Complex Interaction between Resident Microbiota and Misfolded Proteins: Role in Neuroinflammation and Neurodegeneration

 , , and
, , and 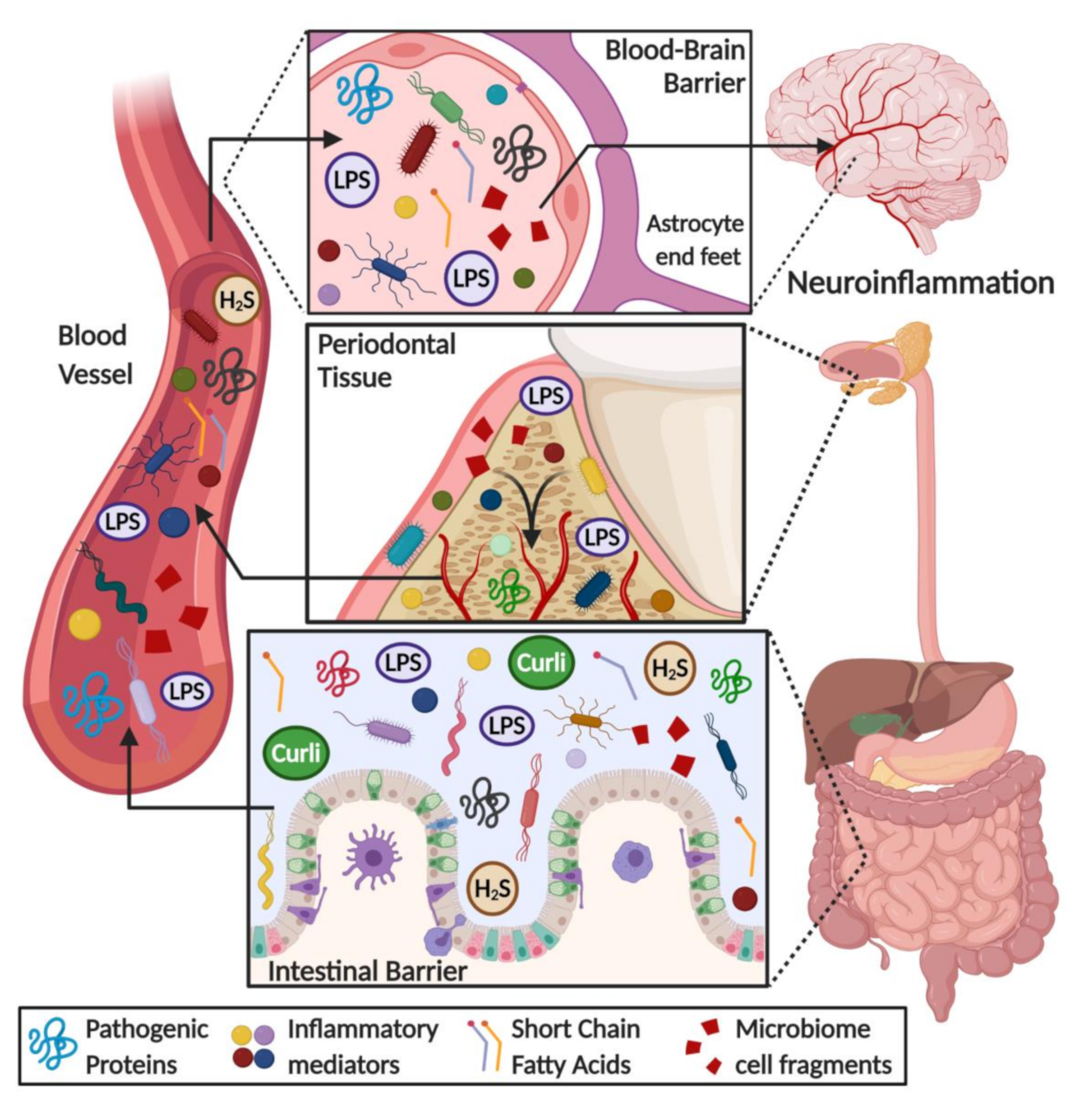{kind=link}
{kind=link}
{kind=link}
Abstract
1. The Burden of Neurodegenerative Diseases
2. Protein Misfolding and Its Accumulation in Neurodegenerative Diseases
3. Protein-Induced Membrane Damage as a Central and Ubiquitous Player in Neurotoxicity
4. Neuroinflammation as a Common Factor across Neurodegenerative Diseases
5. Human Microbiome Dysbiosis as a Source of a Systemic Chronic Inflammatory State
5.1. Human Biofilms: 3D Microbial Structures in Health and Disease
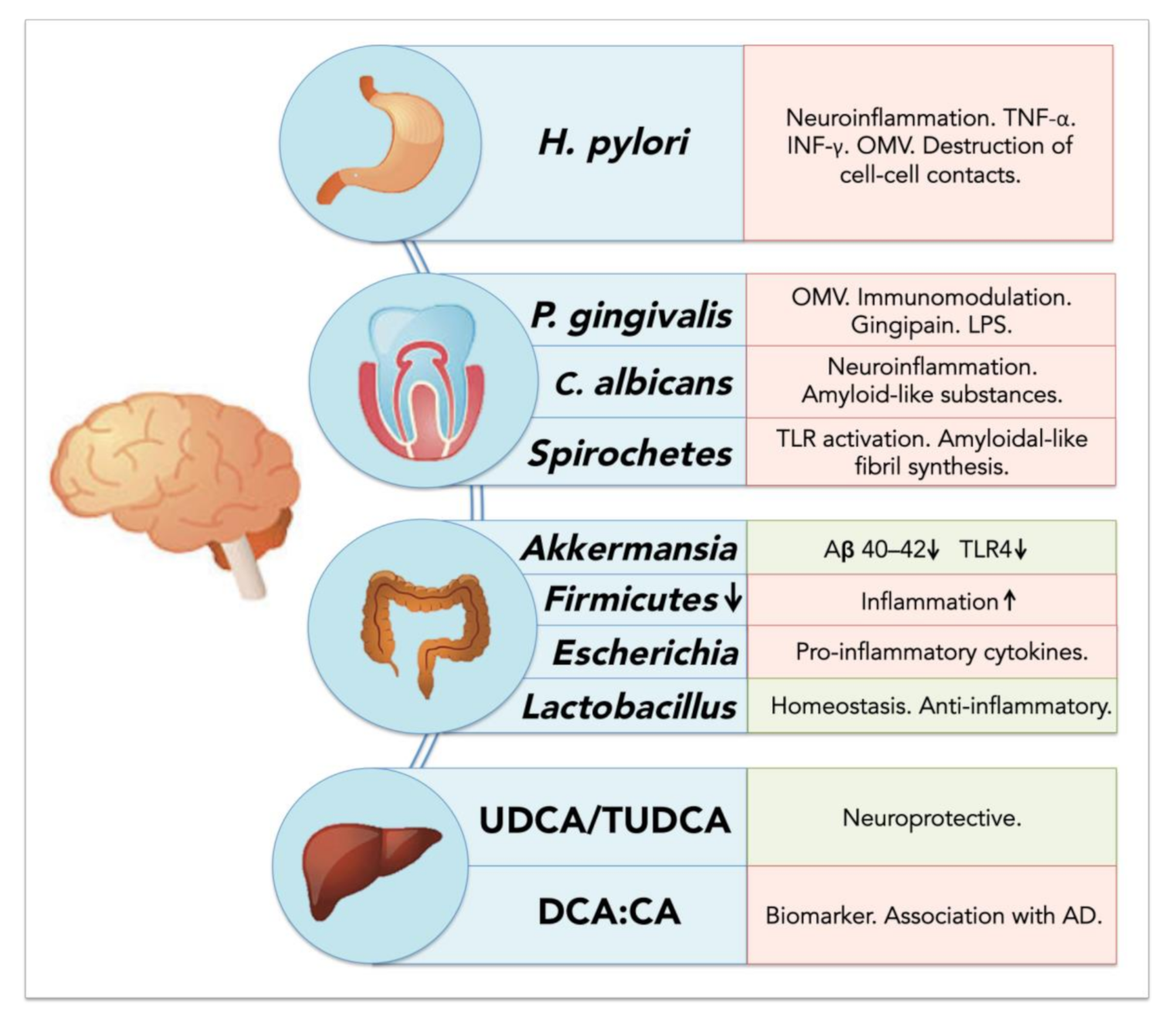
5.2. Resident Oral Microorganisms and Their Association with AD and Neuroinflammation
5.2.1. Porphyromonas gingivalis: Link between Periodontal Disease and Neurodegeneration?
5.2.2. Oral Spirochetes and Brain Infection
5.2.3. Oral Fungi and Brain Infection and Inflammation
5.3. Resident Gut Bacteria and Their Association with Neuroinflammation and Neurodegeneration
5.3.1. Gut Microbiota Dysbiosis Generates a Pro-Inflammatory State
5.3.2. Helicobacter pylori: A Crucial Species for Chronic Inflammation and AD
5.3.3. Akkermansia muciniphila: An Important Regulator of Inflammation in the Gut
5.3.4. Bile Acids and Their Potential Role in Neurodegenerative Diseases
6. Direct and Indirect Effects of Microbiota on the Brain: Role of Barrier Evasion and Permeability
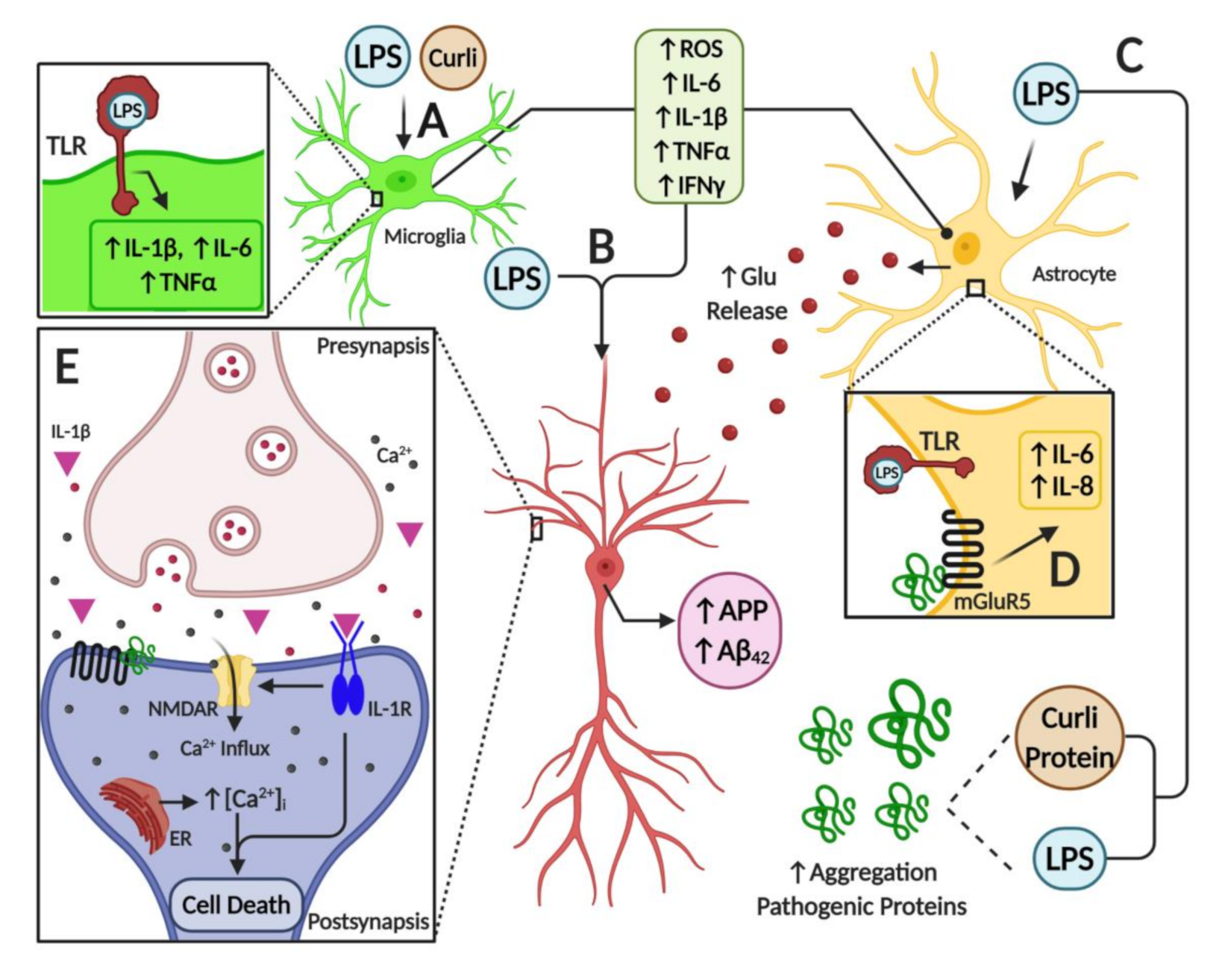
7. Microbiome Dysbiosis and Neuroinflammation: A Complex “Toxic” Mixture Affecting the Brain during NDD
8. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Alzheimer’s Disease International. World Alzheimer Report 2019: Attitudes to Dementia 2019; Alzheimer’s Disease International: London, UK, 2019. [Google Scholar]
- Hebert, L.E.; Weuve, J.; Scherr, P.A.; Evans, D.A. Alzheimer disease in the United States (2010–2050) estimated using the 2010 census. Neurology 2013, 80, 1778–1783. [Google Scholar] [CrossRef] [PubMed]
- Stefanacci, R.G. The costs of Alzheimer’s disease and the value of effective therapies. Am. J. Manag. Care 2011, 17, S356. [Google Scholar] [PubMed]
- Alzheimer’s, A. 2018 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2018, 14, 367–429. [Google Scholar] [CrossRef]
- Panegyres, P.K.; Chen, H.Y. Coalition against Major, D. Early-onset A lzheimer’s disease: A global cross-sectional analysis. Eur. J. Neurol. 2014, 21, 1149–1154. [Google Scholar] [CrossRef] [PubMed]
- Pierce, A.L.; Bullain, S.S.; Kawas, C.H. Late-onset Alzheimer disease. Neurol. Clin. 2017, 35, 283–293. [Google Scholar] [CrossRef]
- Mroczko, B.; Groblewska, M.; Litman-Zawadzka, A.; Kornhuber, J.; Lewczuk, P. Amyloid β oligomers (AβOs) in Alzheimer’s disease. J. Neural. Transm. 2017, 125, 177–191. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer’s disease is a synaptic failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef]
- MacLeod, R.; Hillert, E.-K.; Cameron, R.T.; Baillie, G.S. The role and therapeutic targeting of α-, β-and γ-secretase in Alzheimer’s disease. Future Sci. OA 2015, 1. [Google Scholar] [CrossRef]
- Fernandez-Perez, E.J.; Peters, C.; Aguayo, L.G. Membrane damage induced by amyloid beta and a potential link with neuroinflammation. Curr. Pharm. Des. 2016, 22, 1295–1304. [Google Scholar] [CrossRef]
- Sepulveda, F.J.; Parodi, J.; Peoples, R.W.; Opazo, C.; Aguayo, L.G. Synaptotoxicity of Alzheimer beta amyloid can be explained by its membrane perforating property. PLoS ONE 2010, 5, e11820. [Google Scholar] [CrossRef]
- Diaz, J.C.; Simakova, O.; Jacobson, K.A.; Arispe, N.; Pollard, H.B. Small molecule blockers of the Alzheimer Aβ calcium channel potently protect neurons from Aβ cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 3348–3353. [Google Scholar] [CrossRef] [PubMed]
- Sepúlveda, F.J.; Fierro, H.; Fernandez, E.; Castillo, C.; Peoples, R.W.; Opazo, C.; Aguayo, L.G. Nature of the neurotoxic membrane actions of amyloid-β on hippocampal neurons in Alzheimer’s disease. Neurobiol. Aging 2014, 35, 472–481. [Google Scholar] [CrossRef] [PubMed]
- Killin, L.O.J.; Starr, J.M.; Shiue, I.J.; Russ, T.C. Environmental risk factors for dementia: A systematic review. BMC Geriatr. 2016, 16, 175. [Google Scholar] [CrossRef] [PubMed]
- Dorsey, E.R.; Elbaz, A.; Nichols, E.; Abd-Allah, F.; Abdelalim, A.; Adsuar, J.C.; Ansha, M.G.; Brayne, C.; Choi, J.-Y.J.; Collado-Mateo, D. Global, regional, and national burden of Parkinson’s disease, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018, 17, 939–953. [Google Scholar] [CrossRef]
- Tysnes, O.-B.; Storstein, A. Epidemiology of Parkinson’s disease. J. Neural. Transm. 2017, 124, 901–905. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.-E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Prim. 2017, 3, 1–21. [Google Scholar] [CrossRef]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Fields, C.R.; Bengoa-Vergniory, N.; Wade-Martins, R. Targeting Alpha-Synuclein as a Therapy for Parkinson’s Disease. Front. Mol. Neurosci. 2019, 12, 299. [Google Scholar] [CrossRef]
- Uttley, L.; Carroll, C.; Wong, R.; Hilton, D.A.; Stevenson, M. Creutzfeldt-Jakob disease: A systematic review of global incidence, prevalence, infectivity, and incubation. Lancet Infect. Dis. 2020, 20, e2–e10. [Google Scholar] [CrossRef]
- National Institute of Neurological Disorders and Stroke. Creutzfeldt-Jakob Disease Fact Sheet; National Institute of Neurological Disorders and Stroke: Bethesda, MD, USA, 2018. [Google Scholar]
- Betarbet, R.; Sherer, T.B.; MacKenzie, G.; Garcia-Osuna, M.; Panov, A.V.; Greenamyre, J.T. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat. Neurosci. 2000, 3, 1301–1306. [Google Scholar] [CrossRef]
- Leemans, M. Prion diseases. Anaesth. Intensive Care Med. 2019, 21, 56–59. [Google Scholar] [CrossRef]
- Taylor, J.P.; Hardy, J.; Fischbeck, K.H. Toxic proteins in neurodegenerative disease. Science 2002, 296, 1991–1995. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.G.; Lee, H.; Lee, S.B. Mechanisms of protein toxicity in neurodegenerative diseases. Cell. Mol. Life Sci. 2018, 75, 3159–3180. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J. A hundred years of Alzheimer’s disease research. Neuron 2006, 52, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Bloodgood, B.L.; Townsend, M.; Walsh, D.M.; Selkoe, D.J.; Sabatini, B.L. Natural oligomers of the Alzheimer amyloid-β protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci. 2007, 27, 2866–2875. [Google Scholar] [CrossRef] [PubMed]
- Brindle, N.; George-Hyslop, P.S. The genetics of Alzheimer’s disease. Methods Mol. Med. 2000, 32, 23–43. [Google Scholar] [CrossRef] [PubMed]
- Lesné, S.; Koh, M.T.; Kotilinek, L.; Kayed, R.; Glabe, C.G.; Yang, A.; Gallagher, M.; Ashe, K.H. A specific amyloid-β protein assembly in the brain impairs memory. Nature 2006, 440, 352–357. [Google Scholar] [CrossRef]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A. Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef]
