Revealing Temozolomide Resistance Mechanisms via Genome-Wide CRISPR Libraries

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture Conditions
2.2. Pooled Genome-Wide CRISPR Screen
2.3. Computational Analysis of the Screens
2.4. Cell Survival Measurement
2.5. Gene Silencing
2.6. Establishing Cell Line Overexpression Target Genes
2.7. Real-Time PCR
2.8. Cell Migration
2.9. Bioinformatics
2.10. Statistical Analysis
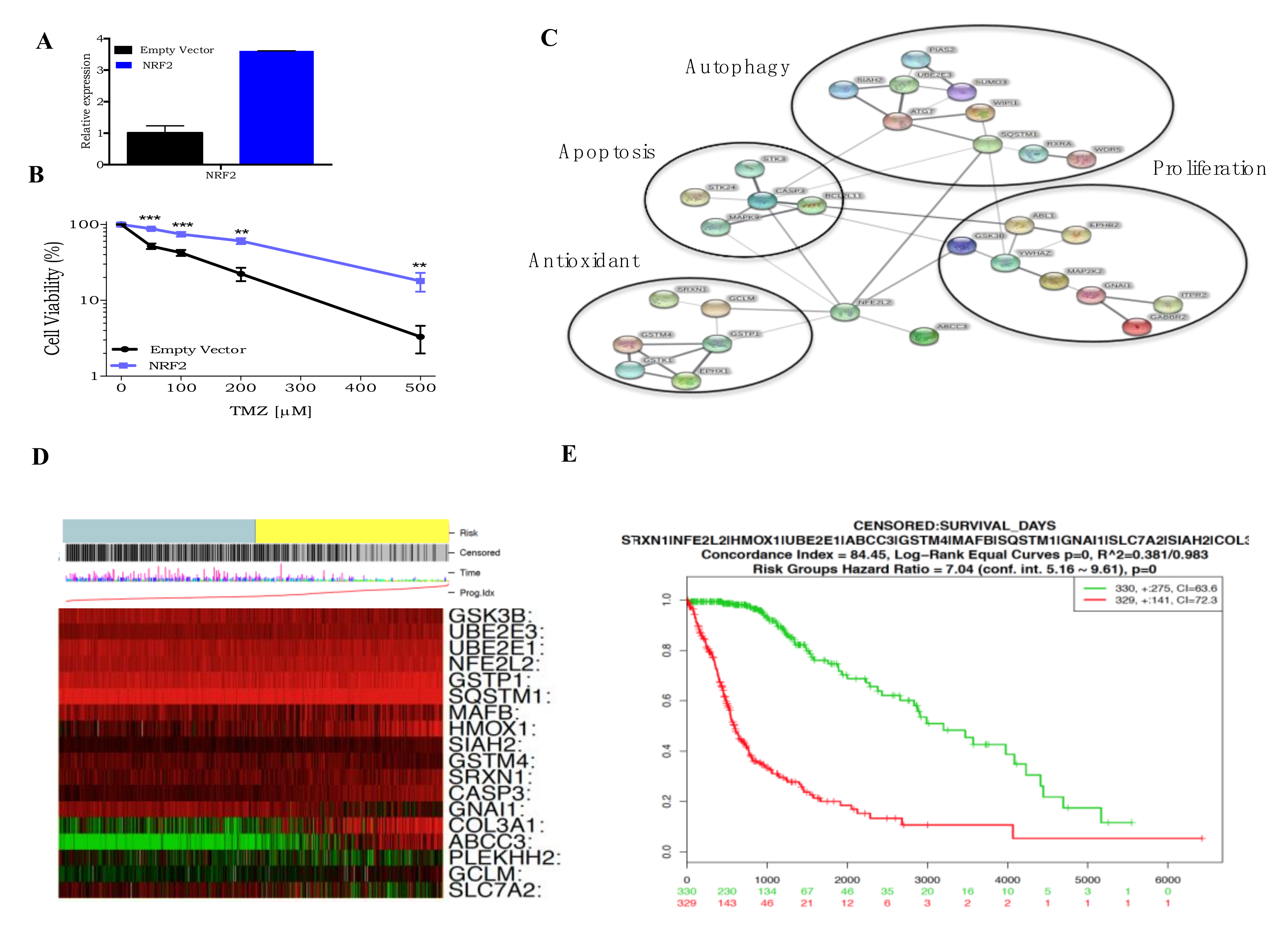
3. Results
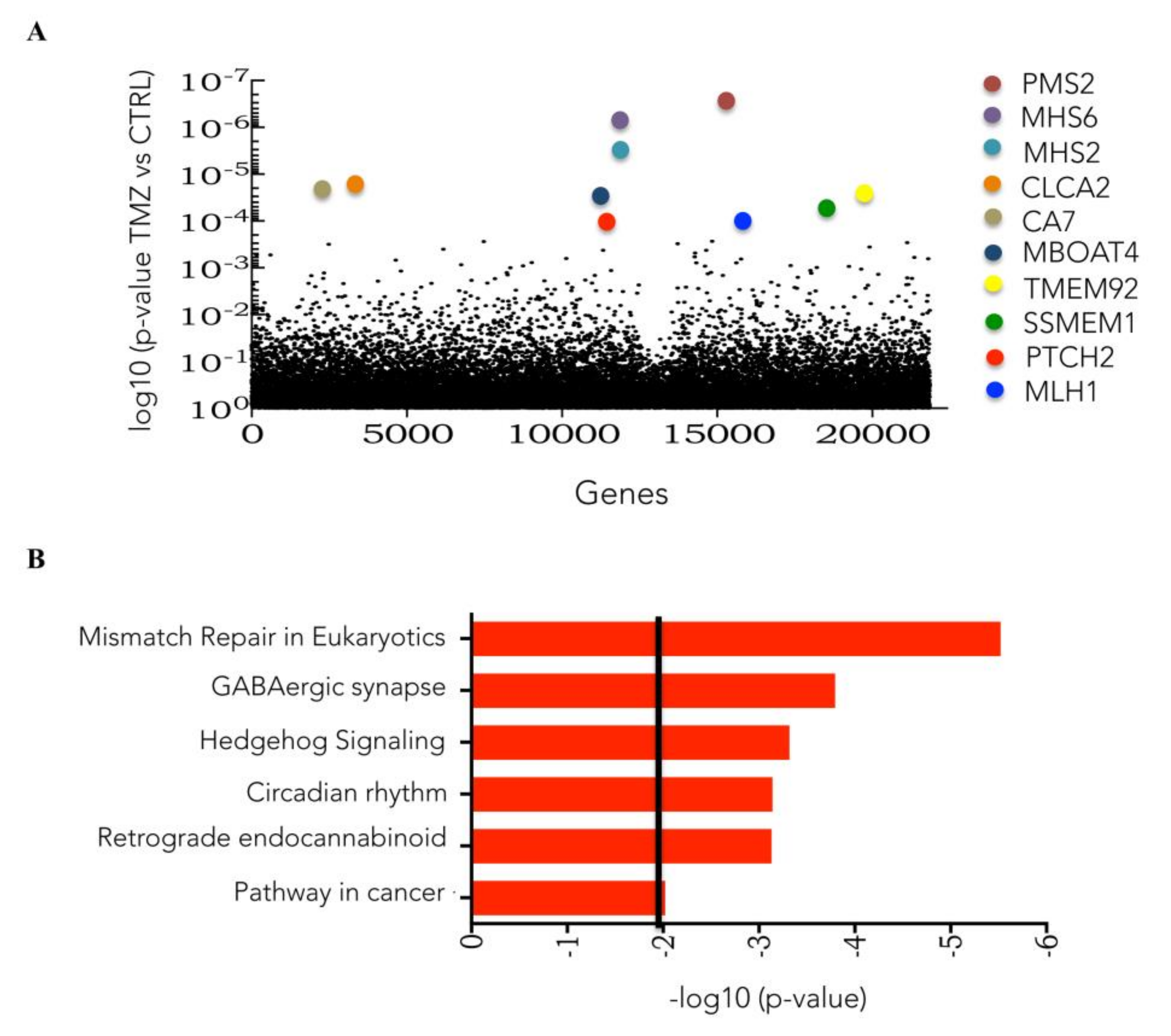
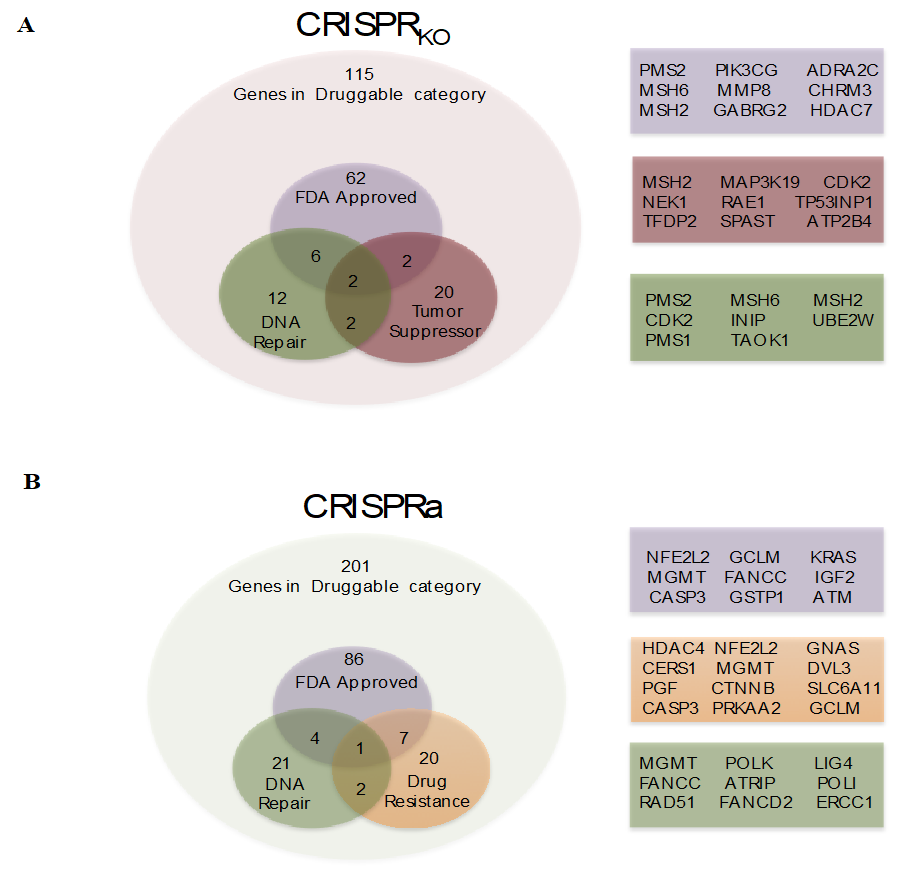
3.1. Genome-Wide CRISPR/Cas9 Knockout Screen in Glioblastoma Cancer Cell
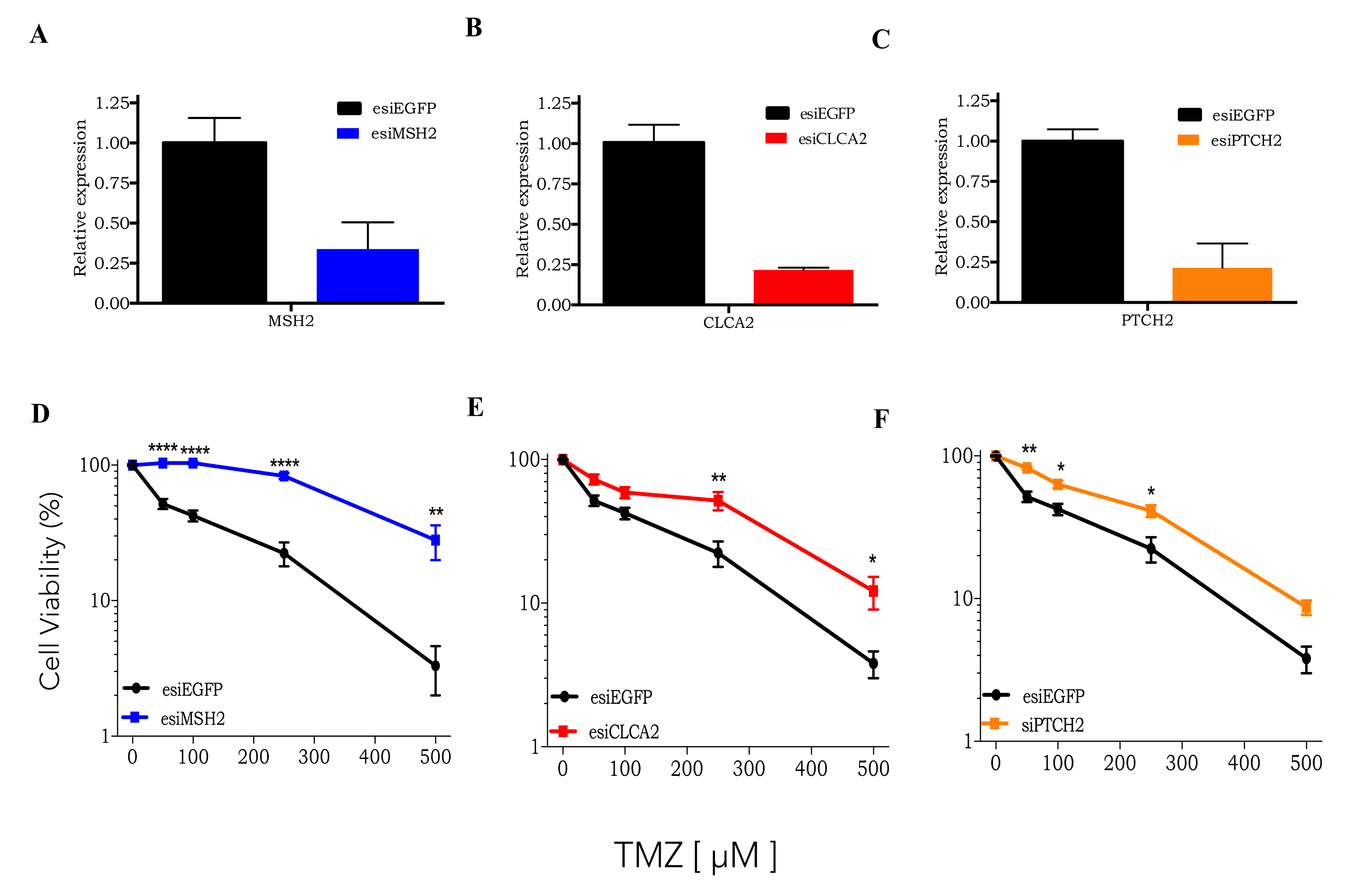
3.2. CRISPRKO Screen Gene Candidates Validation
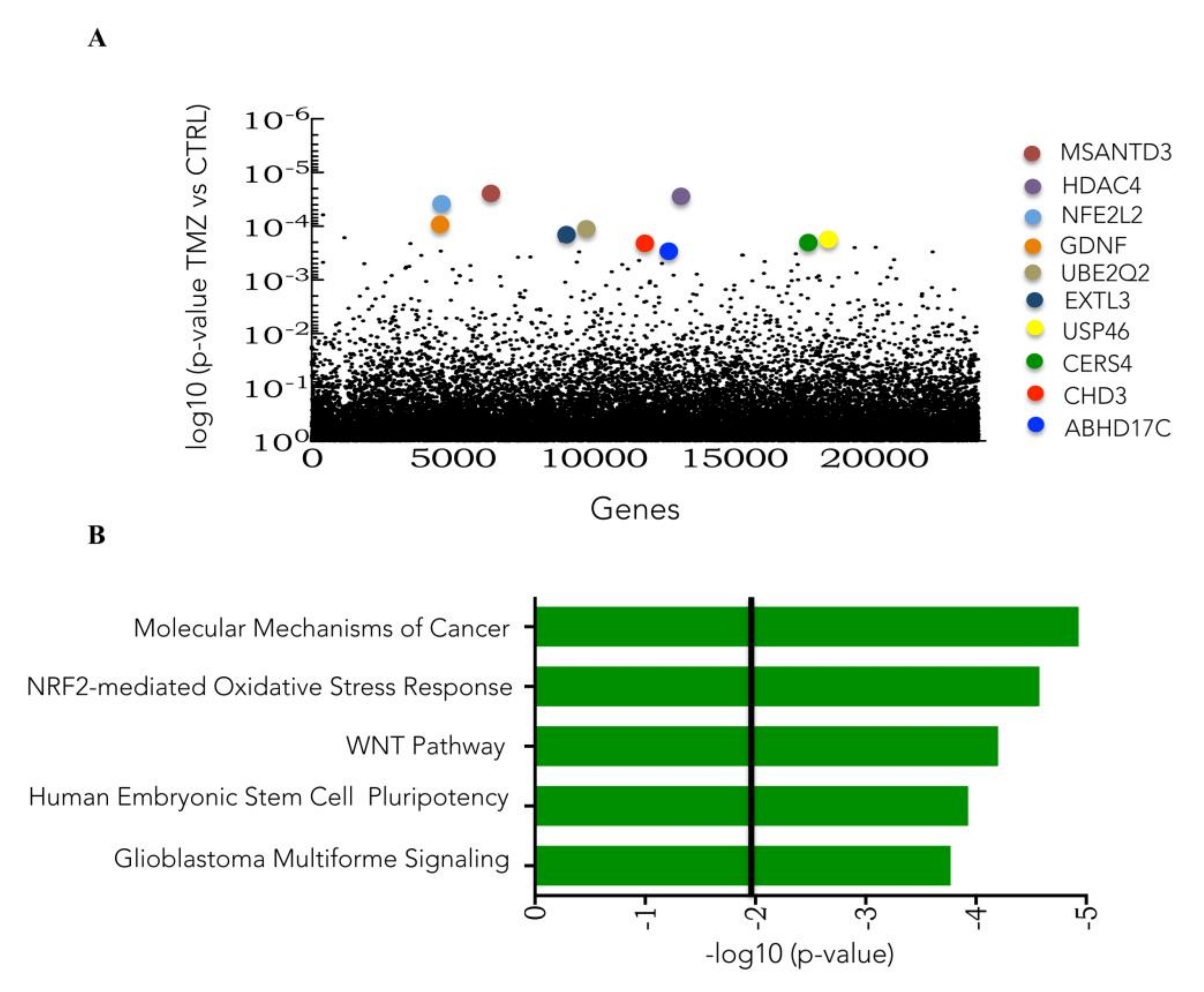
3.3. Genome-Wide CRISPR/Cas9 Activation Screen in Glioblastoma Cancer Cell
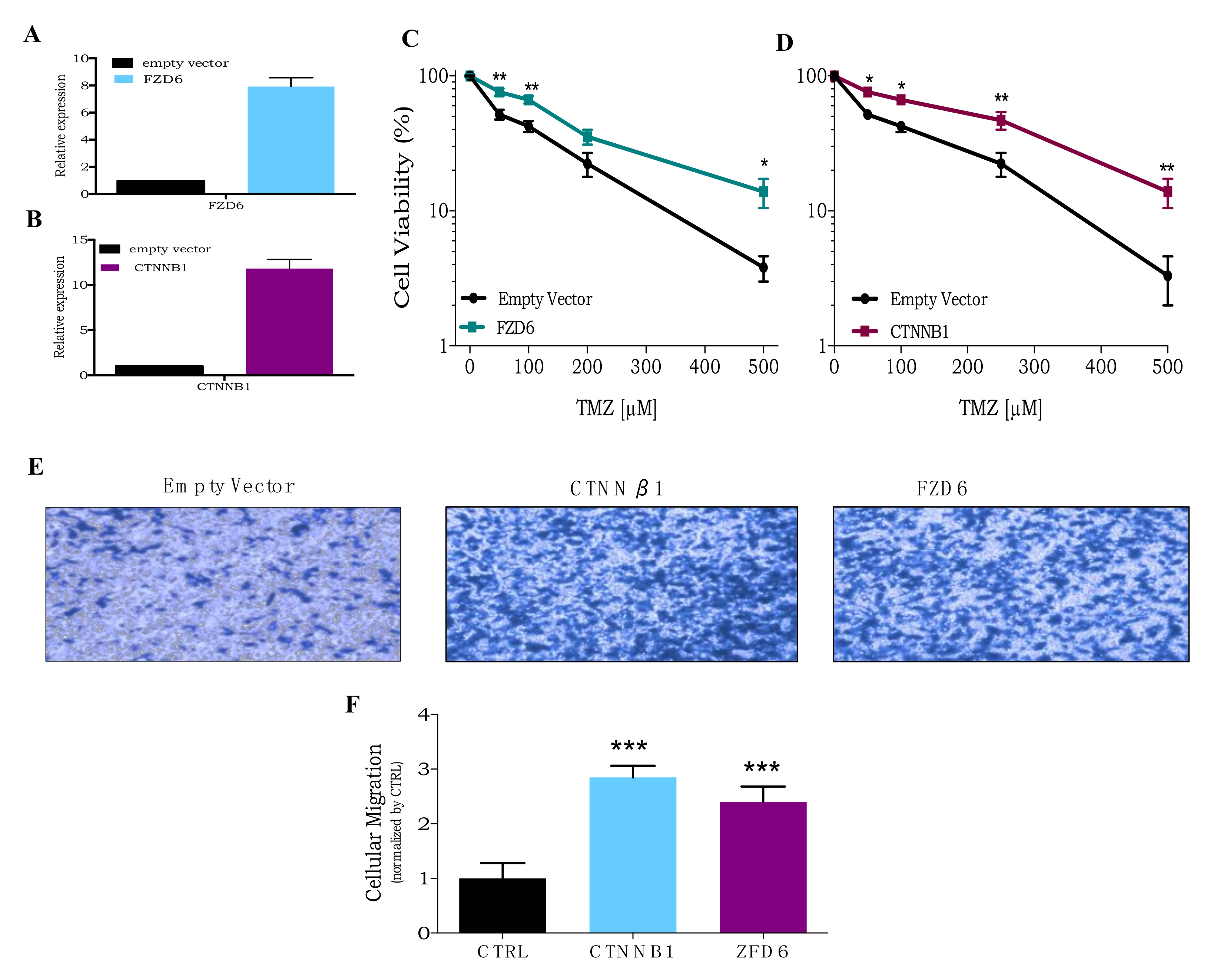
3.4. CRISPR Activation Screen Gene Candidates Validation
3.5. Roles of NRF2 on TMZ Resistance
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Lim, M.; Xia, Y.; Bettegowda, C.; Weller, M. Current state of immunotherapy for glioblastoma. Nat. Rev. Clin. Oncol. 2018, 15, 422–442. [Google Scholar] [CrossRef]
- Stupp, R.; Tonn, J.C.; Brada, M.; Pentheroudakis, G.; ESMO Gudelines Working Group. High-grade malignant glioma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2010, 21, v190–v193. [Google Scholar] [CrossRef]
- Wen, P.Y.; Kesari, S. Malignant gliomas in adults. N. Engl. J. Med. 2008, 359, 492–507. [Google Scholar] [CrossRef]
- Agarwala, S.S.; Kirkwood, J.M. Temozolomide, a Novel Alkylating Agent with Activity in the Central Nervous System, May Improve the Treatment of Advanced Metastatic Melanoma. Oncologist 2000, 5, 144–151. [Google Scholar] [CrossRef]
- Zhang, J.; Stevens, M.F.G.; Bradshaw, T.D. Temozolomide: Mechanisms of Action, Repair and Resistance. Curr. Mol. Pharmacol. 2012, 5, 102–114. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Johannessen, T.C.A.; Bjerkvig, R. Molecular mechanisms of temozolomide resistance in glioblastoma multiforme. Expert Rev. Anticancer Ther. 2012, 12, 635–642. [Google Scholar] [CrossRef]
- Woo, P.; Li, Y.; Chan, A.; Ng, S.; Loong, H.; Chan, D.; Wong, G.; Poon, W. A multifaceted review of temozolomide resistance mechanisms in glioblastoma beyond O-6-methylguanine-DNA methyltransferase. Glioma 2019, 2, 68–82. [Google Scholar]
- Vasan, N.; Baselga, J.; Hyman, D.M. A view on drug resistance in cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef]
