Phase I/II Clinical Trial of the Anti-Podoplanin Monoclonal Antibody Therapy in Dogs with Malignant Melanoma

 , ,
, ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Antibody
2.3. Enzyme-Linked Immunosorbent Assay (ELISA)
2.4. Flow Cytometry
2.5. Evaluation of Antibody-Dependent Cellular Cytotoxicity (ADCC)
2.6. P38Bf Injection into a Healthy Dog
2.7. Immunohistochemistry
2.8. Clinical Trial in Dogs with Malignant Tumors
2.9. Statistics
3. Results
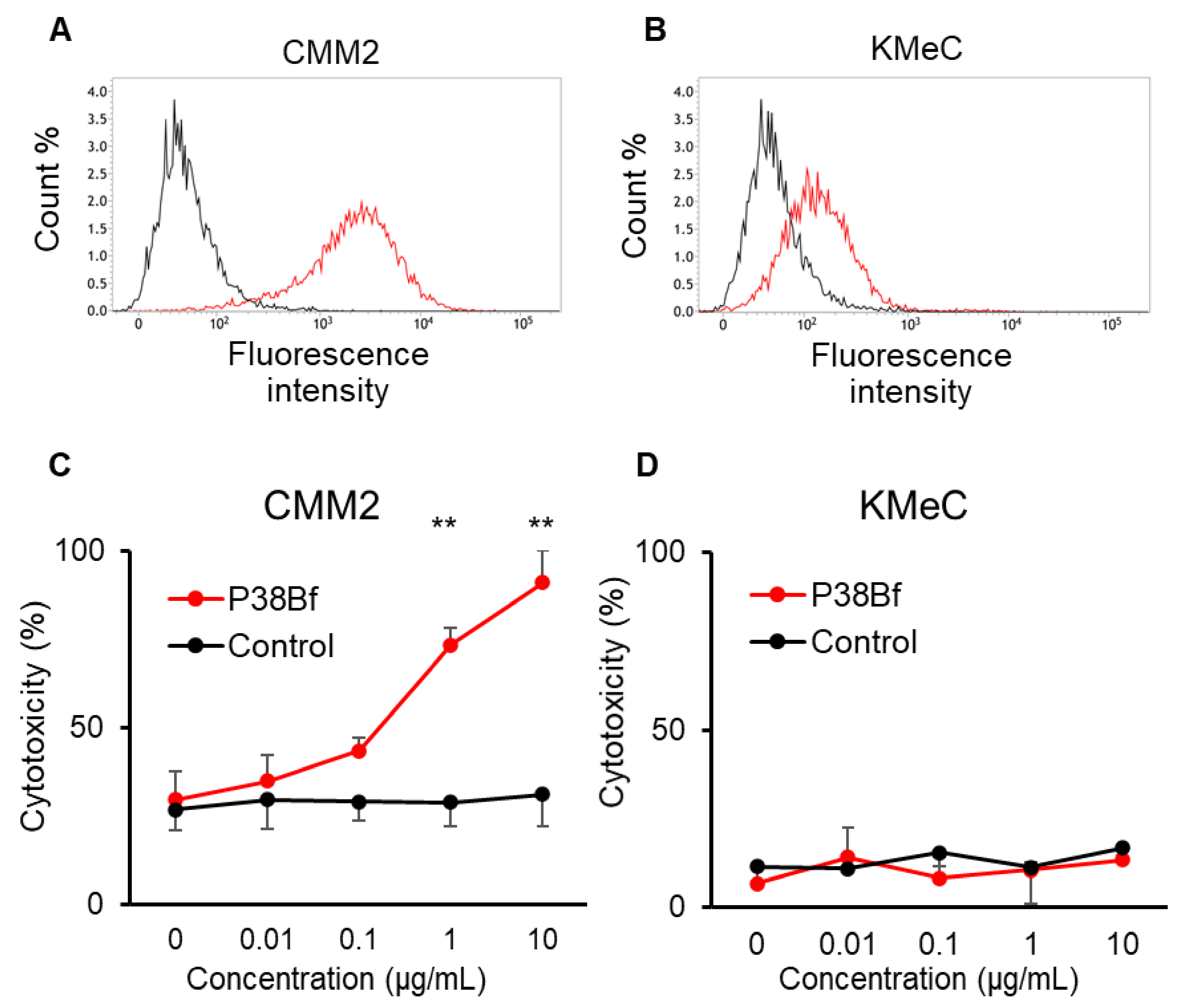
3.1. Antibody-Dependent Cellular Cytotoxicity (ADCC) Induced by a Cancer-Specific Mouse–Dog Chimeric anti-PDPN Antibody (P38Bf) against Canine Melanoma Cells
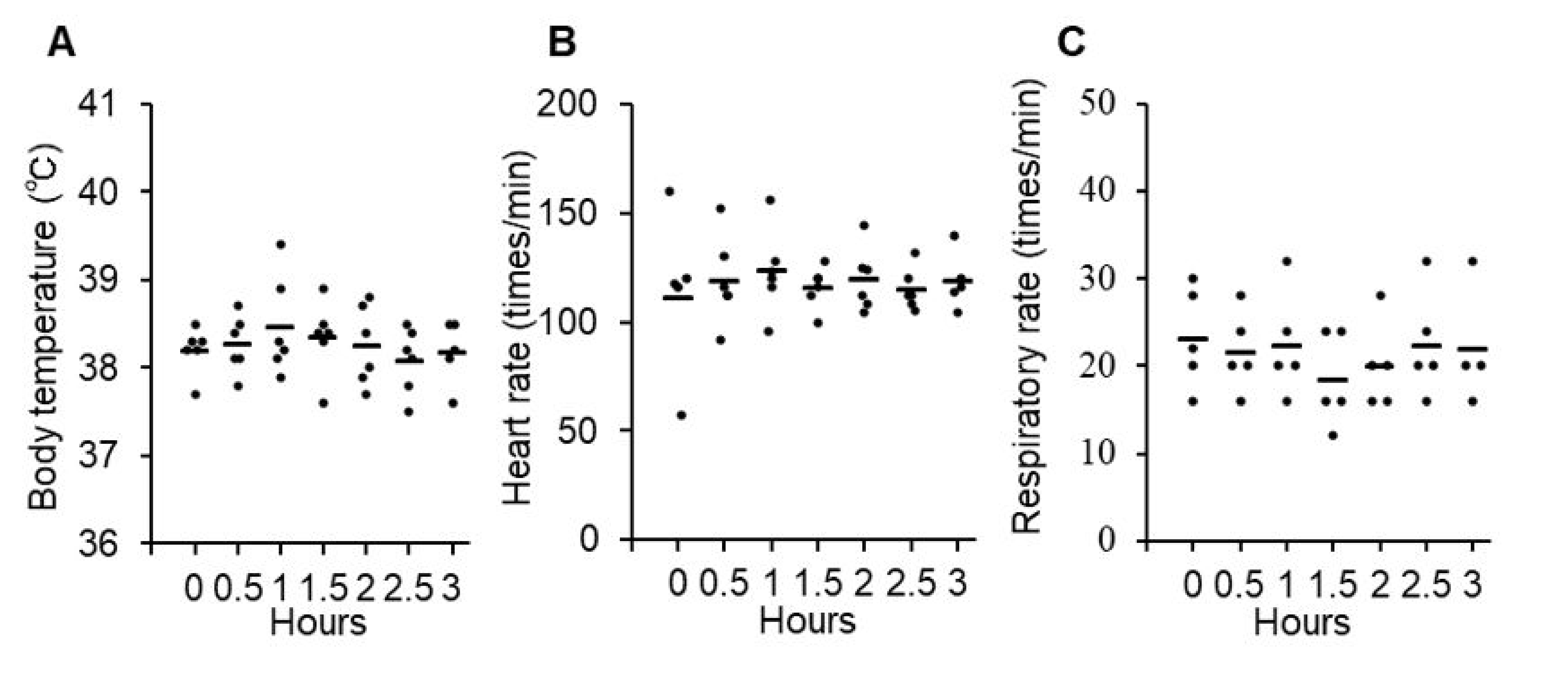
3.2. Safety and Toxicity of P38Bf
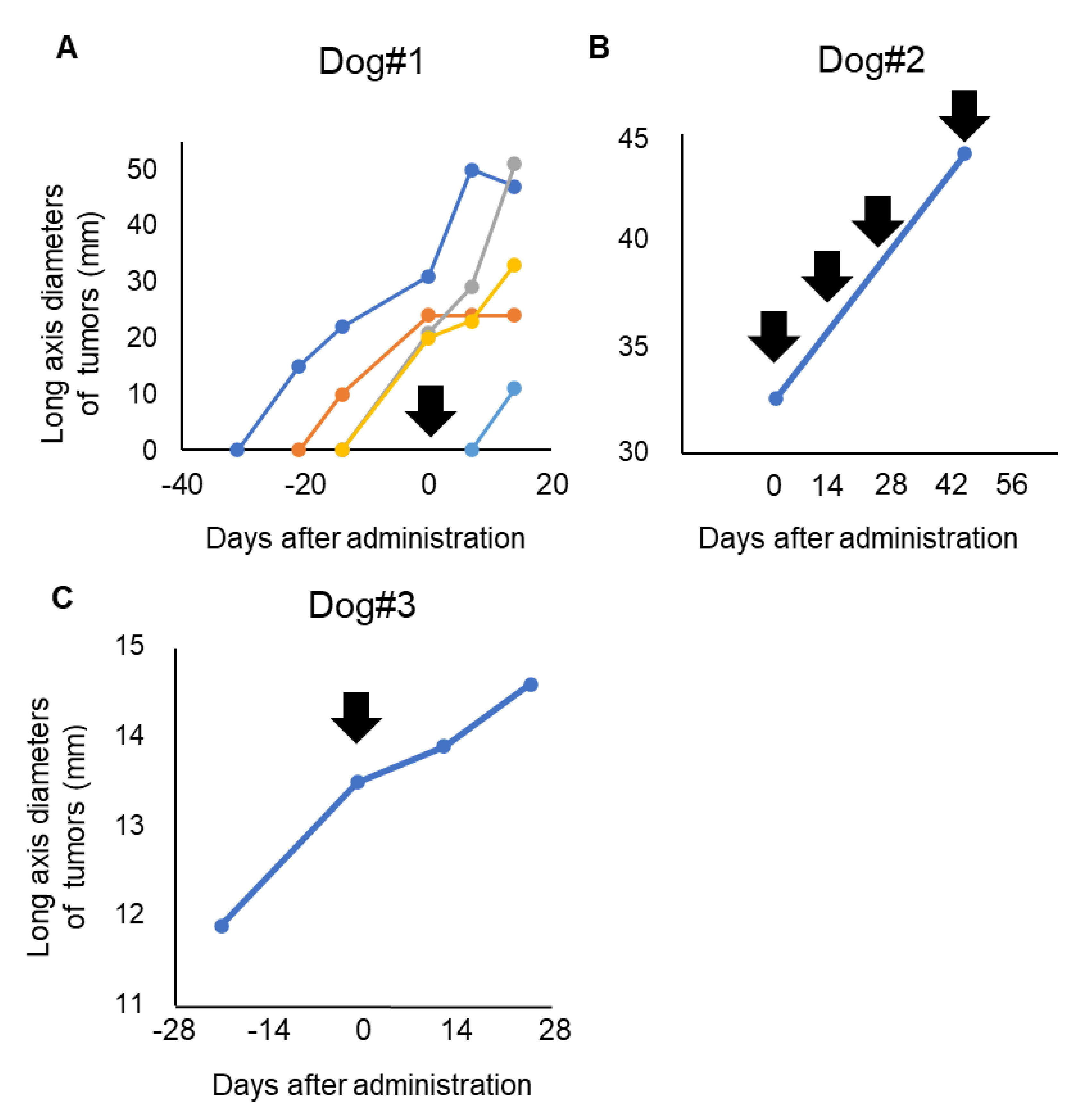
3.3. Clinical Response of P38Bf
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| (PDPN) | podoplanin |
| (CAFs) | cancer-associated fibroblasts |
| (EMT) | epithelial-mesenchymal transition |
| (ADCC) | antibody-dependent cellular cytotoxic |
| (CHO) | chinese hamster ovary |
| (CasMab) | cancer-specific monoclonal antibody |
| (dPDPN) | canine PDPN |
| (mAb) | monoclonal antibody |
| (P38Bf) | a cancer-specific mouse–dog chimeric anti-PDPN antibody |
| (FBS) | fetal bovine serum |
| (PSA) | penicillin, streptomycin, and amphotericin B |
| (CHO/dPDPN) | canine PDPN-expressing CHO-K1 with N-terminal PA tag |
| (IgG) | immunoglobulin G |
| (ELISA) | enzyme-linked immunosorbent assay |
| (FACS) | fluorescence-activated cell sorting |
| (PBS) | phosphate buffered saline |
| (LAK) | lymphokine-activated killer |
| (IL-2) | interleukin-2 |
| (RLU) | relative light units |
| (CBC) | complete blood count |
| (ALB) | albumin |
| (ALP) | alkaline phosphatase |
| (BUN) | blood urea nitrogen |
| (Ca) | calcium |
| (CRE) | creatinine |
| (CRP) | c-reactive protein |
| (Na, K, Cl) | electrolytes |
| (GFR) | glomerular filtration rate |
| (GGT) | γ-glutamyltransferase |
| (GLU) | glucose |
| (GOT) | glutamic oxaloacetic transaminase; glutamic-oxaloacetic transaminase |
| (GPT) | glutamic pyruvic transaminase |
| (TBIL) | total bilirubin |
| (TCHO) | total cholesterol |
| (TG) | triglycerides |
| (TP) | total protein |
| (vLIP) | lipase |
| (iP) | inorganic phosphorus |
| (SpO2) | percutaneous oxygen saturation |
| (CT) | computed tomography |
| (VCOG-CTCAE) | Veterinary Cooperative Oncology Group—Common Terminology Criteria for Adverse Events |
| (TBST) | tris-buffered saline with 0.1% Tween® 20 detergent |
| (VMC) | Veterinary Medical Center |
| (RECIST) | response evaluation criteria for solid tumors |
| (SE) | standard error |
| (CR) | complete response |
| (PR) | partial response |
| (SD) | stable disease |
| (PD) | progressive disease |
| (NK) | natural killer |
| (ADC) | Antibody–drug conjugate |
| (CAR) | chimeric antigen receptor |
References
- Quintanilla, M.; Montero, L.M.; Renart, J.; Villar, E.M. Podoplanin in inflammation and cancer. Int. J. Mol. Sci. 2019, 20, 707. [Google Scholar] [CrossRef]
