SIRT3 Deficiency Sensitizes Angiotensin-II-Induced Renal Fibrosis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
2.1. Experimental Animal Model and Treatment
2.2. Histological and Immunofluorescence Analysis
2.3. Western Blot Analysis
2.4. Endothelial Cell Glycolysis Analysis
2.5. Statistical Analysis
3. Results
3.1. Deficiency of SIRT3 Enhanced Ang-II-Induced Renal Fibrosis
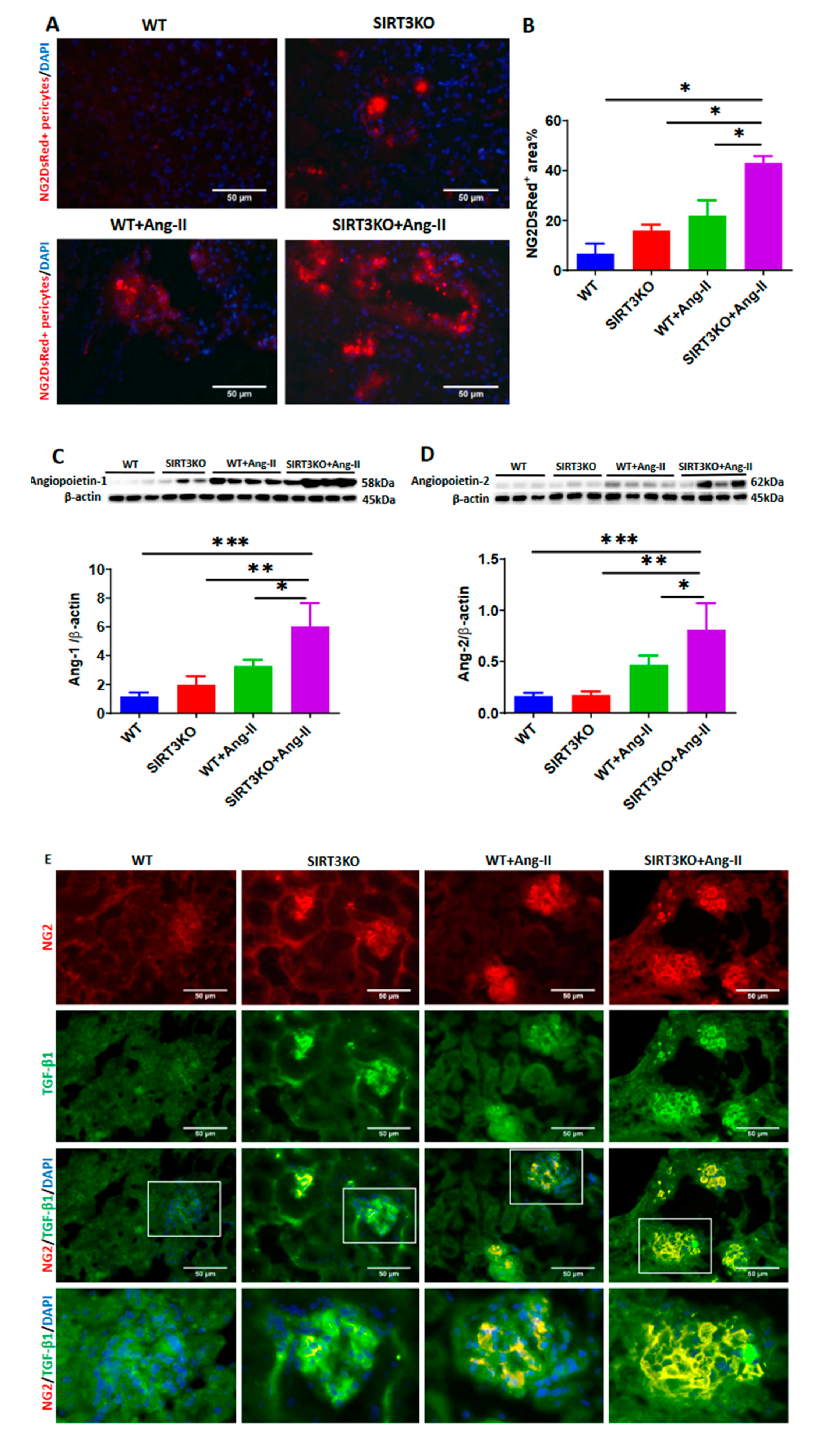
3.2. Deficiency of SIRT3 Promoted Ang-II-Induced Recruitment and Differentiation of Pericytes
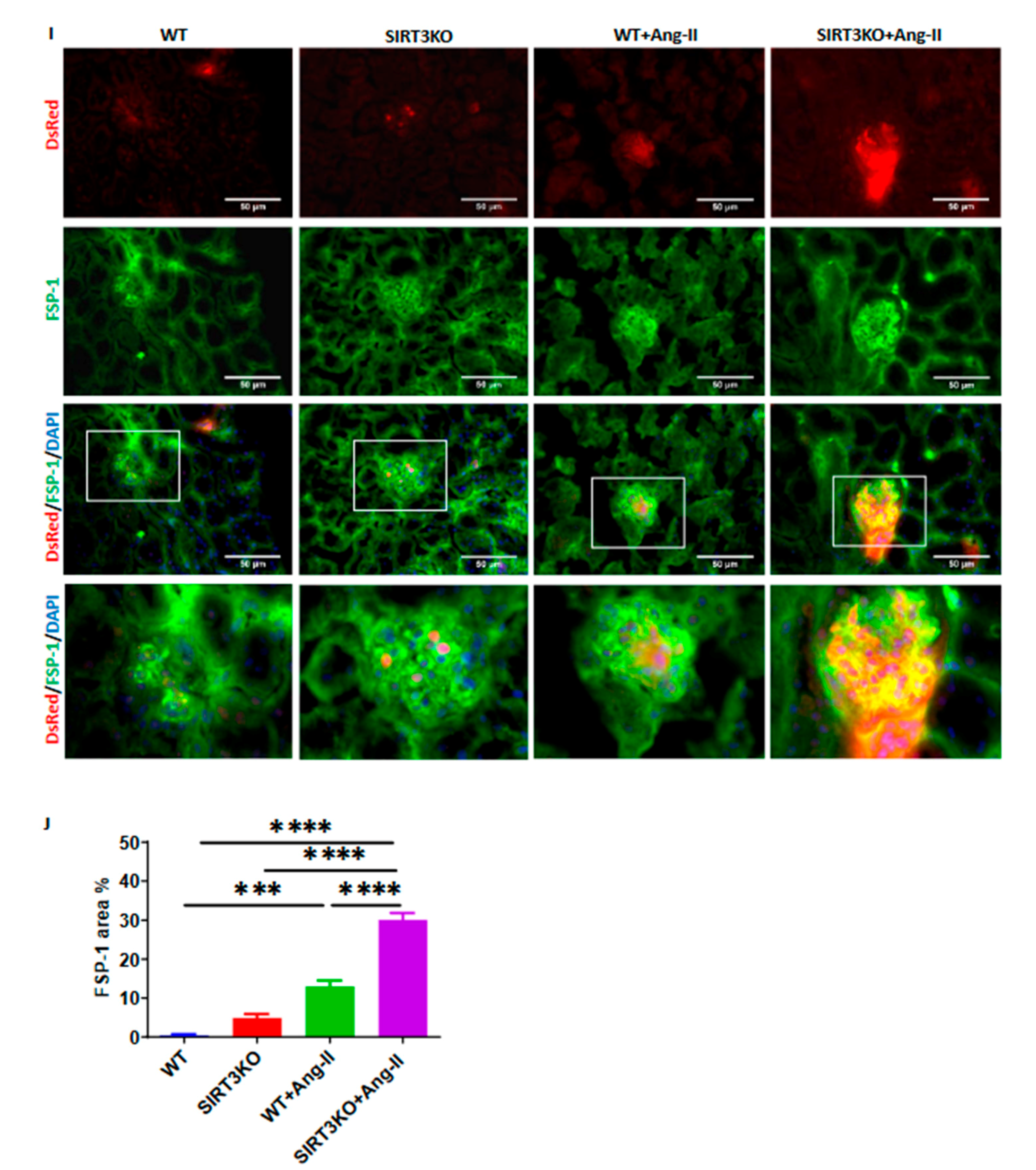
3.3. Deficiency of SIRT3 Exacerbated Ang-II-Induced Iron Overload
3.4. Deficiency of SIRT3 Accelerated Ang-II-Induced Endothelial Dysfunction
4. Discussion
Author Contributions
Funding
Conflicts of Interest
Abbreviation
| Ang-II | angiotensin-II |
| DHE | dihydroethidium |
| FSP-1 | fibroblast-specific protein 1 |
| GLUT1 | glucose transporter 1 |
| HO-1 | heme oxygenase 1 |
| NAD | nicotinamide adenine dinucleotide |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| NG2 | neural/glial antigen 2 |
| PFKFB3 | 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 |
| ROS | reactive oxygen species |
| SIRT3 | sirtuin 3 |
| SIRT3KO | SIRT3 knockout |
| TGF-β1 | transforming growth factor-β1 |
| WT | wild type |
References
- Xia, Y.; Entman, M.L.; Wang, Y. Critical role of CXCL16 in hypertensive kidney injury and fibrosis. Hypertension 2013, 62, 1129–1137. [Google Scholar] [CrossRef] [PubMed]
- Nair, A.R.; Ebenezer, P.J.; Saini, Y.; Francis, J. Angiotensin II-induced hypertensive renal inflammation is mediated through HMGB1-TLR4 signaling in rat tubulo-epithelial cells. Exp. Cell Res. 2015, 335, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Burlaka, I.; Nilsson, L.M.; Scott, L.; Holtback, U.; Eklof, A.C.; Fogo, A.B.; Brismar, H.; Aperia, A. Prevention of apoptosis averts glomerular tubular disconnection and podocyte loss in proteinuric kidney disease. Kidney Int. 2016, 90, 135–148. [Google Scholar] [CrossRef] [PubMed]