- Prelich, G. Gene Overexpression: Uses, Mechanisms, and Interpretation. Genetics 2012, 190, 841–854. [Google Scholar] [CrossRef]
- Jackson, A.L.; Linsley, P.S. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat. Rev. Drug Discov. 2010, 9, 57. [Google Scholar] [CrossRef]
- Kurata, M.; Yamamoto, K.; Moriarity, B.S.; Kitagawa, M.; Largaespada, D.A. CRISPR/Cas9 library screening for drug target discovery. J. Hum. Genet. 2018, 63, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Fellmann, C.; Gowen, B.G.; Lin, P.C.; Doudna, J.A.; Corn, J.E. Cornerstones of CRISPR-Cas in drug discovery and therapy. Nat. Rev. Drug Discov. 2016, 16, 89. [Google Scholar] [CrossRef] [PubMed]
- Pettitt, S.J.; Krastev, D.B.; Brandsma, I.; Dréan, A.; Song, F.; Aleksandrov, R.; Harrell, M.I.; Menon, M.; Brough, R.; Campbell, J.; et al. Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nat. Commun. 2018, 9, 1849. [Google Scholar] [CrossRef] [PubMed]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelsen, T.S.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-Scale CRISPR-Cas9 Knockout Screening in Human Cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Konermann, S.; Brigham, M.D.; Trevino, A.E.; Joung, J.; Abudayyeh, O.O.; Barcena, C.; Hsu, P.D.; Habib, N.; Gootenberg, J.S.; Nishimasu, H.; et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 2015, 517, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Köster, J.; Xu, H.; Chen, C.-H.; Xiao, T.; Liu, J.S.; Brown, M.; Liu, X.S. Quality control, modeling, and visualization of CRISPR screens with MAGeCK-VISPR. Genome Biol. 2015, 16, 281. [Google Scholar] [CrossRef]
- Gomes, L.R.; Rocha, C.R.R.; Martins, D.J.; Fiore, A.P.Z.P.; Kinker, G.S.; Bruni-Cardoso, A.; Menck, C.F.M. ATR mediates cisplatin resistance in 3D-cultured breast cancer cells via translesion DNA synthesis modulation. Cell Death Dis. 2019, 10, 459. [Google Scholar] [CrossRef]
- Ma, Q. Role of Nrf2 in Oxidative Stress and Toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef]
- Jaramillo, M.C.; Zhang, D.D. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013, 27, 2179–2191. [Google Scholar] [CrossRef]
- Sporn, M.B.; Liby, K.T. NRF2 and cancer: The good, the bad and the importance of context. Nat. Rev. Cancer 2012, 12. [Google Scholar] [CrossRef] [PubMed]
- Hanif, F.; Muzaffar, K.; Perveen, K.; Malhi, S.M.; Simjee, S.U. Glioblastoma Multiforme: A Review of its Epidemiology and Pathogenesis through Clinical Presentation and Treatment. Asian Pac. J. Cancer Prev. 2017, 18, 3–9. [Google Scholar] [PubMed]