- Krishnan, H.; Rayes, J.; Miyashita, T.; Ishii, G.; Retzbach, E.P.; Sheehan, S.A.; Takemoto, A.; Chang, Y.W.; Yoneda, K.; Asai, J.; et al. Podoplanin: An emerging cancer biomarker and therapeutic target. Cancer Sci. 2018, 109, 1292–1299. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, G.; Oeffner, F.; Von Messling, V.; Tschernig, T.; Gröne, H.J.; Klenk, H.D.; Herrler, G. Cloning and characterization of gp36, a human mucin-type glycoprotein preferentially expressed in vascular endothelium. Biochem. J. 1999, 341, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Breiteneder-Geleff, S.; Matsui, K.; Soleiman, A.; Meraner, P.; Poczewski, H.; Kalt, R.; Schaffner, G.; Kerjaschki, D. Podoplanin, novel 43-kd membrane protein of glomerular epithelial cells, is down-regulated in puromycin nephrosis. Am. J. Pathol. 1997, 151, 1141–1152. [Google Scholar] [PubMed]
- Martín-Villar, E.; Scholl, F.G.; Gamallo, C.; Yurrita, M.M.; Muñoz-Guerra, M.; Cruces, J.; Quintanilla, M. Characterization of human PA2.26 antigen (T1α-2, podoplanin), a small membrane mucin induced in oral squamous cell carcinomas. Int. J. Cancer 2005, 113, 899–910. [Google Scholar] [CrossRef] [PubMed]
- Mishima, K.; Kato, Y.; Kaneko, M.K.; Nishikawa, R.; Hirose, T.; Matsutani, M. Increased expression of podoplanin in malignant astrocytic tumors as a novel molecular marker of malignant progression. Acta Neuropathol. 2006, 111, 483–488. [Google Scholar] [CrossRef]
- Ordóñez, N.G. D2-40 and podoplanin are highly specific and sensitive immunohistochemical markers of epithelioid malignant mesothelioma. Hum. Pathol. 2005, 36, 372–380. [Google Scholar] [CrossRef]
- Kato, Y.; Kaneko, M.K.; Kuno, A.; Uchiyama, N.; Amano, K.; Chiba, Y.; Hasegawa, Y.; Hirabayashi, J.; Narimatsu, H.; Mishima, K.; et al. Inhibition of tumor cell-induced platelet aggregation using a novel anti-podoplanin antibody reacting with its platelet-aggregation-stimulating domain. Biochem. Biophys. Res. Commun. 2006, 349, 1301–1307. [Google Scholar] [CrossRef]
- Ariizumi, T.; Ogose, A.; Kawashima, H.; Hotta, T.; Li, G.; Xu, Y.; Umezu, H.; Sugai, M.; Endo, N. Expression of podoplanin in human bone and bone tumors: New marker of osteogenic and chondrogenic bone tumors. Pathol. Int. 2010, 60, 193–202. [Google Scholar] [CrossRef]
- Marks, A.; Sutherland, D.R.; Bailey, D.; Iglesias, J.; Law, J.; Lei, M.; Yeger, H.; Banerjee, D.; Baumal, R. Characterization and distribution of an oncofetal antigen (M2A antigen) expressed on testicular germ cell tumours. Br. J. Cancer 1999, 80, 569–578. [Google Scholar] [CrossRef]
- Kitano, H.; Kageyama, S.I.; Hewitt, S.M.; Hayashi, R.; Doki, Y.; Ozaki, Y.; Fujino, S.; Takikita, M.; Kubo, H.; Fukuoka, J. Podoplanin expression in cancerous stroma induces lymphangiogenesis and predicts lymphatic spread and patient survival. Arch. Pathol. Lab. Med. 2010, 134, 1520–1527. [Google Scholar] [CrossRef] [PubMed]