- Peters, C.; Fernandez-Perez, E.J.; Burgos, C.F.; Espinoza, M.P.; Castillo, C.; Urrutia, J.C.; Streltsov, V.A.; Opazo, C.; Aguayo, L.G. Inhibition of amyloid beta-induced synaptotoxicity by a pentapeptide derived from the glycine zipper region of the neurotoxic peptide. Neurobiol. Aging 2013, 34, 2805–2814. [Google Scholar] [CrossRef]
- Liebert, A.; Bicknell, B.; Adams, R. Prion protein signaling in the nervous system—A review and perspective. Sign. Transduct. Insights 2014, 3, STI-S12319. [Google Scholar] [CrossRef]
- Pacheco, C.R.; Morales, C.N.; Ramírez, A.E.; Muñoz, F.J.; Gallegos, S.S.; Caviedes, P.A.; Aguayo, L.G.; Opazo, C.M. Extracellular α-synuclein alters synaptic transmission in brain neurons by perforating the neuronal plasma membrane. J. Neurochem. 2015, 132, 731–741. [Google Scholar] [CrossRef] [PubMed]
- Ghio, S.; Camilleri, A.; Caruana, M.; Ruf, V.C.; Schmidt, F.; Leonov, A.; Ryazanov, S.; Griesinger, C.; Cauchi, R.J.; Kamp, F.; et al. Cardiolipin promotes pore-forming activity of alpha-synuclein oligomers in mitochondrial membranes. ACS Chem. Neurosci. 2019, 10, 3815–3829. [Google Scholar] [CrossRef] [PubMed]
- Österlund, N.; Moons, R.; Ilag, L.L.; Sobott, F.; Graslund, A. Native ion mobility-mass spectrometry reveals the formation of β-barrel shaped amyloid-β hexamers in a membrane-mimicking environment. J. Am. Chem. Soc. 2019, 141, 10440–10450. [Google Scholar] [CrossRef] [PubMed]
- Chich, J.-F.; Chapuis, C.; Henry, C.; Vidic, J.; Rezaei, H.; Noinville, S. Vesicle permeabilization by purified soluble oligomers of prion protein: A comparative study of the interaction of oligomers and monomers with lipid membranes. J. Mol. Biol. 2010, 397, 1017–1030. [Google Scholar] [CrossRef]
- Combet, S.; Cousin, F.; Rezaei, H.; Noinville, S. Membrane interaction of off-pathway prion oligomers and lipid-induced on-pathway intermediates during prion conversion: A clue for neurotoxicity. Biochim. Biophys. Acta-Biomembr. 2019, 1861, 514–523. [Google Scholar] [CrossRef]
- Pan, J.; Sahoo, P.K.; Dalzini, A.; Hayati, Z.; Aryal, C.M.; Teng, P.; Cai, J.; Gutierrez, H.R.; Song, L. Membrane disruption mechanism of a Prion Peptide (106-126) investigated by atomic force microscopy, raman and electron paramagnetic resonance spectroscopy. J. Phys. Chem. B 2017, 121, 5058–5071. [Google Scholar] [CrossRef]
- Paulis, D.; Maras, B.; Schinin, M.E.; Di Francesco, L.; Principe, S.; Galeno, R.; Abdel-Haq, H.; Cardone, F.; Florio, T.; Pocchiari, M.; et al. The pathological prion protein forms ionic conductance in lipid bilayer. Neurochem. Int. 2011, 59, 168–174. [Google Scholar] [CrossRef]
- Stephenson, J.; Nutma, E.; van der Valk, P.; Amor, S. Inflammation in CNS neurodegenerative diseases. Immunology 2018, 154, 204–219. [Google Scholar] [CrossRef]
- Abu-Rumeileh, S.; Oeckl, P.; Baiardi, S.; Halbgebauer, S.; Steinacker, P.; Capellari, S.; Otto, M.; Parchi, P. CSF ubiquitin levels are higher in Alzheimer’s disease than in frontotemporal dementia and reflect the molecular subtype in prion disease. Biomolecules 2020, 10, 497. [Google Scholar] [CrossRef]
- Tanaka, M.; Toldi, J.; Vecsei, L. Exploring the etiological links behind neurodegenerative diseases: Inflammatory cytokines and bioactive kynurenines. Int J. Mol. Sci. 2020, 21, 2431. [Google Scholar] [CrossRef]
- Nelson, P.T.; Soma, L.A.; Lavi, E. Microglia in diseases of the central nervous system. Ann. Med. 2002, 34, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Hong, J.S. Role of microglia in inflammation-mediated neurodegenerative diseases: Mechanisms and strategies for therapeutic intervention. J. Pharmacol. Exp. Ther. 2003, 304, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Garcao, P.; Oliveira, C.R.; Agostinho, P. Comparative study of microglia activation induced by amyloid-beta and prion peptides: Role in neurodegeneration. J. Neurosci. Res. 2006, 84, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Paranjape, G.S.; Gouwens, L.K.; Osborn, D.C.; Nichols, M.R. Isolated amyloid-beta(1-42) protofibrils, but not isolated fibrils, are robust stimulators of microglia. ACS Chem. Neurosci. 2012, 3, 302–311. [Google Scholar] [CrossRef] [PubMed]
- Tu, J.; Chen, B.; Yang, L.; Qi, K.; Lu, J.; Zhao, D. Amyloid-beta activates microglia and regulates protein expression in a manner similar to prions. J. Mol. Neurosci. 2015, 56, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Vom Berg, J.; Prokop, S.; Miller, K.R.; Obst, J.; Kälin, R.E.; Lopategui-Cabezas, I.; Wegner, A.; Mair, F.; Schipke, C.G.; Peters, O.; et al. Inhibition of IL-12/IL-23 signaling reduces Alzheimer’s diseasea-like pathology and cognitive decline. Nat. Med. 2012, 18, 1812–1819. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, Y.; Zhou, J. Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl. Neurodegener. 2015, 4, 19. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, T.; Pei, Z.; Miller, D.S.; Wu, X.; Block, M.L.; Wilson, B.; Zhang, W.; Zhou, Y.; Hong, J.S.; et al. Aggregated alpha-synuclein activates microglia: A process leading to disease progression in Parkinson’s disease. FASEB J. 2005, 19, 533–542. [Google Scholar] [CrossRef]
- Saitgareeva, A.R.; Bulygin, K.V.; Gareev, I.F.; Beylerli, O.A.; Akhmadeeva, L.R. The role of microglia in the development of neurodegeneration. Neurol. Sci. 2020. [Google Scholar] [CrossRef]
- Puoti, G.; Giaccone, G.; Mangieri, M.; Limido, L.; Fociani, P.; Zerbi, P.; Suardi, S.; Rossi, G.; Iussich, S.; Capobianco, R.; et al. Sporadic Creutzfeldt-Jakob disease: The extent of microglia activation is dependent on the biochemical type of PrPSc. J. Neuropathol. Exp. Neurol. 2005, 64, 902–909. [Google Scholar] [CrossRef] [PubMed]
- Franceschini, A.; Strammiello, R.; Capellari, S.; Giese, A.; Parchi, P. Regional pattern of microgliosis in sporadic Creutzfeldt-Jakob disease in relation to phenotypic variants and disease progression. Neuropathol. Appl. Neurobiol. 2018, 44, 574–589. [Google Scholar] [CrossRef] [PubMed]
- Makarava, N.; Chang, J.C.; Molesworth, K.; Baskakov, I. V Region-specific glial homeostatic signature in prion diseases is replaced by a uniform neuroinflammation signature, common for brain regions and prion strains with different cell tropism. Neurobiol. Dis. 2020, 137, 104783. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A.; Liu, Y. A role for astroglia in prion diseases. J. Exp. Med. 2017, 214, 3477–3479. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A.; Zhu, C. Microglia in prion diseases. J. Clin. Invest. 2017, 127, 3230–3239. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef] [PubMed]
- Taipa, R.; das Neves, S.P.; Sousa, A.L.; Fernandes, J.; Pinto, C.; Correia, A.P.; Santos, E.; Pinto, P.S.; Carneiro, P.; Costa, P.; et al. Proinflammatory and anti-inflammatory cytokines in the CSF of patients with Alzheimer’s disease and their correlation with cognitive decline. Neurobiol. Aging 2019, 76, 125–132. [Google Scholar] [CrossRef]