- Higgins, C.E.; Tang, J.; Mian, B.M.; Higgins, S.P.; Gifford, C.C.; Conti, D.J.; Meldrum, K.K.; Samarakoon, R.; Higgins, P.J. TGF-beta1-p53 cooperativity regulates a profibrotic genomic program in the kidney: Molecular mechanisms and clinical implications. FASEB J. 2019, 33, 10596–10606. [Google Scholar] [CrossRef]
- Han, W.Q.; Xu, L.; Tang, X.F.; Chen, W.D.; Wu, Y.J.; Gao, P.J. Membrane rafts-redox signalling pathway contributes to renal fibrosis via modulation of the renal tubular epithelial-mesenchymal transition. J. Physiol. 2018, 596, 3603–3616. [Google Scholar] [CrossRef]
- Wang, S.; Zeng, H.; Chen, S.T.; Zhou, L.; Xie, X.J.; He, X.; Tao, Y.K.; Tuo, Q.H.; Deng, C.; Liao, D.F.; et al. Ablation of endothelial prolyl hydroxylase domain protein-2 promotes renal vascular remodelling and fibrosis in mice. J. Cell. Mol. Med. 2017, 21, 1967–1978. [Google Scholar] [CrossRef]
- Schrimpf, C.; Duffield, J.S. Mechanisms of fibrosis: The role of the pericyte. Curr. Opin. Nephrol. Hypertens. 2011, 20, 297–305. [Google Scholar] [CrossRef]
- Peppiatt-Wildman, C.M. The evolving role of renal pericytes. Curr. Opin. Nephrol. Hypertens. 2013, 22, 10–16. [Google Scholar] [CrossRef]
- Cavdar, Z.; Oktan, M.A.; Ural, C.; Calisir, M.; Kocak, A.; Heybeli, C.; Yildiz, S.; Arici, A.; Ellidokuz, H.; Celik, A.; et al. Renoprotective Effects of Alpha Lipoic Acid on Iron Overload-Induced Kidney Injury in Rats by Suppressing NADPH Oxidase 4 and p38 MAPK Signaling. Biol. Trace Elem. Res. 2020, 193, 483–493. [Google Scholar] [CrossRef]
- Macdougall, I.C.; Bircher, A.J.; Eckardt, K.U.; Obrador, G.T.; Pollock, C.A.; Stenvinkel, P.; Swinkels, D.W.; Wanner, C.; Weiss, G.; Chertow, G.M.; et al. Iron management in chronic kidney disease: Conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) Controversies Conference. Kidney Int. 2016, 89, 28–39. [Google Scholar] [CrossRef]