- Schoppmann, S.F.; Berghoff, A.; Dinhof, C.; Jakesz, R.; Gnant, M.; Dubsky, P.; Jesch, B.; Heinzl, H.; Birner, P. Podoplanin-expressing cancer-associated fibroblasts are associated with poor prognosis in invasive breast cancer. Breast Cancer Res. Treat. 2012, 134, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Kan, S.; Konishi, E.; Arita, T.; Ikemoto, C.; Takenaka, H.; Yanagisawa, A.; Katoh, N.; Asai, J. Podoplanin expression in cancer-associated fibroblasts predicts aggressive behavior in melanoma. J. Cutan. Pathol. 2014, 41, 561–567. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Itai, S.; Kaneko, M.K.; Kato, Y. PMab-48 Recognizes Dog Podoplanin of Lymphatic Endothelial Cells. Monoclon. Antib. Immunodiagn. Immunother. 2018, 37, 63–66. [Google Scholar] [CrossRef] [PubMed]
- Honma, R.; Kaneko, M.K.; Ogasawara, S.; Fujii, Y.; Konnai, S.; Takagi, M.; Kato, Y. Specific Detection of Dog Podoplanin Expressed in Renal Glomerulus by a Novel Monoclonal Antibody PMab-38 in Immunohistochemistry. Monoclon. Antib. Immunodiagn. Immunother. 2016, 35, 212–216. [Google Scholar] [CrossRef]
- Kaneko, M.K.; Honma, R.; Ogasawara, S.; Fujii, Y.; Nakamura, T.; Saidoh, N.; Takagi, M.; Kagawa, Y.; Konnai, S.; Kato, Y. PMab-38 Recognizes Canine Podoplanin of Squamous Cell Carcinomas. Monoclon. Antib. Immunodiagn. Immunother. 2016, 35, 263–266. [Google Scholar] [CrossRef]
- Kiname, K.; Yoshimoto, S.; Kato, D.; Tsuboi, M.; Tanaka, Y.; Yoshitake, R.; Eto, S.; Shinada, M.; Chambers, J.; Saeki, K.; et al. Evaluation of immunohistochemical staining with PMab-38, an anti-dog podoplanin monoclonal antibody, in various canine tumor tissues. Jpn. J. Vet. Res. 2019, 67, 25–34. [Google Scholar] [CrossRef]
- Shinada, M.; Kato, D.; Kamoto, S.; Yoshimoto, S.; Tsuboi, M.; Yoshitake, R.; Eto, S.; Ikeda, N.; Saeki, K.; Hashimoto, Y.; et al. PDPN Is Expressed in Various Types of Canine Tumors and Its Silencing Induces Apoptosis and Cell Cycle Arrest in Canine Malignant Melanoma. Cells 2020, 9, 1136. [Google Scholar] [CrossRef]
- Martín-Villar, E.; Megías, D.; Castel, S.; Yurrita, M.M.; Vilaró, S.; Quintanilla, M. Podoplanin binds ERM proteins to activate RhoA and promote epithelial-mesenchymal transition. J. Cell Sci. 2006, 119, 4541–4553. [Google Scholar] [CrossRef]
- Wicki, A.; Lehembre, F.; Wick, N.; Hantusch, B.; Kerjaschki, D.; Christofori, G. Tumor invasion in the absence of epithelial-mesenchymal transition: Podoplanin-mediated remodeling of the actin cytoskeleton. Cancer Cell 2006, 9, 261–272. [Google Scholar] [CrossRef]
- Miyata, K.; Takemoto, A.; Okumura, S.; Nishio, M.; Fujita, N. Podoplanin enhances lung cancer cell growth in vivo by inducing platelet aggregation. Sci. Rep. 2017, 7, 4059. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Zhang, L.; Yu, J.; Zhang, Y.; Pang, X.; Ma, C.; Shen, M.; Ruan, S.; Wasan, H.S.; Qiu, S. Clinical efficacy and safety of anti-PD-1/PD-L1 inhibitors for the treatment of advanced or metastatic cancer: A systematic review and meta-analysis. Sci. Rep. 2020, 10, 2083. [Google Scholar] [CrossRef] [PubMed]