- Stoeck, K.; Bodemer, M.; Ciesielczyk, B.; Meissner, B.; Bartl, M.; Heinemann, U.; Zerr, I. Interleukin 4 and interleukin 10 levels are elevated in the cerebrospinal fluid of patients with Creutzfeldt-Jakob disease. Arch. Neurol. 2005, 62, 1591–1594. [Google Scholar] [CrossRef][Green Version]
- Stoeck, K.; Bodemer, M.; Zerr, I. Pro- and anti-inflammatory cytokines in the CSF of patients with Creutzfeldt-Jakob disease. J. Neuroimmunol. 2006, 172, 175–181. [Google Scholar] [CrossRef]
- Garcia-Revilla, J.; Alonso-Bellido, I.M.; Burguillos, M.A.; Herrera, A.J.; Espinosa-Oliva, A.M.; Ruiz, R.; Cruz-Hernandez, L.; Garcia-Dominguez, I.; Roca-Ceballos, M.A.; Santiago, M.; et al. Reformulating pro-oxidant microglia in neurodegeneration. J. Clin. Med. 2019, 8, 1719. [Google Scholar] [CrossRef]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.L.; Tan, C.C.; Hou, X.H.; Cao, X.P.; Tan, L.; Yu, J.T. TREM2 variants and neurodegenerative diseases: A systematic review and meta-analysis. J. Alzheimers Dis. 2019, 68, 1171–1184. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wu, X.; Li, X.; Jiang, L.L.; Gui, X.; Liu, Y.; Sun, Y.; Zhu, B.; Pina-Crespo, J.C.; Zhang, M.; et al. TREM2 is a receptor for beta-amyloid that mediates microglial function. Neuron 2018, 97, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Wang, Z.; Wang, D.; Wang, Z.; Martens, Y.A.; Wu, L.; Xu, Y.; Wang, K.; Li, J.; Huang, R.; et al. Amyloid-beta modulates microglial responses by binding to the triggering receptor expressed on myeloid cells 2 (TREM2). Mol. Neurodegener. 2018, 13, 15. [Google Scholar] [CrossRef] [PubMed]
- Jana, M.; Palencia, C.A.; Pahan, K. Fibrillar amyloid-beta peptides activate microglia via TLR2: Implications for Alzheimer’s disease. J. Immunol. 2008, 181, 7254–7262. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Ho, D.H.; Suk, J.E.; You, S.; Michael, S.; Kang, J.; Joong Lee, S.; Masliah, E.; Hwang, D.; Lee, H.J.; et al. Neuron-released oligomeric alpha-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat. Commun. 2013, 4, 1562. [Google Scholar] [CrossRef] [PubMed]
- Lukiw, W.J. Gastrointestinal (GI) tract microbiome-derived neurotoxins-potent neuro-inflammatory signals from the GI tract via the systemic circulation into the brain. Front. Cell Infect. Microbiol. 2020, 10, 22. [Google Scholar] [CrossRef]
- Daws, M.R.; Sullam, P.M.; Niemi, E.C.; Chen, T.T.; Tchao, N.K.; Seaman, W.E. Pattern recognition by TREM-2: Binding of anionic ligands. J. Immunol. 2003, 171, 594–599. [Google Scholar] [CrossRef]
- Batista, I.A.; Melo, S.A. Exosomes and the future of immunotherapy in pancreatic cancer. Int. J. Mol. Sci. 2019, 20, 567. [Google Scholar] [CrossRef]
- Cunningham, C.; Campion, S.; Lunnon, K.; Murray, C.L.; Woods, J.F.; Deacon, R.M.; Rawlins, J.N.; Perry, V.H. Systemic inflammation induces acute behavioral and cognitive changes and accelerates neurodegenerative disease. Biol. Psychiatry 2009, 65, 304–312. [Google Scholar] [CrossRef]
- Sama, D.M.; Norris, C.M. Calcium dysregulation and neuroinflammation: Discrete and integrated mechanisms for age-related synaptic dysfunction. Ageing Res. Rev. 2013, 12, 982–995. [Google Scholar] [CrossRef] [PubMed]
- Sender, R.; Fuchs, S.; Milo, R. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. 2016, 14, e1002533. [Google Scholar] [CrossRef] [PubMed]
- Carding, S.; Verbeke, K.; Vipond, D.T.; Corfe, B.M.; Owen, L.J. Dysbiosis of the gut microbiota in disease. Microb. Ecol. Heal. Dis. 2015, 26. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, L.J.; Monga, M.; Miller, A.W. Defining dysbiosis for a cluster of chronic diseases. Sci. Rep. 2019, 9, 12918. [Google Scholar] [CrossRef]
- Donlan, R.M. Biofilms: Microbial life on surfaces. Emerg. Infect. Dis. 2002, 8, 881–890. [Google Scholar] [CrossRef]
- Van Acker, H.; Van Dijck, P.; Coenye, T. Molecular mechanisms of antimicrobial tolerance and resistance in bacterial and fungal biofilms. Trends Microbiol. 2014, 22, 326–333. [Google Scholar] [CrossRef]
- Costerton, J.W.; Lewandowski, Z.; Caldwell, D.E.; Korber, D.R.; Lappin-Scott, H.M. Microbial biofilms. Annu. Rev. Microbiol. 1995, 49, 711–745. [Google Scholar] [CrossRef]
- Lebeaux, D.; Ghigo, J.-M.; Beloin, C. Biofilm-related infections: Bridging the gap between clinical management and fundamental aspects of recalcitrance toward antibiotics. Microbiol. Mol. Biol. Rev. 2014, 78, 510–543. [Google Scholar] [CrossRef]
- Pitts, N.B.; Zero, D.T.; Marsh, P.D.; Ekstrand, K.; Weintraub, J.A.; Ramos-Gomez, F.; Tagami, J.; Twetman, S.; Tsakos, G.; Ismail, A. Dental caries. Nat. Rev. Dis. Prim. 2017, 3, 17030. [Google Scholar] [CrossRef]
- Gupta, P.; Gupta, N.; Pawar, A.P.; Birajdar, S.S.; Natt, A.S.; Singh, H.P. Role of sugar and sugar substitutes in dental caries: A review. ISRN Dent. 2013, 2013, 519421. [Google Scholar] [CrossRef]
- van Loveren, C. Sugar restriction for caries prevention: Amount and frequency. Which is more important? Caries Res. 2019, 53, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, G.; Darveau, R.P.; Curtis, M.A. The keystone-pathogen hypothesis. Nat. Rev. Microbiol. 2012, 10, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, G. Immune evasion strategies of Porphyromonas gingivalis. J. Oral Biosci. 2011, 53, 233–240. [Google Scholar] [CrossRef]
- Abdi, K.; Chen, T.; Klein, B.A.; Tai, A.K.; Coursen, J.; Liu, X.; Skinner, J.; Periasamy, S.; Choi, Y.; Kessler, B.M.; et al. Mechanisms by which Porphyromonas gingivalis evades innate immunity. PLoS ONE 2017, 12, e0182164. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.S. Oral microbiota and systemic disease. Anaerobe 2013, 24, 90–93. [Google Scholar] [CrossRef]
- Li, X.; Kolltveit, K.M.; Tronstad, L.; Olsen, I. Systemic diseases caused by oral infection. Clin. Microbiol. Rev. 2000, 13, 547–558. [Google Scholar] [CrossRef]
- Bui, F.Q.; Almeida-da-Silva, C.L.C.; Huynh, B.; Trinh, A.; Liu, J.; Woodward, J.; Asadi, H.; Ojcius, D.M. Association between periodontal pathogens and systemic disease. Biomed. J. 2019, 42, 27–35. [Google Scholar] [CrossRef]
- Aguayo, S.; Schuh, C.M.A.P.; Vicente, B.; Aguayo, L.G. Association between Alzheimer’s disease and oral and gut microbiota: Are pore forming proteins the missing link? J. Alzheimer’s Dis. 2018, 65. [Google Scholar] [CrossRef]
- Saini, R.; Marawar, P.P.; Shete, S.; Saini, S. Periodontitis, a true infection. J. Glob. Infect. Dis. 2009, 1, 149–150. [Google Scholar] [CrossRef]
- Mysak, J.; Podzimek, S.; Sommerova, P.; Lyuya-Mi, Y.; Bartova, J.; Janatova, T.; Prochazkova, J.; Duskova, J. Porphyromonas gingivalis: Major Periodontopathic Pathogen Overview. J. Immunol. Res. 2014, 2014, 476068. [Google Scholar] [CrossRef]