- Bahadur, S.; Sahu, A.K.; Baghel, P.; Saha, S. Current promising treatment strategy for glioblastoma multiform: A review. Oncol. Rev. 2019, 13, 417. [Google Scholar] [CrossRef]
- Kaina, B.; Christmann, M. DNA repair in personalized brain cancer therapy with temozolomide and nitrosoureas. DNA Repair Amst. 2019, 78, 128–141. [Google Scholar] [CrossRef] [PubMed]
- Karran, P.; Bignami, M. DNA damage tolerance, mismatch repair and genome instability. Bioessays 1994, 16, 833–839. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016, 3, 198–210. [Google Scholar] [CrossRef] [PubMed]
- MacLeod, G.; Bozek, D.A.; Rajakulendran, N.; Monteiro, V.; Ahmadi, M.; Steinhart, Z.; Kushida, M.M.; Yu, H.; Coutinho, F.J.; Cavalli, F.M.G.; et al. Genome-Wide CRISPR-Cas9 Screens Expose Genetic Vulnerabilities and Mechanisms of Temozolomide Sensitivity in Glioblastoma Stem Cells. Cell Rep. 2019, 27, 971–986. [Google Scholar] [CrossRef]
- Huang, K.; Liu, X.; Li, Y.; Wang, Q.; Zhou, J.; Wang, Y.; Dong, F.; Yang, C.; Sun, Z.; Fang, C.; et al. Genome-Wide CRISPR-Cas9 Screening Identifies NF-κB/E2F6 Responsible for EGFRvIII-Associated Temozolomide Resistance in Glioblastoma. Adv. Sci. 2019, 6, 1900782. [Google Scholar] [CrossRef]
- Ostermann, S.; Csajka, C.; Buclin, T.; Leyvraz, S.; Lejeune, F.; Decosterd, L.A.; Stupp, R. Plasma and cerebrospinal fluid population pharmacokinetics of temozolomide in malignant glioma patients. Clin. Cancer Res. 2004, 10, 3728–3736. [Google Scholar] [CrossRef]
- Patel, M.; McCully, C.; Godwin, K.; Balis, F.M. Plasma and Cerebrospinal Fluid Pharmacokinetics of Intravenous Temozolomide in Non-human Primates. J. Neurooncol. 2003, 61, 203–207. [Google Scholar] [CrossRef]
- Skoda, A.M.; Simovic, D.; Karin, V.; Kardum, V.; Vranic, S.; Serman, L. The role of the Hedgehog signaling pathway in cancer: A comprehensive review. Bosn. J. Basic Med. Sci. 2018, 18, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Carballo, G.B.; Honorato, J.R.; de Lopes, G.P.F.; de Sampaio e Spohr, T.C.L. A highlight on Sonic hedgehog pathway. Cell Commun. Signal. 2018, 16, 11. [Google Scholar] [CrossRef] [PubMed]
- Ulasov, I.V.; Nandi, S.; Dey, M.; Sonabend, A.M.; Lesniak, M.S. Inhibition of Sonic hedgehog and Notch pathways enhances sensitivity of CD133(+) glioma stem cells to temozolomide therapy. Mol. Med. 2011, 17, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Melamed, J.R.; Morgan, J.T.; Ioele, S.A.; Gleghorn, J.P.; Sims-Mourtada, J.; Day, E.S. Investigating the role of Hedgehog/GLI1 signaling in glioblastoma cell response to temozolomide. Oncotarget 2018, 9, 27000–27015. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.; Zwick, A.; Kissel, S.; Forster, C.U.; Pfeifer, D.; Follo, M.; Illert, A.L.; Decker, S.; Benkler, T.; Pahl, H.; et al. Ptch2 loss drives myeloproliferation and myeloproliferative neoplasm progression. J. Exp. Med. 2016, 213, 273–290. [Google Scholar] [CrossRef]
- Pekovic-Vaughan, V.; Gibbs, J.; Yoshitane, H.; Yang, N.; Pathiranage, D.; Guo, B.; Sagami, A.; Taguchi, K.; Bechtold, D.; Loudon, A.; et al. The circadian clock regulates rhythmic activation of the NRF2/glutathione-mediated antioxidant defense pathway to modulate pulmonary fibrosis. Genes Dev. 2014, 28, 548–560. [Google Scholar] [CrossRef]
- Kang, T.H.; Reardon, J.T.; Kemp, M.; Sancar, A. Circadian oscillation of nucleotide excision repair in mammalian brain. Proc. Natl. Acad. Sci. USA 2009, 106, 2864–2867. [Google Scholar] [CrossRef]
- Feillet, C.; Krusche, P.; Tamanini, F.; Janssens, R.C.; Downey, M.J.; Martin, P.; Teboul, M.; Saito, S.; Lévi, F.A.; Bretschneider, T.; et al. Phase locking and multiple oscillating attractors for the coupled mammalian clock and cell cycle. Proc. Natl. Acad. Sci. USA 2014, 111, 9828–9833. [Google Scholar] [CrossRef]
- Walia, V.; Ding, M.; Kumar, S.; Nie, D.; Premkumar, L.S.; Elble, R.C. hCLCA2 Is a p53-Inducible Inhibitor of Breast Cancer Cell Proliferation. Cancer Res. 2009, 69, 6624–6632. [Google Scholar] [CrossRef]
- Sasaki, Y.; Koyama, R.; Maruyama, R.; Hirano, T.; Tamura, M.; Sugisaka, J.; Suzuki, H.; Idogawa, M.; Shinomura, Y.; Tokino, T. CLCA2, a target of the p53 family, negatively regulates cancer cell migration and invasion. Cancer Biol. Ther. 2012, 13, 1512–1521. [Google Scholar] [CrossRef]
- Li, X.; Cowell, J.K.; Sossey-Alaoui, K. CLCA2 tumour suppressor gene in 1p31 is epigenetically regulated in breast cancer. Oncogene 2004, 23, 1474–1480. [Google Scholar] [CrossRef] [PubMed]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- Ackers, I.; Malgor, R. Interrelationship of canonical and non-canonical Wnt signalling pathways in chronic metabolic diseases. Diabetes Vasc. Dis. Res. 2018, 15, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Wickström, M.; Dyberg, C.; Milosevic, J.; Einvik, C.; Calero, R.; Sveinbjörnsson, B.; Sandén, E.; Darabi, A.; Siesjö, P.; Kool, M.; et al. Wnt/β-catenin pathway regulates MGMT gene expression in cancer and inhibition of Wnt signalling prevents chemoresistance. Nat. Commun. 2015, 6, 8904. [Google Scholar] [CrossRef] [PubMed]