- Dougan, M.; Dranoff, G.; Dougan, S.K. Cancer immunotherapy: Beyond checkpoint blockade. Annu. Rev. Cancer Biol. 2019, 3, 55–75. [Google Scholar] [CrossRef]
- Lu, R.M.; Hwang, Y.C.; Liu, I.J.; Lee, C.C.; Tsai, H.Z.; Li, H.J.; Wu, H.C. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 2020, 27, 1. [Google Scholar] [CrossRef]
- Esfahani, K.; Roudaia, L.; Buhlaiga, N.; Del Rincon, S.V.; Papneja, N.; Miller, W.H. A review of cancer immunotherapy: From the past, to the present, to the future. Curr. Oncol. 2020, 27, 87–97. [Google Scholar] [CrossRef]
- Lee, S.; Lee, H.; Kim, E.Y. Comparative Efficacy and Safety of Biosimilar Rituximab and Originator Rituximab in Rheumatoid Arthritis and Non-Hodgkin’s Lymphoma: A Systematic Review and Meta-analysis. BioDrugs 2019, 33, 469–483. [Google Scholar] [CrossRef] [PubMed]
- Pondé, N.; Aftimos, P.; Piccart, M. Antibody-Drug Conjugates in Breast Cancer: A Comprehensive Review. Curr. Treat. Options Oncol. 2019, 20, 37. [Google Scholar] [CrossRef]
- Takagi, S.; Sato, S.; Oh-hara, T.; Takami, M.; Koike, S.; Mishima, Y.; Hatake, K.; Fujita, N. Platelets Promote Tumor Growth and Metastasis via Direct Interaction between Aggrus/Podoplanin and CLEC-2. PLoS ONE 2013, 8, e73609. [Google Scholar] [CrossRef]
- Xu, M.; Wang, X.; Pan, Y.; Zhao, X.; Yan, B.; Ruan, C.; Xia, L.; Zhao, Y. Blocking podoplanin suppresses growth and pulmonary metastasis of human malignant melanoma. BMC Cancer 2019, 19, 599. [Google Scholar] [CrossRef]
- Abe, S.; Kaneko, M.K.; Tsuchihashi, Y.; Izumi, T.; Ogasawara, S.; Okada, N.; Sato, C.; Tobiume, M.; Otsuka, K.; Miyamoto, L.; et al. Antitumor effect of novel anti-podoplanin antibody NZ-12 against malignant pleural mesothelioma in an orthotopic xenograft model. Cancer Sci. 2016, 107, 1198–1205. [Google Scholar] [CrossRef]
- Kato, Y.; Kaneko, M.K. A cancer-specific monoclonal antibody recognizes the aberrantly glycosylated podoplanin. Sci. Rep. 2014, 4, 5924. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, M.K.; Yamada, S.; Nakamura, T.; Abe, S.; Nishioka, Y.; Kunita, A.; Fukayama, M.; Fujii, Y.; Ogasawara, S.; Kato, Y. Antitumor activity of chLpMab-2, a human–mouse chimeric cancer-specific antihuman podoplanin antibody, via antibody-dependent cellular cytotoxicity. Cancer Med. 2017, 6, 768–777. [Google Scholar] [CrossRef] [PubMed]
- Thamm, D.H. Canine Cancer: Strategies in Experimental Therapeutics. Front. Oncol. 2019, 9, 1257. [Google Scholar] [CrossRef] [PubMed]