- Meka, A.; Bakthavatchalu, V.; Sathishkumar, S.; Lopez, M.C.; Verma, R.K.; Wallet, S.M.; Bhattacharyya, I.; Boyce, B.F.; Handfield, M.; Lamont, R.J.; et al. Porphyromonas gingivalis infection-induced tissue and bone transcriptional profiles. Mol. Oral Microbiol. 2010, 25, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, G.; Lambris, J.D. Complement and dysbiosis in periodontal disease. Immunobiology 2012, 217, 1111–1116. [Google Scholar] [CrossRef] [PubMed]
- Chaves, E.S.; Jeffcoat, M.K.; Ryerson, C.C.; Snyder, B. Persistent bacterial colonization of Porphyromonas gingivalis, Prevotella intermedia, and Actinobacillus actinomycetemcomitans in periodontitis and its association with alveolar bone loss after 6 months of therapy. J. Clin. Periodontol. 2000, 27, 897–903. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, G.; Lamont, R.J. Breaking bad: Manipulation of the host response by Porphyromonas gingivalis. Eur. J. Immunol. 2014, 44, 328–338. [Google Scholar] [CrossRef]
- Popadiak, K.; Potempa, J.; Riesbeck, K.; Blom, A.M. Biphasic effect of gingipains from porphyromonas gingivalis on the human complement system. J. Immunol. 2007, 178, 7242–7250. [Google Scholar] [CrossRef]
- Dominy, S.S.; Lynch, C.; Ermini, F.; Benedyk, M.; Marczyk, A.; Konradi, A.; Nguyen, M.; Haditsch, U.; Raha, D.; Griffin, C.; et al. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci. Adv. 2019, 5, eaau3333. [Google Scholar] [CrossRef]
- Poole, S.; Singhrao, S.K.; Kesavalu, L.; Curtis, M.A.; Crean, S.J. Determining the presence of periodontopathic virulence factors in short-term postmortem Alzheimer’s disease brain tissue. In Handbook of Infection and Alzheimer’s Disease; IOS Press: Amsterdam, The Netherlands, 2017; pp. 105–117. ISBN 9781614997061. [Google Scholar]
- Hauss-Wegrzyniak, B.; Wenk, G.L. Beta-amyloid deposition in the brains of rats chronically infused with thiorphan or lipopolysaccharide: The role of ascorbic acid in the vehicle. Neurosci. Lett. 2002, 322, 75–78. [Google Scholar] [CrossRef]
- Lee, J.; Lee, Y.; Yuk, D.; Choi, D.; Ban, S.; Oh, K.; Hong, J. Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. J. Neuroinflamm. 2008, 5, 37. [Google Scholar] [CrossRef]
- Zhan, X.; Stamova, B.; Sharp, F.R. Lipopolysaccharide associates with amyloid plaques, neurons and oligodendrocytes in Alzheimer’s disease brain: A review. Front. Aging Neurosci. 2018, 10, 42. [Google Scholar] [CrossRef]
- Haditsch, U.; Roth, T.; Rodriguez, L.; Hancock, S.; Cecere, T.; Nguyen, M.; Arastu-Kapur, S.; Broce, S.; Raha, D.; Lynch, C.C.; et al. Alzheimer’s disease-like neurodegeneration in porphyromonas gingivalis infected neurons with persistent expression of active gingipains. J. Alzheimer’s Dis. 2020, 1–16. [Google Scholar] [CrossRef]
- Poole, S.; Singhrao, S.K.; Chukkapalli, S.; Rivera, M.; Velsko, I.; Kesavalu, L.; Crean, S. Active invasion of porphyromonas gingivalis and infection-induced complement activation in ApoE-/-Mice brains. J. Alzheimer’s Dis. 2015, 43, 67–80. [Google Scholar] [CrossRef] [PubMed]
- Ilievski, V.; Zuchowska, P.K.; Green, S.J.; Toth, P.T.; Ragozzino, M.E.; Le, K.; Aljewari, H.W.; O’Brien-Simpson, N.M.; Reynolds, E.C.; Watanabe, K. Chronic oral application of a periodontal pathogen results in brain inflammation, neurodegeneration and amyloid beta production in wild type mice. PLoS ONE 2018, 13, e0204941. [Google Scholar] [CrossRef] [PubMed]
- Carter, C.J.; France, J.; Crean, S.J.; Singhrao, S.K. The porphyromonas gingivalis/host interactome shows enrichment in GWASdb genes related to alzheimer’s disease, diabetes and cardiovascular diseases. Front. Aging Neurosci. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Kamer, A.R.; Craig, R.G.; Pirraglia, E.; Dasanayake, A.P.; Norman, R.G.; Boylan, R.J.; Nehorayoff, A.; Glodzik, L.; Brys, M.; de Leon, M.J. TNF-alpha and antibodies to periodontal bacteria discriminate between Alzheimer’s disease patients and normal subjects. J. Neuroimmunol. 2009, 216, 92–97. [Google Scholar] [CrossRef]
- Nagakubo, T.; Nomura, N.; Toyofuku, M. Cracking open bacterial membrane vesicles. Front. Microbiol. 2019, 10, 3026. [Google Scholar] [CrossRef]
- Beveridge, T.J. Structures of gram-negative cell walls and their derived membrane vesicles. J. Bacteriol. 1999, 181, 4725–4733. [Google Scholar] [CrossRef]
- McBroom, A.J.; Johnson, A.P.; Vemulapalli, S.; Kuehn, M.J. Outer membrane vesicle production by Escherichia coli is independent of membrane instability. J. Bacteriol. 2006, 188, 5385–5392. [Google Scholar] [CrossRef]
- Bielig, H.; Dongre, M.; Zurek, B.; Wai, S.N.; Kufer, T.A. A role for quorum sensing in regulating innate immune responses mediated by Vibrio cholerae outer membrane vesicles (OMVs). Gut Microbes 2011, 2, 274–279. [Google Scholar] [CrossRef]
- Donato, G.M.; Goldsmith, C.S.; Paddock, C.D.; Eby, J.C.; Gray, M.C.; Hewlett, E.L. Delivery of Bordetella pertussis adenylate cyclase toxin to target cells via outer membrane vesicles. FEBS Lett. 2012, 586, 459–465. [Google Scholar] [CrossRef]
- Dutta, S.; Iida, K.I.; Takade, A.; Meno, Y.; Nair, G.B.; Yoshida, S.I. Release of Shiga toxin by membrane vesicles in Shigella dysenteriae serotype 1 strains and in vitro effects of antimicrobials on toxin production and release. Microbiol. Immunol. 2004, 48, 965–969. [Google Scholar] [CrossRef]
- Rumbo, C.; Fernández-Moreira, E.; Merino, M.; Poza, M.; Mendez, J.A.; Soares, N.C.; Mosquera, A.; Chaves, F.; Bou, G. Horizontal transfer of the OXA-24 carbapenemase gene via outer membrane vesicles: A new mechanism of dissemination of carbapenem resistance genes in Acinetobacter baumannii. Antimicrob. Agents Chemother. 2011, 55, 3084–3090. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Torchia, M.L.G.; Lawson, G.W.; Karp, C.L.; Ashwell, J.D.; Mazmanian, S.K. Outer membrane vesicles of a human commensal mediate immune regulation and disease protection. Cell Host Microbe 2012, 12, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Stentz, R.; Osborne, S.; Horn, N.; Li, A.W.H.; Hautefort, I.; Bongaerts, R.; Rouyer, M.; Bailey, P.; Shears, S.B.; Hemmings, A.M.; et al. A bacterial homolog of a eukaryotic inositol phosphate signaling enzyme mediates cross-kingdom dialog in the mammalian gut. Cell Rep. 2014. [Google Scholar] [CrossRef] [PubMed]
- Cañas, M.A.; Fábrega, M.J.; Giménez, R.; Badia, J.; Baldomà, L. Outer membrane vesicles from probiotic and commensal Escherichia coli activate NOD1-mediated immune responses in intestinal epithelial cells. Front. Microbiol. 2018. [Google Scholar] [CrossRef]
- Gui, M.J.; Dashper, S.G.; Slakeski, N.; Chen, Y.-Y.; Reynolds, E.C. Spheres of influence: Porphyromonas gingivalis outer membrane vesicles. Mol. Oral Microbiol. 2016, 31, 365–378. [Google Scholar] [CrossRef]
- Waller, T.; Kesper, L.; Hirschfeld, J.; Dommisch, H.; Kölpin, J.; Oldenburg, J.; Uebele, J.; Hoerauf, A.; Deschner, J.; Jepsen, S.; et al. Porphyromonas gingivalis outer membrane vesicles induce selective tumor necrosis factor tolerance in a toll-like receptor 4- and mTOR-dependent manner. Infect. Immun. 2016, 84, 1194–1204. [Google Scholar] [CrossRef]