- Yun, E.J.; Kim, S.; Hsieh, J.T.; Baek, S.T. Wnt/β-catenin signaling pathway induces autophagy-mediated temozolomide-resistance in human glioblastoma. Cell Death Dis. 2020, 11, 771. [Google Scholar] [CrossRef]
- Tomar, V.S.; Patil, V.; Somasundaram, K. Temozolomide induces activation of Wnt/β-catenin signaling in glioma cells via PI3K/Akt pathway: Implications in glioma therapy. Cell Biol. Toxicol. 2020, 36, 273–278. [Google Scholar] [CrossRef]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2017, 29, 1727–1745. [Google Scholar] [CrossRef]
- Jiang, T.; Harder, B.; Rojo de la Vega, M.; Wong, P.K.; Chapman, E.; Zhang, D.D. p62 links autophagy and Nrf2 signaling. Free Radic. Biol. Med. 2015, 88, 199–204. [Google Scholar] [CrossRef]
- Pajares, M.; Jiménez-Moreno, N.; García-Yagüe, Á.J.; Escoll, M.; de Ceballos, M.L.; Van Leuven, F.; Rábano, A.; Yamamoto, M.; Rojo, A.I.; Cuadrado, A. Transcription factor NFE2L2/NRF2 is a regulator of macroautophagy genes. Autophagy 2016, 12, 1902–1916. [Google Scholar] [CrossRef]
- Hyttinen, J.M.T.; Kannan, R.; Felszeghy, S.; Niittykoski, M.; Salminen, A.; Kaarniranta, K. The Regulation of NFE2L2 (NRF2) Signalling and Epithelial-to-Mesenchymal Transition in Age-Related Macular Degeneration Pathology. Int. J. Mol. Sci. 2019, 20, 5800. [Google Scholar] [CrossRef]
- Pölönen, P.; Jawahar Deen, A.; Leinonen, H.M.; Jyrkkänen, H.K.; Kuosmanen, S.; Mononen, M.; Jain, A.; Tuomainen, T.; Pasonen-Seppänen, S.; Hartikainen, J.M.; et al. Nrf2 and SQSTM1/p62 jointly contribute to mesenchymal transition and invasion in glioblastoma. Oncogene 2019, 38, 7473–7490. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.M.; Rocha, C.R.R.; Kinker, G.S.; Pelegrini, A.L.; Menck, C.F.M. The balance between NRF2/GSH antioxidant mediated pathway and DNA repair modulates cisplatin resistance in lung cancer cells. Sci. Rep. 2019, 9, 17639. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Lu, H.; Bai, Y. Nrf2 in cancers: A double-edged sword. Cancer Med. 2019, 8, 2252–2267. [Google Scholar] [CrossRef] [PubMed]
- Rocha, C.R.R.; Kajitani, G.S.; Quinet, A.; Fortunato, R.S.; Menck, C.F.M. NRF2 and glutathione are key resistance mediators to temozolomide in glioma and melanoma cells. Oncotarget 2016, 7, 48081–48092. [Google Scholar] [CrossRef] [PubMed]
- Rocha, C.R.R.; Garcia, C.C.M.; Vieira, D.B.; Quinet, A.; de Andrade-Lima, L.C.; Munford, V.; Belizario, J.E.; Menck, C.F.M. Glutathione depletion sensitizes cisplatin- and temozolomide-resistant glioma cells in vitro and in vivo. Cell Death Dis 2014, 5, e1505. [Google Scholar] [CrossRef]
