at ER-plasma membrane junctions. J. Cell Biol. 2006, 174, 815–825. [Google Scholar] [CrossRef]
- Ambudkar, I.S.; Ong, H.L.; Liu, X.; Bandyopadhyay, B.C.; Cheng, K.T. TRPC1: The link between functionally distinct store-operated calcium channels. Cell Calcium 2007, 42, 213–223. [Google Scholar] [CrossRef]
- Chen, X.; Sooch, G.; Demaree, I.S.; White, F.A.; Obukhov, A.G. Transient Receptor Potential Canonical (TRPC) Channels: Then and Now. Cells 2020, 9, 1983. [Google Scholar] [CrossRef]
- Moccia, F.; Zuccolo, E.; Soda, T.; Tanzi, F.; Guerra, G.; Mapelli, L.; Lodola, F.; D’Angelo, E. Stim and Orai proteins in neuronal Ca(2+) signaling and excitability. Front. Cell Neurosci. 2015, 9, 153. [Google Scholar] [CrossRef]
- Wegierski, T.; Kuznicki, J. Neuronal calcium signaling via store-operated channels in health and disease. Cell Calcium 2018, 74, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Heine, M.; Heck, J.; Ciuraszkiewicz, A.; Bikbaev, A. Dynamic compartmentalization of calcium channel signalling in neurons. Neuropharmacology 2020, 169, 107556. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Zhang, H.; Liu, J.; Popugaeva, E.; Xu, N.J.; Feske, S.; White, C.L., 3rd; Bezprozvanny, I. Reduced synaptic STIM2 expression and impaired store-operated calcium entry cause destabilization of mature spines in mutant presenilin mice. Neuron 2014, 82, 79–93. [Google Scholar] [CrossRef] [PubMed]
- Tong, B.C.; Lee, C.S.; Cheng, W.H.; Lai, K.O.; Foskett, J.K.; Cheung, K.H. Familial Alzheimer’s disease-associated presenilin 1 mutants promote gamma-secretase cleavage of STIM1 to impair store-operated Ca2+ entry. Sci. Signal. 2016, 9, ra89. [Google Scholar] [CrossRef]
- Ryazantseva, M.; Goncharova, A.; Skobeleva, K.; Erokhin, M.; Methner, A.; Georgiev, P.; Kaznacheyeva, E. Presenilin-1 Delta E9 Mutant Induces STIM1-Driven Store-Operated Calcium Channel Hyperactivation in Hippocampal Neurons. Mol. Neurobiol. 2018, 55, 4667–4680. [Google Scholar] [CrossRef]
- Linde, C.I.; Baryshnikov, S.G.; Mazzocco-Spezzia, A.; Golovina, V.A. Dysregulation of Ca2+ signaling in astrocytes from mice lacking amyloid precursor protein. Am. J. Physiol. Cell Physiol. 2011, 300, C1502–C1512. [Google Scholar] [CrossRef]
- Wang, J.; Lu, R.; Yang, J.; Li, H.; He, Z.; Jing, N.; Wang, X.; Wang, Y. TRPC6 specifically interacts with APP to inhibit its cleavage by gamma-secretase and reduce Abeta production. Nat. Commun. 2015, 6, 8876. [Google Scholar] [CrossRef]
- Lessard, C.B.; Lussier, M.P.; Cayouette, S.; Bourque, G.; Boulay, G. The overexpression of presenilin2 and Alzheimer’s-disease-linked presenilin2 variants influences TRPC6-enhanced Ca2+ entry into HEK293 cells. Cell Signal. 2005, 17, 437–445. [Google Scholar] [CrossRef]
- Prikhodko, V.; Chernyuk, D.; Sysoev, Y.; Zernov, N.; Okovityi, S.; Popugaeva, E. Potential Drug Candidates to Treat TRPC6 Channel Deficiencies in the Pathophysiology of Alzheimer’s Disease and Brain Ischemia. Cells 2020, 9, 2351. [Google Scholar] [CrossRef]
- Giorgi, C.; Missiroli, S.; Patergnani, S.; Duszynski, J.; Wieckowski, M.R.; Pinton, P. Mitochondria-associated membranes: Composition, molecular mechanisms, and physiopathological implications. Antioxid. Redox Signal. 2015, 22, 995–1019. [Google Scholar] [CrossRef]
- Area-Gomez, E.; de Groof, A.; Bonilla, E.; Montesinos, J.; Tanji, K.; Boldogh, I.; Pon, L.; Schon, E.A. A key role for MAM in mediating mitochondrial dysfunction in Alzheimer disease. Cell Death Dis. 2018, 9, 335. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chami, M.; Checler, F. Alterations of the Endoplasmic Reticulum (ER) Calcium Signaling Molecular Components in Alzheimer’s Disease. Cells 2020, 9, 2577. https://doi.org/10.3390/cells9122577
Chami M, Checler F. Alterations of the Endoplasmic Reticulum (ER) Calcium Signaling Molecular Components in Alzheimer’s Disease. Cells. 2020; 9(12):2577. https://doi.org/10.3390/cells9122577
Chicago/Turabian StyleChami, Mounia, and Frédéric Checler. 2020. "Alterations of the Endoplasmic Reticulum (ER) Calcium Signaling Molecular Components in Alzheimer’s Disease" Cells 9, no. 12: 2577. https://doi.org/10.3390/cells9122577
APA StyleChami, M., & Checler, F. (2020). Alterations of the Endoplasmic Reticulum (ER) Calcium Signaling Molecular Components in Alzheimer’s Disease. Cells, 9(12), 2577. https://doi.org/10.3390/cells9122577