- Furuta, N.; Takeuchi, H.; Amano, A. Entry of porphyromonas gingivalis outer membrane vesicles into epithelial cells causes cellular functional impairment. Infect. Immun. 2009, 77, 4761–4770. [Google Scholar] [CrossRef]
- Furuta, N.; Tsuda, K.; Omori, H.; Yoshimori, T.; Yoshimura, F.; Amano, A. Porphyromonas gingivalis outer membrane vesicles enter human epithelial cells via an endocytic pathway and are sorted to lysosomal compartments. Infect. Immun. 2009, 77, 4187–4196. [Google Scholar] [CrossRef]
- Han, E.C.; Choi, S.Y.; Lee, Y.; Park, J.W.; Hong, S.H.; Lee, H.J. Extracellular RNAs in periodontopathogenic outer membrane vesicles promote TNF-α production in human macrophages and cross the blood-brain barrier in mice. FASEB J. 2019. [Google Scholar] [CrossRef]
- Adams, B.; Nunes, J.M.; Page, M.J.; Roberts, T.; Carr, J.; Nell, T.A.; Kell, D.B.; Pretorius, E. Parkinson’s Disease: A Systemic Inflammatory Disease Accompanied by Bacterial Inflammagens. Front. Aging Neurosci. 2019, 11, 210. [Google Scholar] [CrossRef]
- Olsen, I.; Kell, D.B.; Pretorius, E. Is Porphyromonas gingivalis involved in Parkinson’s disease? Eur. J. Clin. Microbiol. Infect. Dis. 2020. [Google Scholar] [CrossRef] [PubMed]
- Wolgemuth, C.W. Flagellar motility of the pathogenic spirochetes. Semin. Cell Dev. Biol. 2015, 46, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Miklossy, J. Alzheimer’s disease—A spirochetosis? Neuroreport 1993, 4, 841–848. [Google Scholar] [CrossRef] [PubMed]
- Riviere, G.; Riviere, K.H.; Smith, K.S. Molecular and immunological evidence of oral Treponema in the human brain and their association with Alzheimer’s disease. Oral Microbiol. Immunol. 2002, 17, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Foschi, F.; Izard, J.; Sasaki, H.; Sambri, V.; Prati, C.; Müller, R.; Stashenko, P. Treponema denticola in disseminating endodontic infections. J. Dent. Res. 2006, 85, 761–765. [Google Scholar] [CrossRef]
- Schröder, N.W.J.; Heine, H.; Alexander, C.; Manukyan, M.; Eckert, J.; Hamann, L.; Göbel, U.B.; Schumann, R.R. Lipopolysaccharide binding protein binds to triacylated and diacylated lipopeptides and mediates innate immune responses. J. Immunol. 2004, 173, 2683–2691. [Google Scholar] [CrossRef]
- Sellati, T.J.; Bouis, D.A.; Kitchens, R.L.; Darveau, R.P.; Pugin, J.; Ulevitch, R.J.; Gangloff, S.C.; Goyert, S.M.; Norgard, M.V.; Radolf, J.D. Treponema pallidum and borrelia burgdorferi lipoproteins and synthetic lipopeptides activate monocytic cells via a CD14-dependent pathway distinct from that used by lipopolysaccharide. J. Immunol. 1998, 160, 5455–5464. [Google Scholar]
- Ohnishi, S.; Koide, A.; Koide, S. Solution conformation and amyloid-like fibril formation of a polar peptide derived from a β-hairpin in the OspA single-layer β-sheet11. J. Mol. Biol. 2000, 301, 477–489. [Google Scholar] [CrossRef]
- Soscia, S.J.; Kirby, J.E.; Washicosky, K.J.; Tucker, S.M.; Ingelsson, M.; Hyman, B.; Burton, M.A.; Goldstein, L.E.; Duong, S.; Tanzi, R.E.; et al. The Alzheimer’s disease-associated amyloid β-protein is an antimicrobial peptide. PLoS ONE 2010, 5. [Google Scholar] [CrossRef]
- Bertolini, M.; Ranjan, A.; Thompson, A.; Diaz, P.I.; Sobue, T.; Maas, K.; Dongari-Bagtzoglou, A. Candida albicans induces mucosal bacterial dysbiosis that promotes invasive infection. PLoS Pathog. 2019, 15, e1007717. [Google Scholar] [CrossRef]
- Alonso, R.; Pisa, D.; Marina, A.I.; Morato, E.; Rábano, A.; Carrasco, L. Fungal infection in patients with Alzheimer’s disease. J. Alzheimer’s Dis. 2014, 41, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Alonso, R.; Pisa, D.; Rábano, A.; Carrasco, L. Alzheimer’s disease and disseminated mycoses. Eur. J. Clin. Microbiol. Infect. Dis. 2014, 33, 1125–1132. [Google Scholar] [CrossRef] [PubMed]
- Alonso, R.; Pisa, D.; Fernández-Fernández, A.M.; Carrasco, L. Infection of fungi and bacteria in brain tissue from elderly persons and patients with Alzheimer’s disease. Front. Aging Neurosci. 2018, 10, 159. [Google Scholar] [CrossRef] [PubMed]
- Pisa, D.; Alonso, R.; Rábano, A.; Rodal, I.; Carrasco, L. Different brain regions are infected with fungi in Alzheimer’s disease. Sci. Rep. 2015, 5, 15015. [Google Scholar] [CrossRef]
- Parady, B. Innate Immune and fungal model of Alzheimer’s disease. J. Alzheimer’s Dis. Rep. 2018, 2, 139–152. [Google Scholar] [CrossRef]
- Romani, L. Immunity to fungal infections. Nat. Rev. Immunol. 2004, 4, 11–24. [Google Scholar] [CrossRef]
- Gilchrist, K.B.; Garcia, M.C.; Sobonya, R.; Lipke, P.N.; Klotz, S.A. New features of invasive candidiasis in humans: Amyloid formation by fungi and deposition of serum amyloid P component by the host. J. Infect. Dis. 2012, 206, 1473–1478. [Google Scholar] [CrossRef]
- Wu, Y.; Du, S.; Johnson, J.L.; Tung, H.-Y.; Landers, C.T.; Liu, Y.; Seman, B.G.; Wheeler, R.T.; Costa-Mattioli, M.; Kheradmand, F.; et al. Microglia and amyloid precursor protein coordinate control of transient Candida cerebritis with memory deficits. Nat. Commun. 2019, 10, 58. [Google Scholar] [CrossRef]
- Bekkering, P.; Jafri, I.; van Overveld, F.J.; Rijkers, G.T. The intricate association between gut microbiota and development of Type 1, Type 2 and Type 3 diabetes. Expert Rev. Clin. Immunol. 2013, 9, 1031–1041. [Google Scholar] [CrossRef]
- Kim, D.; Zeng, M.Y.; Núñez, G. The interplay between host immune cells and gut microbiota in chronic inflammatory diseases. Exp. Mol. Med. 2017, 49, e339. [Google Scholar] [CrossRef]
- Cryan, J.F.; Dinan, T.G. Mind-altering microorganisms: The impact of the gut microbiota on brain and behaviour. Nat. Rev. Neurosci. 2012, 13, 701–712. [Google Scholar] [CrossRef] [PubMed]
- Collins, S.M.; Surette, M.; Bercik, P. The interplay between the intestinal microbiota and the brain. Nat. Rev. Microbiol. 2012, 10, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Vogt, N.M.; Kerby, R.L.; Dill-McFarland, K.A.; Harding, S.J.; Merluzzi, A.P.; Johnson, S.C.; Carlsson, C.M.; Asthana, S.; Zetterberg, H.; Blennow, K.; et al. Gut microbiome alterations in Alzheimer’s disease. Sci. Rep. 2017, 7, 13537. [Google Scholar] [CrossRef]
- Sanguinetti, E.; Collado, M.C.; Marrachelli, V.G.; Monleon, D.; Selma-Royo, M.; Pardo-Tendero, M.M.; Burchielli, S.; Iozzo, P. Microbiome-metabolome signatures in mice genetically prone to develop dementia, fed a normal or fatty diet. Sci. Rep. 2018, 8, 4907. [Google Scholar] [CrossRef] [PubMed]
- Minter, M.R.; Zhang, C.; Leone, V.; Ringus, D.L.; Zhang, X.; Oyler-Castrillo, P.; Musch, M.W.; Liao, F.; Ward, J.F.; Holtzman, D.M.; et al. Antibiotic-induced perturbations in gut microbial diversity influences neuro-inflammation and amyloidosis in a murine model of Alzheimer’s disease. Sci. Rep. 2016, 6, 30028. [Google Scholar] [CrossRef]
- Harach, T.; Marungruang, N.; Duthilleul, N.; Cheatham, V.; Mc Coy, K.D.; Frisoni, G.; Neher, J.J.; Fåk, F.; Jucker, M.; Lasser, T.; et al. Reduction of Abeta amyloid pathology in APPPS1 transgenic mice in the absence of gut microbiota. Sci. Rep. 2017, 7, 41802. [Google Scholar] [CrossRef]