- Da Silva, P.B.G.; dos Santos, M.C.T.; Rodini, C.O.; Kaid, C.; Pereira, M.C.L.; Furukawa, G.; da Cruz, D.S.G.; Goldfeder, M.B.; Rocha, C.R.R.; Rosenberg, C.; et al. High OCT4A levels drive tumorigenicity and metastatic potential of medulloblastoma cells. Oncotarget 2017, 8, 19192. [Google Scholar] [CrossRef]
- Zhu, Z.; Du, S.; Du, Y.; Ren, J.; Ying, G.; Yan, Z. Glutathione reductase mediates drug resistance in glioblastoma cells by regulating redox homeostasis. J. Neurochem. 2018, 144, 93–104. [Google Scholar] [CrossRef]
- Yi, G.; Liu, Y.; Xiang, W.; Wang, H.; Chen, Z.; Xie, S.; Qi, S. Akt and β-catenin contribute to TMZ resistance and EMT of MGMT negative malignant glioma cell line. J. Neurol. Sci. 2016, 367, 101–106. [Google Scholar] [CrossRef]
- McFaline-Figueroa, J.L.; Braun, C.J.; Stanciu, M.; Nagel, Z.D.; Mazzucato, P.; Sangaraju, D.; Cerniauskas, E.; Barford, K.; Vargas, A.; Chen, Y.; et al. Minor Changes in Expression of the Mismatch Repair Protein MSH2 Exert a Major Impact on Glioblastoma Response to Temozolomide. Cancer Res. 2015, 75, 3127–3138. [Google Scholar] [CrossRef]
- Wang, K.; Chen, D.; Qian, Z.; Cui, D.; Gao, L.; Lou, M. Hedgehog/Gli1 signaling pathway regulates MGMT expression and chemoresistance to temozolomide in human glioblastoma. Cancer Cell Int. 2017, 17, 117. [Google Scholar] [CrossRef]
- Yang, W.; Liu, Y.; Gao, R.; Yu, H.; Sun, T. HDAC6 inhibition induces glioma stem cells differentiation and enhances cellular radiation sensitivity through the SHH/Gli1 signaling pathway. Cancer Lett. 2018, 415, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.T.; Nguyen, N.T.; Choi, Y.J.; Zhang, G.; Nguyen, H.N.; Filka, E.; Green, S.; Yong, W.H.; Liau, L.M.; Green, R.M.; et al. Phase 2 Study of Bortezomib Combined With Temozolomide and Regional Radiation Therapy for Upfront Treatment of Patients With Newly Diagnosed Glioblastoma Multiforme: Safety and Efficacy Assessment. Int. J. Radiat. Oncol. Biol. Phys. 2018, 100, 1195–1203. [Google Scholar] [CrossRef] [PubMed]
- Lesueur, P.; Lequesne, J.; Grellard, J.M.; Dugué, A.; Coquan, E.; Brachet, P.E.; Geffrelot, J.; Kao, W.; Emery, E.; Berro, D.H.; et al. Phase I/IIa study of concomitant radiotherapy with olaparib and temozolomide in unresectable or partially resectable glioblastoma: OLA-TMZ-RTE-01 trial protocol. BMC Cancer 2019, 19, 198. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| sgRNA | Gene | Control_count | Treatment_count | FDR | Rank |
