Early Treatment of Interleukin-33 can Attenuate Lupus Development in Young NZB/W F1 Mice



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Approval
2.2. Animals and Treatment
2.3. Measurement of Proteinuria and Anti-dsDNA Antibodies
2.4. Renal Histology
2.5. Flow Cytometry Analysis
2.6. RNA Extraction and Sequencing Analysis
2.7. Statistical Analysis
3. Results
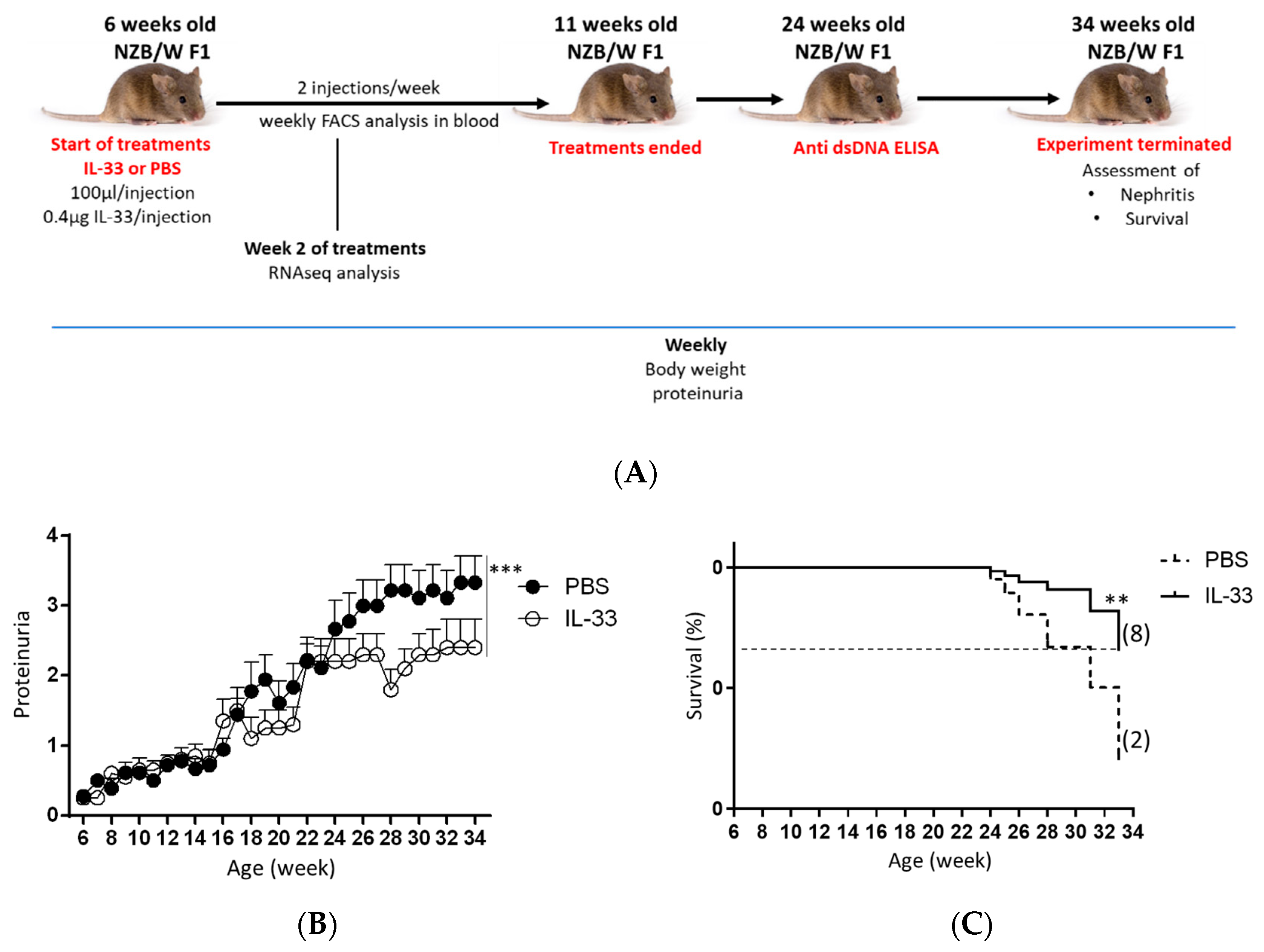
3.1. Early IL-33 Treatments Reduced Proteinuria Level and Prolonged Survival in NZB/W F1 Mice