- Forsyth, C.B.; Shannon, K.M.; Kordower, J.H.; Voigt, R.M.; Shaikh, M.; Jaglin, J.A.; Estes, J.D.; Dodiya, H.B.; Keshavarzian, A. Increased intestinal permeability correlates with sigmoid mucosa alpha-synuclein staining and endotoxin exposure markers in early Parkinson’s disease. PLoS ONE 2011, 6, e28032. [Google Scholar] [CrossRef]
- Keshavarzian, A.; Green, S.J.; Engen, P.A.; Voigt, R.M.; Naqib, A.; Forsyth, C.B.; Mutlu, E.; Shannon, K.M. Colonic bacterial composition in Parkinson’s disease. Mov. Disord. 2015, 30, 1351–1360. [Google Scholar] [CrossRef]
- Sampson, T.R.; Challis, C.; Jain, N.; Moiseyenko, A.; Ladinsky, M.S.; Shastri, G.G.; Thron, T.; Needham, B.D.; Horvath, I.; Debelius, J.W.; et al. A gut bacterial amyloid promotes α-synuclein aggregation and motor impairment in mice. Elife 2020, 9, e53111. [Google Scholar] [CrossRef]
- Uemura, N.; Yagi, H.; Uemura, M.T.; Hatanaka, Y.; Yamakado, H.; Takahashi, R. Inoculation of α-synuclein preformed fibrils into the mouse gastrointestinal tract induces Lewy body-like aggregates in the brainstem via the vagus nerve. Mol. Neurodegener. 2018, 13, 21. [Google Scholar] [CrossRef]
- Cattaneo, A.; Cattane, N.; Galluzzi, S.; Provasi, S.; Lopizzo, N.; Festari, C.; Ferrari, C.; Guerra, U.P.; Paghera, B.; Muscio, C.; et al. Association of brain amyloidosis with pro-inflammatory gut bacterial taxa and peripheral inflammation markers in cognitively impaired elderly. Neurobiol. Aging 2017, 49, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Wang, T.; Hu, X.; Luo, J.; Li, W.; Wu, X.; Duan, Y.; Jin, F. Administration of Lactobacillus helveticus NS8 improves behavioral, cognitive, and biochemical aberrations caused by chronic restraint stress. Neuroscience 2015, 310, 561–577. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Hu, X.; Liang, S.; Li, W.; Wu, X.; Wang, L.; Jin, F. Lactobacillus fermentum NS9 restores the antibiotic induced physiological and psychological abnormalities in rats. Benef. Microbes 2015, 6, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Ohsawa, K.; Uchida, N.; Ohki, K.; Nakamura, Y.; Yokogoshi, H. Lactobacillus helveticus-fermented milk improves learning and memory in mice. Nutr. Neurosci. 2015, 18, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Wang, T.; Liang, S.; Hu, X.; Li, W.; Jin, F. Ingestion of Lactobacillus strain reduces anxiety and improves cognitive function in the hyperammonemia rat. Sci. China Life Sci. 2014, 57, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Savignac, H.M.; Tramullas, M.; Kiely, B.; Dinan, T.G.; Cryan, J.F. Bifidobacteria modulate cognitive processes in an anxious mouse strain. Behav. Brain Res. 2015, 287, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Allen, A.P.; Hutch, W.; Borre, Y.E.; Kennedy, P.J.; Temko, A.; Boylan, G.; Murphy, E.; Cryan, J.F.; Dinan, T.G.; Clarke, G. Bifidobacterium longum 1714 as a translational psychobiotic: Modulation of stress, electrophysiology and neurocognition in healthy volunteers. Transl. Psychiatry 2016, 6. [Google Scholar] [CrossRef]
- Lew, L.-C.; Hor, Y.-Y.; Yusoff, N.A.A.; Choi, S.-B.; Yusoff, M.S.B.; Roslan, N.S.; Ahmad, A.; Mohammad, J.A.M.; Abdullah, M.F.I.L.; Zakaria, N.; et al. Probiotic Lactobacillus plantarum P8 alleviated stress and anxiety while enhancing memory and cognition in stressed adults: A randomised, double-blind, placebo-controlled study. Clin. Nutr. 2019, 38, 2053–2064. [Google Scholar] [CrossRef]
- Bonfili, L.; Cecarini, V.; Berardi, S.; Scarpona, S.; Suchodolski, J.S.; Nasuti, C.; Fiorini, D.; Boarelli, M.C.; Rossi, G.; Eleuteri, A.M. Microbiota modulation counteracts Alzheimer’s disease progression influencing neuronal proteolysis and gut hormones plasma levels. Sci. Rep. 2017, 7, 2426. [Google Scholar] [CrossRef]
- Wang, F.; Xu, T.; Zhang, Y.; Zheng, T.; He, Y.; He, F.; Jiang, Y. Long-term combined administration of Bifidobacterium bifidum TMC3115 and Lactobacillus plantarum 45 alleviates spatial memory impairment and gut dysbiosis in APP/PS1 mice. FEMS Microbiol. Lett. 2020, 367. [Google Scholar] [CrossRef]
- Wroblewski, L.E.; Peek, R.M.; Wilson, K.T. Helicobacter pylori and gastric cancer: Factors that modulate disease risk. Clin. Microbiol. Rev. 2010, 23, 713–739. [Google Scholar] [CrossRef] [PubMed]
- Cárdenas, M.V.; Boller, F.; Román, C.G. Helicobacter pylori, vascular risk factors and cognition in U.S. older adults. Brain Sci. 2019, 9, 370. [Google Scholar] [CrossRef] [PubMed]
- Doulberis, M.; Kotronis, G.; Thomann, R.; Polyzos, S.A.; Boziki, M.; Gialamprinou, D.; Deretzi, G.; Katsinelos, P.; Kountouras, J. Review: Impact of Helicobacter pylori on Alzheimer’s disease: What do we know so far? Helicobacter 2018, 23, e12454. [Google Scholar] [CrossRef]
- Chang, Y.-P.; Chiu, G.-F.; Kuo, F.-C.; Lai, C.-L.; Yang, Y.-H.; Hu, H.-M.; Chang, P.-Y.; Chen, C.-Y.; Wu, D.-C.; Yu, F.-J. Eradication of Helicobacter pylori is associated with the progression of dementia: A population-based study. Gastroenterol. Res. Pract. 2013, 2013, 175729. [Google Scholar] [CrossRef] [PubMed]
- Kountouras, J.; Boziki, M.; Gavalas, E.; Zavos, C.; Grigoriadis, N.; Deretzi, G.; Tzilves, D.; Katsinelos, P.; Tsolaki, M.; Chatzopoulos, D.; et al. Eradication of helicobacter pylori may be beneficial in the management of Alzheimer’s disease. J. Neurol. 2009, 256, 758–767. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Liu, L.; Ji, H.-F. Alzheimer’s disease histological and behavioral manifestations in transgenic mice correlate with specific gut microbiome state. J. Alzheimer’s Dis. 2017, 56, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Romero-Adrián, T.B.; Leal-Montiel, J.; Monsalve-Castillo, F.; Mengual-Moreno, E.; McGregor, E.G.; Perini, L.; Antúnez, A. Helicobacter pylori: Bacterial factors and the role of cytokines in the immune response. Curr. Microbiol. 2010, 60, 143–155. [Google Scholar] [CrossRef]
- Wyss-Coray, T.; Rogers, J. Inflammation in Alzheimer disease—A brief review of the basic science and clinical literature. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef]
- Roubaud-Baudron, C.; Krolak-Salmon, P.; Quadrio, I.; Mégraud, F.; Salles, N. Impact of chronic Helicobacter pylori infection on Alzheimer’s disease: Preliminary results. Neurobiol. Aging 2012, 33, 1009. e11–1009. e19. [Google Scholar] [CrossRef]
- Felice, C.; Federico, C.; Andrea, F.; Riccardo, A.C.; Walter, S.; Domenico, I.; Chiara, M.; Rosanna, C. The hypothesis that Helicobacter pylori predisposes to Alzheimer’s disease is biologically plausible. Sci. Rep. 2017, 7, 7817. [Google Scholar] [CrossRef]
- Noto, J.M.; Peek, R.M. Helicobacter pylori makes a molecular incision to gain epithelial entry. Cell Host Microbe 2017, 22, 434–436. [Google Scholar] [CrossRef]