|---|---|---|---|---|---|
| HGLibA_56959 | PMS2 | 10.1/8.3 | 1644.2/1039.1 | 0 | 1 |
| HGLibA_29380 | MLH1 | 71.8/75.3 | 10,516.1/8493.7 | 0 | 2 |
| HGLibB_45493 | SLPI | 62.3/60.9 | 1612.5/22,063.1 | 3.3 × 10−283 | 3 |
| HGLibA_30203 | MSH6 | 145.4/314.7 | 21,025.1/17,627.1 | 1.5 × 10−135 | 4 |
| HGLibB_30160 | MSH6 | 56.9/51.6 | 4275.1/3345.3 | 1.8 × 10−154 | 5 |
| HGLibB_30150 | MSH2 | 109.2/147.5 | 8836.2/8593.6 | 4.6 × 10−100 | 6 |
| HGLibB_30148 | MSH2 | 40.9/5.2 | 963.5/914.5 | 5.1 × 10−240 | 7 |
| HGLibA_53112 | UQCRC1 | 22.5/14.4 | 1063.1/995.6 | 2.2 × 10−170 | 8 |
| HGLibB_53357 | USPL1 | 21.4/16.5 | 1051.1/997.9 | 6.3 × 10−153 | 9 |
| HGLibB_40798 | REPS1 | 31.5/3.1 | 471.9/531.3 | 3.0 × 10−172 | 10 |
| HGLibB_09799 | CLCA2 | 18.9/9.3 | 845.6/510.1 | 3.7 × 10−146 | 11 |
| HGLibB_37031 | PLEKHA6 | 49.3/2.1 | 391.1/577.1 | 5.7 × 10−144 | 12 |
| HGLibA_56902 | ZNF90 | 21.9/25.8 | 989.8/1188.4 | 5.5 × 10−95 | 13 |
| HGLibA_06820 | CA7 | 31.5/10.3 | 886.1/747.6 | 9.2 × 10−106 | 14 |
| HGLibA_29915 | MRPL18 | 10.7/55.7 | 1234.5/965.1 | 4.3 × 10−90 | 15 |
| HGLibA_14727 | EIF2AK2 | 25.5/13.4 | 746.2/778.1 | 1.6 × 10−86 | 16 |
| HGLibB_44264 | SLAMF7 | 238.1/408.7 | 13,929.1/11,287.1 | 1.3 × 10−21 | 17 |
| HGLibA_61495 | PER2 | 79.5/72.2 | 2574.1/3308.8 | 6.2 × 10−39 | 18 |
| HGLibA_00885 | ADAT3 | 16.6/13.4 | 729.8/405.5 | 4.6 × 10−74 | 19 |
| HGLibA_25835 | KRTAP9-3 | 22.5/29.9 | 1108.9/794.6 | 1.6 × 10−56 | 20 |
| HGLibA_60473 | PER1 | 42.1/18.5 | 1010.6/980.3 | 7.9 × 10−53 | 21 |
| HGLibA_20486 | GSTO1 | 30.3/6.2 | 512.4/444.3 | 8.1 × 10−70 | 22 |
| HGLibB_45186 | SLC51A | 26.1/29.9 | 867.4/1028.5 | 1.6 × 10−47 | 23 |
| HGLibA_36058 | PER2 | 33.3/21.7 | 1025.9/779.3 | 5.0 × 10−47 | 24 |
| HGLibA_05746 | C1orf111 | 22.5/25.8 | 773.5/795.7 | 5.1 × 10−47 | 25 |
| sgRNA Sequence | Gene | Control_count | Treatment_count | FDR | Rank |
|---|---|---|---|---|---|
| GTTATCACGGACAGGGAATA | MSANTD3 | 4.9/5.4 | 1289.4/1079.7 | 0 | 1 |
| CCCGCACTCAAGGAGGTCTT | NFE2L2 | 241.1/555.1 | 55,614.1/69,280.1 | 0 | 2 |
| GACGCCGGCCCGCAAGTGAC | GCLM | 4.9/9.9 | 750.1/892.1 | 8.90 × 10−291 | 3 |
| CGCAAGGCTGCCGCAGCCAA | HDAC4 | 9.7/7.8 | 991.2/829.8 | 2.60 × 10−283 | 4 |
| CGGGGTGGCCTATCGCCGCC | HDAC4 | 9.7/6.1 | 653.6/730.7 | 6.30 × 10−278 | 5 |
| CCCGCACTCAAGGAGGTCTT | NFE2L2 | 16.7/13.4 | 1376.3/1028.8 | 2.90 × 10−75 | 6 |
| GCGGCAGGGGCCAGCCCGCC | UBE2E3 | 10.8/12.2 | 1029.5/795.2 | 3.00 × 10−238 | 7 |
| GGCCAGGACAAAGAGCGCGG | UBE2E3 | 22.6/1.6 | 1013.1/877.5 | 1.70 × 10−183 | 8 |