- Mennuni, S.; Rubattu, S.; Pierelli, G.; Tocci, G.; Fofi, C.; Volpe, M. Hypertension and kidneys: Unraveling complex molecular mechanisms underlying hypertensive renal damage. J. Hum. Hypertens. 2014, 28, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.R.; Zheng, Y.J.; Zhang, Z.B.; Shen, W.L.; Li, X.D.; Wei, T.; Ruan, C.C.; Chen, X.H.; Zhu, D.L.; Gao, P.J. Suppression of Endothelial-to-Mesenchymal Transition by SIRT (Sirtuin) 3 Alleviated the Development of Hypertensive Renal Injury. Hypertension 2018, 72, 350–360. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Li, Z.; Yue, Z.; Gao, H.; Feng, G.; Wang, P.; Huang, Y.; Luo, W.; Hong, H.; Liang, L.; et al. SIRT3 prevents angiotensin II-induced renal tubular epithelial-mesenchymal transition by ameliorating oxidative stress and mitochondrial dysfunction. Mol. Cell. Endocrinol. 2018, 460, 1–13. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Zeng, H.; Chen, J.X. Ablation of SIRT3 causes coronary microvascular dysfunction and impairs cardiac recovery post myocardial ischemia. Int. J. Cardiol. 2016, 215, 349–357. [Google Scholar] [CrossRef]
- Zeng, H.; Vaka, V.R.; He, X.; Booz, G.W.; Chen, J.X. High-fat diet induces cardiac remodelling and dysfunction: Assessment of the role played by SIRT3 loss. J. Cell. Mol. Med. 2015, 19, 1847–1856. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Zeng, H.; Chen, S.T.; Roman, R.J.; Aschner, J.L.; Didion, S.; Chen, J.X. Endothelial specific SIRT3 deletion impairs glycolysis and angiogenesis and causes diastolic dysfunction. J. Mol. Cell. Cardiol. 2017, 112, 104–113. [Google Scholar] [CrossRef]
- Zeng, H.; Li, L.; Chen, J.X. Loss of Sirt3 limits bone marrow cell-mediated angiogenesis and cardiac repair in post-myocardial infarction. PLoS ONE 2014, 9, e107011. [Google Scholar] [CrossRef]
- Su, H.; Zeng, H.; Liu, B.; Chen, J.X. Sirtuin 3 is essential for hypertension-induced cardiac fibrosis via mediating pericyte transition. J. Cell. Mol. Med. 2020. [Google Scholar] [CrossRef]