- Pinho, S.S.; Carvalho, S.; Cabral, J.; Reis, C.A.; Gärtner, F. Canine tumors: A spontaneous animal model of human carcinogenesis. Transl. Res. 2012, 159, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Rowell, J.L.; McCarthy, D.O.; Alvarez, C.E. Dog models of naturally occurring cancer. Trends Mol. Med. 2011, 17, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Mizuno, T.; Yamada, S.; Nakamura, T.; Itai, S.; Yanaka, M.; Sano, M.; Kaneko, M.K. Establishment of P38Bf, a Core-Fucose-Deficient Mouse-Canine Chimeric Antibody Against Dog Podoplanin. Monoclon. Antib. Immunodiagn. Immunother. 2018, 37, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Ohishi, T.; Kawada, M.; Maekawa, N.; Konnai, S.; Itai, S.; Yamada, S.; Kaneko, M.K. The mouse–canine chimeric anti-dog podoplanin antibody P38B exerts antitumor activity in mouse xenograft models. Biochem. Biophys. Rep. 2019, 17, 23–26. [Google Scholar] [CrossRef]
- Yoshitake, R.; Saeki, K.; Watanabe, M.; Nakaoka, N.; Ong, S.M.; Hanafusa, M.; Choisunirachon, N.; Fujita, N.; Nishimura, R.; Nakagawa, T. Molecular investigation of the direct anti-tumour effects of nonsteroidal anti-inflammatory drugs in a panel of canine cancer cell lines. Vet. J. 2017, 221, 38–47. [Google Scholar] [CrossRef]
- Uchida, M.; Saeki, K.; Maeda, S.; Tamahara, S.; Yonezawa, T.; Matsuki, N. Apoptosis inhibitor of macrophage (AIM) reduces cell number in canine histiocytic sarcoma cell lines. J. Vet. Med. Sci. 2016, 78, 1515–1520. [Google Scholar] [CrossRef]
- Ohashi, E.; Hong, S.H.; Takahashi, T.; Nakagawa, T.; Mochizuki, M.; Nishimura, R.; Sasaki, N. Effect of Retinoids on Growth Inhibition of Two Canine Melanoma Cell Lines. J. Vet. Med. Sci. 2001, 63, 83–86. [Google Scholar] [CrossRef][Green Version]
- Inoue, K.; Ohashi, E.; Kadosawa, T.; Hong, S.H.; Matsunaga, S.; Mochizuki, M.; Nishimura, R.; Sasaki, N. Establishment and characterization of four canine melanoma cell lines. J. Vet. Med. Sci. 2004, 66, 1437–1440. [Google Scholar] [CrossRef] [PubMed]
- Fujii, Y.; Kaneko, M.; Neyazaki, M.; Nogi, T.; Kato, Y.; Takagi, J. PA tag: A versatile protein tagging system using a super high affinity antibody against a dodecapeptide derived from human podoplanin. Protein Expr. Purif. 2014, 95, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Reference, Q. Veterinary cooperative oncology group—Common terminology criteria for adverse events (VCOG-CTCAE) following chemotherapy or biological antineoplastic therapy in dogs and cats v1.1. Vet. Comp. Oncol. 2016, 14, 417–446. [Google Scholar] [CrossRef]
- Nguyen, S.M.; Thamm, D.H.; Vail, D.M.; London, C.A. Response evaluation criteria for solid tumours in dogs (v1.0): A Veterinary Cooperative Oncology Group (VCOG) consensus document. Vet. Comp. Oncol. 2015, 13, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Ogasawara, S.; Honma, R.; Kaneko, M.K.; Fujii, Y.; Kagawa, Y.; Konnai, S.; Kato, Y. Podoplanin Expression in Canine Melanoma. Monoclon. Antib. Immunodiagn. Immunother. 2016, 35, 304–306. [Google Scholar] [CrossRef]
- Abe, S.; Morita, Y.; Kaneko, M.K.; Hanibuchi, M.; Tsujimoto, Y.; Goto, H.; Kakiuchi, S.; Aono, Y.; Huang, J.; Sato, S.; et al. A Novel Targeting Therapy of Malignant Mesothelioma Using Anti-Podoplanin Antibody. J. Immunol. 2013, 190, 6239–6249. [Google Scholar] [CrossRef]