- Turkina, M.V.; Olofsson, A.; Magnusson, K.E.; Arnqvist, A.; Vikström, E. Helicobacter pylori vesicles carrying CagA localize in the vicinity of cell-cell contacts and induce histone H1 binding to ATP in epithelial cells. FEMS Microbiol. Lett. 2015, 362. [Google Scholar] [CrossRef]
- Ou, Z.; Deng, L.; Lu, Z.; Wu, F.; Liu, W.; Huang, D.; Peng, Y. Protective effects of Akkermansia muciniphila on cognitive deficits and amyloid pathology in a mouse model of Alzheimer’s disease. Nutr. Diabetes 2020, 10, 12. [Google Scholar] [CrossRef]
- Yang, J.; Wise, L.; Fukuchi, K.I. TLR4 cross-talk with NLRP3 inflammasome and complement signaling pathways in Alzheimer’s disease. Front. Immunol. 2020. [Google Scholar] [CrossRef]
- Ashrafian, F.; Shahriary, A.; Behrouzi, A.; Moradi, H.R.; Keshavarz Azizi Raftar, S.; Lari, A.; Hadifar, S.; Yaghoubfar, R.; Ahmadi Badi, S.; Khatami, S.; et al. Akkermansia muciniphila-derived extracellular vesicles as a mucosal delivery vector for amelioration of obesity in mice. Front. Microbiol. 2019. [Google Scholar] [CrossRef]
- Ashrafian, F.; Behrouzi, A.; Shahriary, A.; Badi, S.A.; Davari, M.; Khatami, S.; Jamnani, F.R.; Fateh, A.; Vaziri, F.; Siadat, S.D. Comparative study of effect of Akkermansia muciniphila and its extracellular vesicles on toll-like receptors and tight junction. Gastroenterol. Hepatol. Bed Bench 2019. [Google Scholar] [CrossRef]
- Siniscalco, D.; Schultz, S.; Brigida, A.L.; Antonucci, N. Inflammation and neuro-immune dysregulations in autism spectrum disorders. Pharmaceuticals 2018, 11, 56. [Google Scholar] [CrossRef]
- Wang, L.; Christophersen, C.T.; Sorich, M.J.; Gerber, J.P.; Angley, M.T.; Conlon, M.A. Low relative abundances of the mucolytic bacterium Akkermansia muciniphila and Bifidobacterium spp. in feces of children with autism. Appl. Environ. Microbiol. 2011. [Google Scholar] [CrossRef]
- McGaughey, K.D.; Yilmaz-Swenson, T.; Elsayed, N.M.; Cruz, D.A.; Rodriguiz, R.M.; Kritzer, M.D.; Peterchev, A.V.; Roach, J.; Wetsel, W.C.; Williamson, D.E. Relative abundance of Akkermansia spp. and other bacterial phylotypes correlates with anxiety- and depressive-like behavior following social defeat in mice. Sci. Rep. 2019, 9, 3281. [Google Scholar] [CrossRef]
- Haikal, C.; Chen, Q.-Q.; Li, J.-Y. Microbiome changes: An indicator of Parkinson’s disease? Transl. Neurodegener. 2019, 8, 38. [Google Scholar] [CrossRef]
- Zhu, L.; Liu, W.; Alkhouri, R.; Baker, R.D.; Bard, J.E.; Quigley, E.M.; Baker, S.S. Structural changes in the gut microbiome of constipated patients. Physiol. Genomics 2014, 46, 679–686. [Google Scholar] [CrossRef]
- Vandeputte, D.; Falony, G.; Vieira-Silva, S.; Tito, R.Y.; Joossens, M.; Raes, J. Stool consistency is strongly associated with gut microbiota richness and composition, enterotypes and bacterial growth rates. Gut 2016, 65. [Google Scholar] [CrossRef]
- Lee, K.M.; Paik, C.N.; Chung, W.C.; Yang, J.M.; Choi, M.G. Breath methane positivity is more common and higher in patients with objectively proven delayed transit constipation. Eur. J. Gastroenterol. Hepatol. 2013, 25, 726–732. [Google Scholar] [CrossRef]
- Heintz-Buschart, A.; Pandey, U.; Wicke, T.; Sixel-Döring, F.; Janzen, A.; Sittig-Wiegand, E.; Trenkwalder, C.; Oertel, W.H.; Mollenhauer, B.; Wilmes, P. The nasal and gut microbiome in Parkinson’s disease and idiopathic rapid eye movement sleep behavior disorder. Mov. Disord. 2018, 46, 679–686. [Google Scholar] [CrossRef]
- Lin, C.H.; Chen, C.C.; Chiang, H.L.; Liou, J.M.; Chang, C.M.; Lu, T.P.; Chuang, E.Y.; Tai, Y.C.; Cheng, C.; Lin, H.Y.; et al. Altered gut microbiota and inflammatory cytokine responses in patients with Parkinson’s disease. J. Neuroinflamm. 2019, 16, 129. [Google Scholar] [CrossRef]
- Li, F.; Wang, P.; Chen, Z.; Sui, X.; Xie, X.; Zhang, J. Alteration of the fecal microbiota in North-Eastern Han Chinese population with sporadic Parkinson’s disease. Neurosci. Lett. 2019, 707, 134297. [Google Scholar] [CrossRef]
- Ridlon, J.M.; Harris, S.C.; Bhowmik, S.; Kang, D.J.; Hylemon, P.B. Consequences of bile salt biotransformations by intestinal bacteria. Gut Microbes 2016, 7, 22–39. [Google Scholar] [CrossRef]
- Kurdi, P.; Kawanishi, K.; Mizutani, K.; Yokota, A. Mechanism of growth inhibition by free bile acids in lactobacilli and bifidobacteria. J. Bacteriol. 2006. [Google Scholar] [CrossRef]
- Ramírez-Pérez, O.; Cruz-Ramón, V.; Chinchilla-López, P.; Méndez-Sánchez, N. The role of the gut microbiota in bile acid metabolism. Ann. Hepatol. 2017, 16, S21–S26. [Google Scholar] [CrossRef]
- Schubring, S.R.; Fleischer, W.; Lin, J.S.; Haas, H.L.; Sergeeva, O.A. The bile steroid chenodeoxycholate is a potent antagonist at NMDA and GABA A receptors. Neurosci. Lett. 2012, 506, 322–326. [Google Scholar] [CrossRef]
- Klindt, C.; Reich, M.; Hellwig, B.; Stindt, J.; Rahnenführer, J.; Hengstler, J.G.; Köhrer, K.; Schoonjans, K.; Häussinger, D.; Keitel, V. The G Protein-coupled bile acid receptor TGR5 (Gpbar1) modulates endothelin-1 signaling in liver. Cells 2019, 8, 1467. [Google Scholar] [CrossRef] [PubMed]
- Hoeke, M.O.; Heegsma, J.; Hoekstra, M.; Moshage, H.; Faber, K.N. Human FXR regulates SHP expression through direct binding to an LRH-1 binding site, independent of an IR-1 and LRH-1. PLoS ONE 2014. [Google Scholar] [CrossRef]
- Chen, Q.; Ma, H.; Guo, X.; Liu, J.; Gui, T.; Gai, Z. Farnesoid X receptor (FXR) aggravates amyloid-β-triggered apoptosis by modulating the cAMP-response element-binding protein (CREB)/brain-derived neurotrophic factor (BDNF) pathway in vitro. Med. Sci. Monit. 2019, 25, 9335–9345. [Google Scholar] [CrossRef] [PubMed]
- Vavassori, P.; Mencarelli, A.; Renga, B.; Distrutti, E.; Fiorucci, S. The bile acid receptor FXR is a modulator of intestinal innate immunity. J. Immunol. 2009, 183, 6251–6261. [Google Scholar] [CrossRef]
- Castro-Caldas, M.; Carvalho, A.N.; Rodrigues, E.; Henderson, C.J.; Wolf, C.R.; Rodrigues, C.M.P.; Gama, M.J. Tauroursodeoxycholic acid prevents MPTP-induced dopaminergic cell death in a mouse model of Parkinson’s disease. Mol. Neurobiol. 2012, 46, 475–486. [Google Scholar] [CrossRef]
- Ramalho, R.M.; Nunes, A.F.; Dias, R.B.; Amaral, J.D.; Lo, A.C.; D’Hooge, R.; Sebastião, A.M.; Rodrigues, C.M.P. Tauroursodeoxycholic acid suppresses amyloid β-induced synaptic toxicity in vitro and in APP/PS1 mice. Neurobiol. Aging 2013, 34, 551–561. [Google Scholar] [CrossRef]
- Lo, A.C.; Callaerts-Vegh, Z.; Nunes, A.F.; Rodrigues, C.M.P.; D’Hooge, R. Tauroursodeoxycholic acid (TUDCA) supplementation prevents cognitive impairment and amyloid deposition in APP/PS1 mice. Neurobiol. Dis. 2013, 50, 21–29. [Google Scholar] [CrossRef]
- Cortez, L.M.; Campeau, J.; Norman, G.; Kalayil, M.; Van der Merwe, J.; McKenzie, D.; Sim, V.L. Bile Acids reduce prion conversion, reduce neuronal loss, and prolong male survival in models of prion disease. J. Virol. 2015, 89, 7660–7672. [Google Scholar] [CrossRef]