- Siddesha, J.M.; Valente, A.J.; Sakamuri, S.S.; Yoshida, T.; Gardner, J.D.; Somanna, N.; Takahashi, C.; Noda, M.; Chandrasekar, B. Angiotensin II stimulates cardiac fibroblast migration via the differential regulation of matrixins and RECK. J. Mol. Cell. Cardiol. 2013, 65, 9–18. [Google Scholar] [CrossRef]
- He, X.; Zeng, H.; Roman, R.J.; Chen, J.X. Inhibition of prolyl hydroxylases alters cell metabolism and reverses pre-existing diastolic dysfunction in mice. Int. J. Cardiol. 2018, 272, 281–287. [Google Scholar] [CrossRef]
- Drawz, P.; Rahman, M. Chronic kidney disease. Ann. Intern. Med. 2015, 162, ITC1–ITC16. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, A.; Pires, M.J.; Oliveira, P.A. Pathophysiological Mechanisms of Renal Fibrosis: A Review of Animal Models and Therapeutic Strategies. In Vivo 2017, 31, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Juchem, G.; Weiss, D.R.; Knott, M.; Senftl, A.; Forch, S.; Fischlein, T.; Kreuzer, E.; Reichart, B.; Laufer, S.; Nees, S. Regulation of coronary venular barrier function by blood borne inflammatory mediators and pharmacological tools: Insights from novel microvascular wall models. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H567–H581. [Google Scholar] [CrossRef]
- Zhao, H.; Chappell, J.C. Microvascular bioengineering: A focus on pericytes. J. Biol. Eng. 2019, 13, 26. [Google Scholar] [CrossRef] [PubMed]
- Verrecchia, F.; Mauviel, A. Transforming growth factor-beta signaling through the Smad pathway: Role in extracellular matrix gene expression and regulation. J. Invest. Dermatol. 2002, 118, 211–215. [Google Scholar] [CrossRef]
- Khan, Z.A.; Barbin, Y.P.; Cukiernik, M.; Adams, P.C.; Chakrabarti, S. Heme-oxygenase-mediated iron accumulation in the liver. Can. J. Physiol. Pharmacol. 2004, 82, 448–456. [Google Scholar] [CrossRef]
- Chiang, S.K.; Chen, S.E.; Chang, L.C. A Dual Role of Heme Oxygenase-1 in Cancer Cells. Int. J. Mol. Sci. 2018, 20, 39. [Google Scholar] [CrossRef]