- Kato, Y.; Kunita, A.; Abe, S.; Ogasawara, S.; Fujii, Y.; Oki, H.; Fukayama, M.; Nishioka, Y.; Kaneko, M.K. The chimeric antibody chLpMab-7 targeting human podoplanin suppresses pulmonary metastasis via ADCC and CDC rather than via its neutralizing activity. Oncotarget 2015, 6, 36003–36018. [Google Scholar] [CrossRef]
- Kaneko, M.K.; Abe, S.; Ogasawara, S.; Fujii, Y.; Yamada, S.; Murata, T.; Uchida, H.; Tahara, H.; Nishioka, Y.; Kato, Y. Chimeric Anti-Human Podoplanin Antibody NZ-12 of Lambda Light Chain Exerts Higher Antibody-Dependent Cellular Cytotoxicity and Complement-Dependent Cytotoxicity Compared with NZ-8 of Kappa Light Chain. Monoclon. Antib. Immunodiagn. Immunother. 2017, 36, 25–29. [Google Scholar] [CrossRef]
- Chames, P.; Van Regenmortel, M.; Weiss, E.; Baty, D. Therapeutic antibodies: Successes, limitations and hopes for the future. Br. J. Pharmacol. 2009, 157, 220–233. [Google Scholar] [CrossRef]
- Guan, M.; Zhou, Y.P.; Sun, J.L.; Chen, S.C. Adverse Events of Monoclonal Antibodies Used for Cancer Therapy. BioMed Res. Int. 2015, 2015. [Google Scholar] [CrossRef]
- Gharwan, H.; Groninger, H. Kinase inhibitors and monoclonal antibodies in oncology: Clinical implications. Nat. Rev. Clin. Oncol. 2016, 13, 209–227. [Google Scholar] [CrossRef] [PubMed]
- Roselló, S.; Blasco, I.; Garća Fabregat, L.; Cervantes, A.; Jordan, K. Management of infusion reactions to systemic anticancer therapy: ESMO Clinical Practice Guidelines. Ann. Oncol. 2017, 28, iv100–iv118. [Google Scholar] [CrossRef] [PubMed]
- Brain, A.B. Adverse events to monoclonal antibodies used for cancer therapy. Focus on hypersensitivity responses. Oncoimmunology 2013, 10, e26333. [Google Scholar] [CrossRef]
- Tarazona, R.; Duran, E.; Solana, R. Natural killer cell recognition of melanoma: New clues for a more effective immunotherapy. Front. Immunol. 2016, 6, 649. [Google Scholar] [CrossRef] [PubMed]
- Nishiya, A.T.; Massoco, C.O.; Felizzola, C.R.; Perlmann, E.; Batschinski, K.; Tedardi, M.V.; Garcia, J.S.; Mendonça, P.P.; Teixeira, T.F.; Dagli, M.L.Z. Comparative aspects of canine melanoma. Vet. Sci. 2016, 3, 7. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, B.; Adissu, H.A.; Wei, B.R.; Michael, H.T.; Merlino, G.; Mark Simpson, R. Naturally occurring canine melanoma as a predictive comparative oncology model for human mucosal and other triple wild-type melanomas. Int. J. Mol. Sci. 2018, 19, 394. [Google Scholar] [CrossRef]
- Prouteau, A.; André, C. Canine melanomas as models for human melanomas: Clinical, histological, and genetic comparison. Genes 2019, 10, 501. [Google Scholar] [CrossRef]
- Simpson, R.M.; Bastian, B.C.; Michael, H.T.; Webster, J.D.; Prasad, M.L.; Conway, C.M.; Prieto, V.M.; Gary, J.M.; Goldschmidt, M.H.; Esplin, D.G.; et al. Sporadic naturally occurring melanoma in dogs as a preclinical model for human melanoma. Pigment Cell Melanoma Res. 2014, 27, 37–47. [Google Scholar] [CrossRef]