| CCGAGCAGCCGCCGCTGCTT | GDNF | 7.3/7.9 | 519.2/407.1 | 3.30 × 10−159 | 9 |
| CCCTGACCCGGGTGTCACCG | EXTL3 | 10.7/12.2 | 662.1/691.5 | 3.50 × 10−130 | 10 |
| TACACACATTTCGTGGGGTG | USP46 | 7.7/5.6 | 403.9/329.3 | 1.90 × 10−73 | 11 |
| GAATGAATGTTATCACGGAC | MSANTD3 | 3.4/11.1 | 480.2/309.1 | 7.70 × 10−127 | 12 |
| CCGAGCAGCCGCCGCTGCTT | GDNF | 23.9/7.9 | 906.6/770.6 | 6.20 × 10−120 | 13 |
| CATATGACTCAGCACATGAC | MSANTD3 | 29.5/15.9 | 1201.4/934.4 | 7.20 × 10−114 | 14 |
| AGTACTGGGAGCGATTCTTG | GNAS | 34.8/19.8 | 1140.1/1025.1 | 2.40 × 10−59 | 15 |
| CGGGGTGGCCTATCGCCGCC | HDAC4 | 32.46/15.4 | 932.8/847.8 | 1.30 × 10−83 | 16 |
| GAATGAATGTTATCACGGAC | MSANTD3 | 13.3/18.3 | 587.3/590.1 | 3.50 × 10−83 | 17 |
| GGGCCGGGCGCCGGGGATTG | EXTL3 | 4.8/10.8 | 591;1/544.6 | 3.60 × 10−78 | 18 |
| CGCCTTCTCCGGGCGCATCC | BOK | 22.9/16.7 | 592.9/795.2 | 2.40 × 10−51 | 19 |
| AGCAGTTCAACTTCCTATTA | FZD6 | 8.6/15.9 | 479.4/364.5 | 5.10 × 10−77 | 20 |
| AAAGCCGGCGGCGGCGTCCA | ABHD17C | 24.8/35.4 | 890.3/954.7 | 7.00 × 10−59 | 21 |
| AGCGGCCCCTGGGCCAAGCC | ABCC3 | 39.6/36.2 | 1030.9/1233.1 | 3.30 × 10−68 | 22 |
| CGTCGAGGGGGGCAAGCGCC | CERS4 | 19.8/13.7 | 501.1/297.3 | 8.00 × 10−72 | 23 |
| CGGCGTGTCGCGTCAGACGC | POLK | 52.5/35.7 | 893.9/1099.3 | 1.40 × 10−69 | 24 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rocha, C.R.R.; Rocha, A.R.; Silva, M.M.; Gomes, L.R.; Latancia, M.T.; Andrade-Tomaz, M.; de Souza, I.; Monteiro, L.K.S.; Menck, C.F.M. Revealing Temozolomide Resistance Mechanisms via Genome-Wide CRISPR Libraries. Cells 2020, 9, 2573. https://doi.org/10.3390/cells9122573
Rocha CRR, Rocha AR, Silva MM, Gomes LR, Latancia MT, Andrade-Tomaz M, de Souza I, Monteiro LKS, Menck CFM. Revealing Temozolomide Resistance Mechanisms via Genome-Wide CRISPR Libraries. Cells. 2020; 9(12):2573. https://doi.org/10.3390/cells9122573
Chicago/Turabian StyleRocha, Clarissa Ribeiro Reily, Alexandre Reily Rocha, Matheus Molina Silva, Luciana Rodrigues Gomes, Marcela Teatin Latancia, Marina Andrade-Tomaz, Izadora de Souza, Linda Karolynne Seregni Monteiro, and Carlos Frederico Martins Menck. 2020. "Revealing Temozolomide Resistance Mechanisms via Genome-Wide CRISPR Libraries" Cells 9, no. 12: 2573. https://doi.org/10.3390/cells9122573
APA StyleRocha, C. R. R., Rocha, A. R., Silva, M. M., Gomes, L. R., Latancia, M. T., Andrade-Tomaz, M., de Souza, I., Monteiro, L. K. S., & Menck, C. F. M. (2020). Revealing Temozolomide Resistance Mechanisms via Genome-Wide CRISPR Libraries. Cells, 9(12), 2573. https://doi.org/10.3390/cells9122573