- Rosa, A.I.; Fonseca, I.; Nunes, M.J.; Moreira, S.; Rodrigues, E.; Carvalho, A.N.; Rodrigues, C.M.P.; Gama, M.J.; Castro-Caldas, M. Novel insights into the antioxidant role of tauroursodeoxycholic acid in experimental models of Parkinson’s disease. Biochim. Biophys. Acta-Mol. Basis Dis. 2017, 1863, 2171–2181. [Google Scholar] [CrossRef]
- Nunes, A.F.; Amaral, J.D.; Lo, A.C.; Fonseca, M.B.; Viana, R.J.S.; Callaerts-Vegh, Z.; D’Hooge, R.; Rodrigues, C.M.P. TUDCA, a bile acid, attenuates amyloid precursor protein processing and amyloid-β deposition in APP/PS1 mice. Mol. Neurobiol. 2012, 45, 440–454. [Google Scholar] [CrossRef]
- Wu, X.; Liu, C.; Chen, L.; Du, Y.-F.; Hu, M.; Reed, M.N.; Long, Y.; Suppiramaniam, V.; Hong, H.; Tang, S.-S. Protective effects of tauroursodeoxycholic acid on lipopolysaccharide-induced cognitive impairment and neurotoxicity in mice. Int. Immunopharmacol. 2019, 72, 166–175. [Google Scholar] [CrossRef] [PubMed]
- MahmoudianDehkordi, S.; Arnold, M.; Nho, K.; Ahmad, S.; Jia, W.; Xie, G.; Louie, G.; Kueider-Paisley, A.; Moseley, M.A.; Thompson, J.W.; et al. Altered bile acid profile associates with cognitive impairment in Alzheimer’s disease—An emerging role for gut microbiome. Alzheimer’s Dement. 2019, 15, 76–92. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Wu, L.; Peng, G.; Han, Y.; Tang, R.; Ge, J.; Zhang, L.; Jia, L.; Yue, S.; Zhou, K.; et al. Altered microbiomes distinguish Alzheimer’s disease from amnestic mild cognitive impairment and health in a Chinese cohort. Brain. Behav. Immun. 2019, 80, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Vital, M.; Rud, T.; Rath, S.; Pieper, D.H.; Schlüter, D. Diversity of bacteria exhibiting bile acid-inducible 7α-dehydroxylation genes in the human gut. Comput. Struct. Biotechnol. J. 2019, 17, 1016–1019. [Google Scholar] [CrossRef] [PubMed]
- Quinn, M.; McMillin, M.; Galindo, C.; Frampton, G.; Pae, H.Y.; DeMorrow, S. Bile acids permeabilize the blood brain barrier after bile duct ligation in rats via Rac1-dependent mechanisms. Dig. Liver Dis. 2014, 46, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Erickson, M.A.; Dohi, K.; Banks, W.A. Neuroinflammation: A common pathway in CNS diseases as mediated at the blood-brain barrier. Neuroimmunomodulation 2012, 19, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Kalakoti, P.; Nanda, A.; Sun, H. Blood-brain barrier disruption during neuroinflammation. In Neuroinflammation; Elsevier: Amsterdam, The Netherlands, 2018; pp. 529–539. [Google Scholar]
- Bhattacharjee, S.; Lukiw, W.J. Alzheimer’s disease and the microbiome. Front. Cell. Neurosci. 2013, 7, 153. [Google Scholar] [CrossRef] [PubMed]
- Asti, A.; Gioglio, L. Can a bacterial endotoxin be a key factor in the kinetics of amyloid fibril formation? J. Alzheimer’s Dis. 2014, 39, 169–179. [Google Scholar] [CrossRef]
- Blasko, I.; Marx, F.; Steiner, E.; Hartmann, T.; Grubeck-Loebenstein, B. TNFα plus IFNγ induce the production of Alzheimer β-amyloid peptides and decrease the secretion of APPs. FASEB J. 1999, 13, 63–68. [Google Scholar] [CrossRef]
- Buxbaum, J.D.; Oishi, M.; Chen, H.I.; Pinkas-Kramarski, R.; Jaffe, E.A.; Gandy, S.E.; Greengard, P. Cholinergic agonists and interleukin 1 regulate processing and secretion of the Alzheimer β/A4 amyloid protein precursor. Proc. Natl. Acad. Sci. USA 1992, 89, 10075–10078. [Google Scholar] [CrossRef]
- Evans, M.L.; Chorell, E.; Taylor, J.D.; Åden, J.; Götheson, A.; Li, F.; Koch, M.; Sefer, L.; Matthews, S.J.; Wittung-Stafshede, P.; et al. The bacterial curli system possesses a potent and selective inhibitor of amyloid formation. Mol. Cell 2015, 57, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.G.; Stribinskis, V.; Rane, M.J.; Demuth, D.R.; Gozal, E.; Roberts, A.M.; Jagadapillai, R.; Liu, R.; Choe, K.; Shivakumar, B.; et al. Exposure to the functional bacterial amyloid protein curli enhances alpha-synuclein aggregation in aged fischer 344 rats and caenorhabditis elegans. Sci. Rep. 2016, 6, 1–10. [Google Scholar] [CrossRef]
- Ho, L.; Ono, K.; Tsuji, M.; Mazzola, P.; Singh, R.; Pasinetti, G.M. Protective roles of intestinal microbiota derived short chain fatty acids in Alzheimer’s disease-type beta-amyloid neuropathological mechanisms. Expert Rev. Neurother. 2018, 18, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Pistollato, F.; Cano, S.S.; Elio, I.; Vergara, M.M.; Giampieri, F.; Battino, M. Role of gut microbiota and nutrients in amyloid formation and pathogenesis of Alzheimer disease. Nutr. Rev. 2016, 74, 624–634. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.; Silverstein, P.S.; Singh, D.P.; Kumar, A. Involvement of metabotropic glutamate receptor 5, AKT/PI3K Signaling and NF-κB pathway in methamphetamine-mediated increase in IL-6 and IL-8 expression in astrocytes. J. Neuroinflamm. 2012, 9, 547. [Google Scholar] [CrossRef]
- Chantong, B.; Kratschmar, D.V.; Lister, A.; Odermatt, A. Inhibition of metabotropic glutamate receptor 5 induces cellular stress through pertussis toxinsensitive Gi-proteins in murine BV-2 microglia cells. J. Neuroinflamm. 2014, 11, 190. [Google Scholar] [CrossRef]
- Renner, M.; Lacor, P.N.; Velasco, P.T.; Xu, J.; Contractor, A.; Klein, W.L.; Triller, A. Deleterious effects of amyloid β oligomers acting as an extracellular scaffold for mGluR5. Neuron 2010, 66, 739–754. [Google Scholar] [CrossRef]
- Shrivastava, A.N.; Aperia, A.; Melki, R.; Triller, A. Physico-pathologic mechanisms involved in neurodegeneration: Misfolded protein-plasma membrane interactions. Neuron 2017, 95, 33–50. [Google Scholar] [CrossRef]
- Yang, T.; Liu, Y.W.; Zhao, L.; Wang, H.; Yang, N.; Dai, S.S.; He, F. Metabotropic glutamate receptor 5 deficiency inhibits neutrophil infiltration after traumatic brain injury in mice. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Sanmiguel, J.; Schuh, C.M.A.P.; Muñoz-Montesino, C.; Contreras-Kallens, P.; Aguayo, L.G.; Aguayo, S. Complex Interaction between Resident Microbiota and Misfolded Proteins: Role in Neuroinflammation and Neurodegeneration. Cells 2020, 9, 2476. https://doi.org/10.3390/cells9112476
González-Sanmiguel J, Schuh CMAP, Muñoz-Montesino C, Contreras-Kallens P, Aguayo LG, Aguayo S. Complex Interaction between Resident Microbiota and Misfolded Proteins: Role in Neuroinflammation and Neurodegeneration. Cells. 2020; 9(11):2476. https://doi.org/10.3390/cells9112476
Chicago/Turabian StyleGonzález-Sanmiguel, Juliana, Christina M. A. P. Schuh, Carola Muñoz-Montesino, Pamina Contreras-Kallens, Luis G. Aguayo, and Sebastian Aguayo. 2020. "Complex Interaction between Resident Microbiota and Misfolded Proteins: Role in Neuroinflammation and Neurodegeneration" Cells 9, no. 11: 2476. https://doi.org/10.3390/cells9112476
APA StyleGonzález-Sanmiguel, J., Schuh, C. M. A. P., Muñoz-Montesino, C., Contreras-Kallens, P., Aguayo, L. G., & Aguayo, S. (2020). Complex Interaction between Resident Microbiota and Misfolded Proteins: Role in Neuroinflammation and Neurodegeneration. Cells, 9(11), 2476. https://doi.org/10.3390/cells9112476