- Goldenberg, M.M. Trastuzumab, a recombinant DNA-derived humanized monoclonal antibody, a novel agent for the treatment of metastatic breast cancer. Clin. Ther. 1999, 21, 309–318. [Google Scholar] [CrossRef]
- Vogel, C.L.; Cobleigh, M.A.; Tripathy, D.; Gutheil, J.C.; Harris, L.N.; Fehrenbacher, L.; Slamon, D.J.; Murphy, M.; Novotny, W.F.; Burchmore, M.; et al. Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J. Clin. Oncol. 2002, 20, 719–726. [Google Scholar] [CrossRef]
- Smith, E. Efficacy and safety of Herceptin in women with metastatic breast cancer: Results from pivotal clinical studies. Anticancer Drugs 2001, 12 (Suppl. 4), S3–S10. [Google Scholar] [CrossRef] [PubMed]
- Sproston, N.R.; Ashworth, J.J. Role of C-reactive protein at sites of inflammation and infection. Front. Immunol. 2018, 9, 754. [Google Scholar] [CrossRef] [PubMed]
- Abolhassani, A.R.; Schuler, G.; Kirchberger, M.C.; Heinzerling, L. C-reactive protein as an early marker of immune-related adverse events. J. Cancer Res. Clin. Oncol. 2019, 145, 2625–2631. [Google Scholar] [CrossRef] [PubMed]
- Le Guenno, G.; Ruivard, M.; Charra, L.; Philippe, P. Rituximab-induced serum sickness in refractory immune thrombocytopenic purpura. Intern. Med. J. 2011, 41, 202–205. [Google Scholar] [CrossRef] [PubMed]
- Maker, J.H.; Stroup, C.M.; Huang, V.; James, S.F. Antibiotic Hypersensitivity Mechanisms. Pharmacy 2019, 7, 122. [Google Scholar] [CrossRef]
- Watanabe, M.; Sugimoto, Y.; Tsuruo, T. Expression of a Mr 41,000 Glycoprotein Associated with Thrombin-independent Platelet Aggregation in High Metastatic Variants of Murine B16 Melanoma. Cancer Res. 1990, 50, 6657–6662. [Google Scholar]
- Manuscript, A.; Proximity, I. T-Cell and NK-Cell Infiltration into Solid Tumors: A Key Limiting Factor for Efficacious Cancer Immunotherapy Ignacio. Cancer Discov. 2011, 4, 522–526. [Google Scholar] [CrossRef]
- Ben-Shmuel, A.; Biber, G.; Barda-Saad, M. Unleashing Natural Killer Cells in the Tumor Microenvironment—The Next Generation of Immunotherapy? Front. Immunol. 2020, 11, 275. [Google Scholar] [CrossRef]
- Horiuchi, Y.; Tominaga, M.; Ichikawa, M.; Yamashita, M.; Okano, K.; Jikumaru, Y.; Nariai, Y.; Nakajima, Y.; Kuwabara, M.; Yukawa, M. Relationship between regulatory and type 1 T cells in dogs with oral malignant melanoma. Microbiol. Immunol. 2010, 54, 152–159. [Google Scholar] [CrossRef]
- Casi, G.; Neri, D. Antibody-drug conjugates: Basic concepts, examples and future perspectives. J. Control. Release 2012, 161, 422–428. [Google Scholar] [CrossRef]
- Lewis Phillips, G.D.; Li, G.; Dugger, D.L.; Crocker, L.M.; Parsons, K.L.; Mai, E.; Blättler, W.A.; Lambert, J.M.; Chari, R.V.J.; Lutz, R.J.; et al. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res. 2008, 68, 9280–9290. [Google Scholar] [CrossRef] [PubMed]