- Lawen, A.; Lane, D.J. Mammalian iron homeostasis in health and disease: Uptake, storage, transport, and molecular mechanisms of action. Antioxid. Redox Signal. 2013, 18, 2473–2507. [Google Scholar] [CrossRef]
- Wang, S.J.; Li, D.; Ou, Y.; Jiang, L.; Chen, Y.; Zhao, Y.; Gu, W. Acetylation Is Crucial for p53-Mediated Ferroptosis and Tumor Suppression. Cell Rep. 2016, 17, 366–373. [Google Scholar] [CrossRef]
- Zeng, H.; Chen, J.X. Sirtuin 3, Endothelial Metabolic Reprogramming, and Heart Failure with Preserved Ejection Fraction. J. Cardiovasc. Pharmacol. 2019, 74, 315–323. [Google Scholar] [CrossRef]
- Lakhal-Littleton, S.; Wolna, M.; Carr, C.A.; Miller, J.J.; Christian, H.C.; Ball, V.; Santos, A.; Diaz, R.; Biggs, D.; Stillion, R.; et al. Cardiac ferroportin regulates cellular iron homeostasis and is important for cardiac function. Proc. Natl. Acad. Sci. USA 2015, 112, 3164–3169. [Google Scholar] [CrossRef] [PubMed]
- Chuaiphichai, S.; Rashbrook, V.S.; Hale, A.B.; Trelfa, L.; Patel, J.; McNeill, E.; Lygate, C.A.; Channon, K.M.; Douglas, G. Endothelial Cell Tetrahydrobiopterin Modulates Sensitivity to Ang (Angiotensin) II-Induced Vascular Remodeling, Blood Pressure, and Abdominal Aortic Aneurysm. Hypertension 2018, 72, 128–138. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feng, X.; Su, H.; He, X.; Chen, J.-X.; Zeng, H. SIRT3 Deficiency Sensitizes Angiotensin-II-Induced Renal Fibrosis. Cells 2020, 9, 2510. https://doi.org/10.3390/cells9112510
Feng X, Su H, He X, Chen J-X, Zeng H. SIRT3 Deficiency Sensitizes Angiotensin-II-Induced Renal Fibrosis. Cells. 2020; 9(11):2510. https://doi.org/10.3390/cells9112510
Chicago/Turabian StyleFeng, Xiaomeng, Han Su, Xiaochen He, Jian-Xiong Chen, and Heng Zeng. 2020. "SIRT3 Deficiency Sensitizes Angiotensin-II-Induced Renal Fibrosis" Cells 9, no. 11: 2510. https://doi.org/10.3390/cells9112510
APA StyleFeng, X., Su, H., He, X., Chen, J.-X., & Zeng, H. (2020). SIRT3 Deficiency Sensitizes Angiotensin-II-Induced Renal Fibrosis. Cells, 9(11), 2510. https://doi.org/10.3390/cells9112510