- Sadelain, M.; Brentjens, R.; Riviere, I. The basic principles of chimeric antigen receptor (CAR) design. October 2008, 141, 520–529. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Dudley, M.E.; Kassim, S.H.; Somerville, R.P.T.; Carpenter, R.O.; Maryalice, S.S.; Yang, J.C.; Phan, G.Q.; Hughes, M.S.; Sherry, R.M.; et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J. Clin. Oncol. 2015, 33, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.M.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [PubMed]
- Shiina, S.; Ohno, M.; Ohka, F.; Kuramitsu, S.; Yamamichi, A.; Kato, A.; Motomura, K.; Tanahashi, K.; Yamamoto, T.; Watanabe, R.; et al. CAR T cells targeting Podoplanin reduce orthotopic glioblastomas in mouse brains. Cancer Immunol. Res. 2016, 4, 259–268. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Case # | Breed | Age (Years) | Sex | Body Weight (kg) | Diagnosis | Primary Site | WHO Stage | Prior Therapy |
|---|---|---|---|---|---|---|---|---|
| 1 | Mix | 14.7 | Male, castrated | 15.5 | Malignant melanoma | Tongue | IV | Surgery, radiation, chemotherapy |
| 2 | Miniature dachshund | 13.7 | Female, spayed | 4.9 | Malignant melanoma | Hard plate | III | Surgery |
| 3 | Beagle | 14.8 | Male | 11 | Malignant melanoma (amelanotic) | Gingiva | I | Surgery |
| Events | Number of Cases | |||||
|---|---|---|---|---|---|---|
| Grade 1 | Grade 2 | Grade 3 | Grade 4 | Grade 5 | Total | |
| General disorders | ||||||
| Vomiting | 0 | 1 | 0 | 0 | 0 | 1 |
| Biochemical parameters | ||||||
| Increased CRP | 3 | 0 | 0 | 0 | 0 | 3 |
| Case # | Dosage | Times Given P38Bf | Treatment Duration | Best overall Response |
|---|---|---|---|---|
| 1 | 2 mg/kg | 1 | 2 weeks | PD * |
| 2 | 2 mg/kg | 4 | 9 weeks | PD |
| 3 | 2 mg/kg | 1 | 2 weeks | SD |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kamoto, S.; Shinada, M.; Kato, D.; Yoshimoto, S.; Ikeda, N.; Tsuboi, M.; Yoshitake, R.; Eto, S.; Hashimoto, Y.; Takahashi, Y.; et al. Phase I/II Clinical Trial of the Anti-Podoplanin Monoclonal Antibody Therapy in Dogs with Malignant Melanoma. Cells 2020, 9, 2529. https://doi.org/10.3390/cells9112529
Kamoto S, Shinada M, Kato D, Yoshimoto S, Ikeda N, Tsuboi M, Yoshitake R, Eto S, Hashimoto Y, Takahashi Y, et al. Phase I/II Clinical Trial of the Anti-Podoplanin Monoclonal Antibody Therapy in Dogs with Malignant Melanoma. Cells. 2020; 9(11):2529. https://doi.org/10.3390/cells9112529
Chicago/Turabian StyleKamoto, Satoshi, Masahiro Shinada, Daiki Kato, Sho Yoshimoto, Namiko Ikeda, Masaya Tsuboi, Ryohei Yoshitake, Shotaro Eto, Yuko Hashimoto, Yosuke Takahashi, and et al. 2020. "Phase I/II Clinical Trial of the Anti-Podoplanin Monoclonal Antibody Therapy in Dogs with Malignant Melanoma" Cells 9, no. 11: 2529. https://doi.org/10.3390/cells9112529
APA StyleKamoto, S., Shinada, M., Kato, D., Yoshimoto, S., Ikeda, N., Tsuboi, M., Yoshitake, R., Eto, S., Hashimoto, Y., Takahashi, Y., Chambers, J., Uchida, K., Kaneko, M. K., Fujita, N., Nishimura, R., Kato, Y., & Nakagawa, T. (2020). Phase I/II Clinical Trial of the Anti-Podoplanin Monoclonal Antibody Therapy in Dogs with Malignant Melanoma. Cells, 9(11), 2529. https://doi.org/10.3390/cells9112529