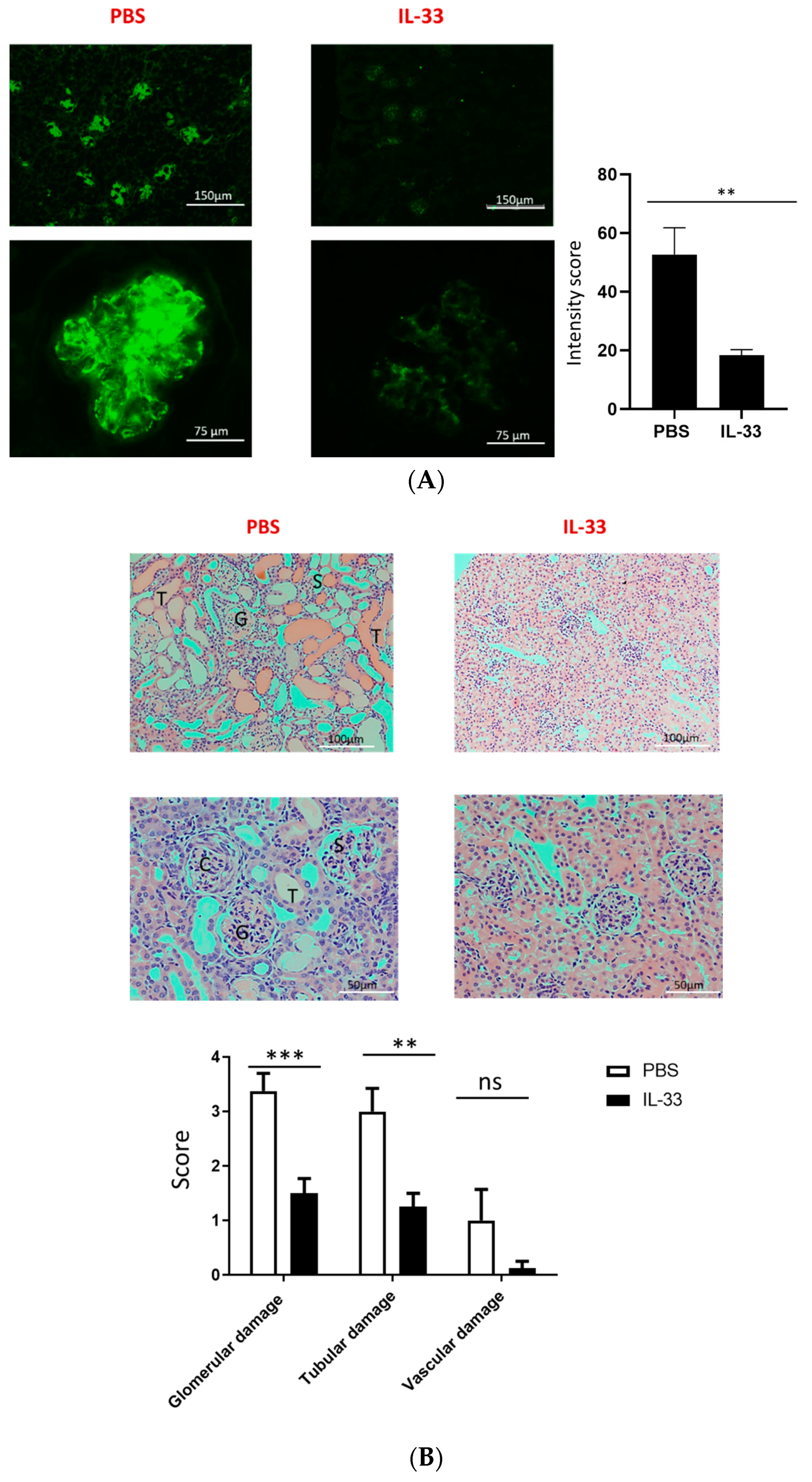
3.2. Early IL-33 Treatments Reduced IgG-Immune-Complex Deposition and Glomerular and Tubular Damage in NZB/W F1 Mice
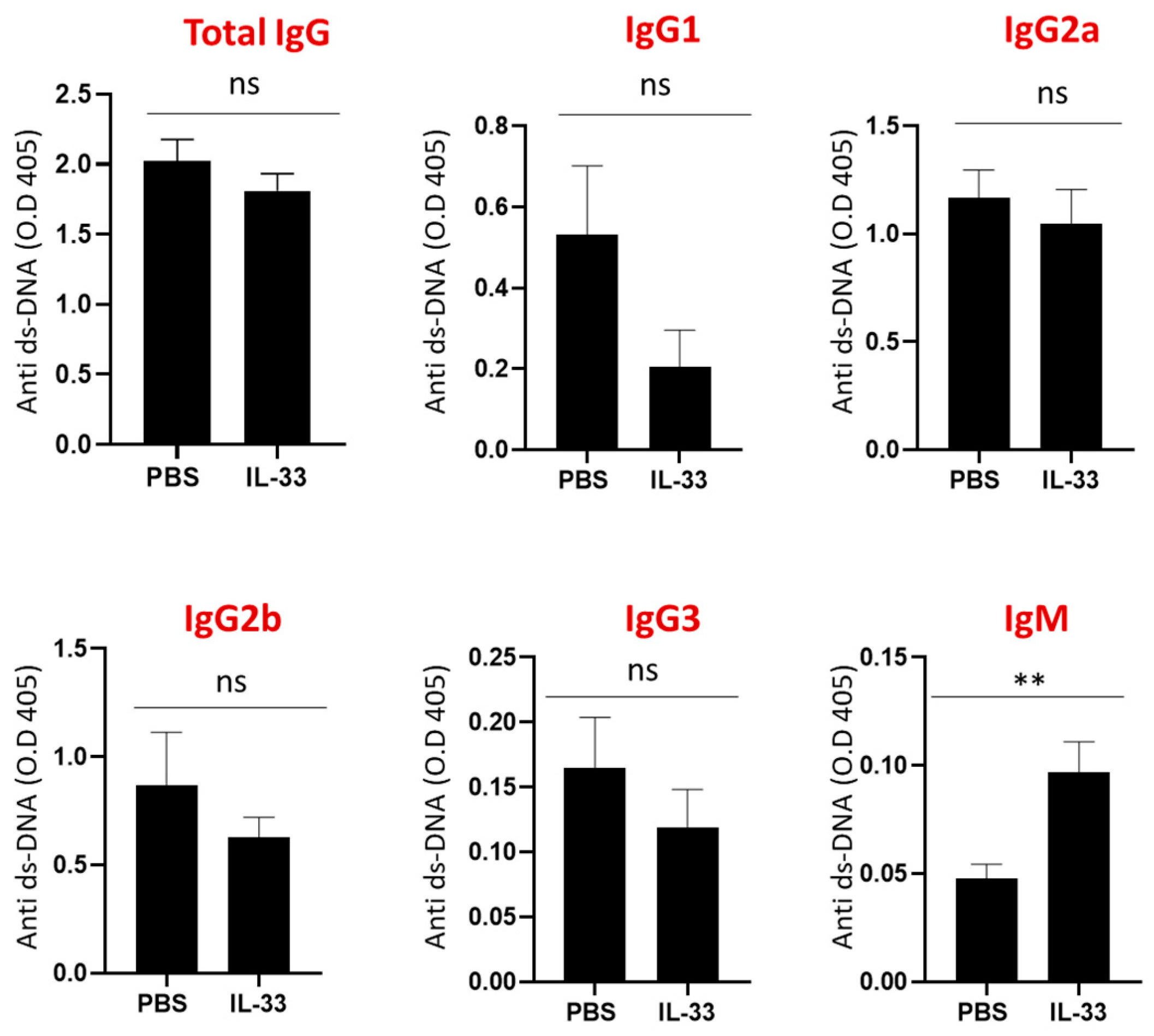
3.3. Early IL-33 Treatments Induced IgM Anti-dsDNA Antibodies in NZB/W F1 Mice
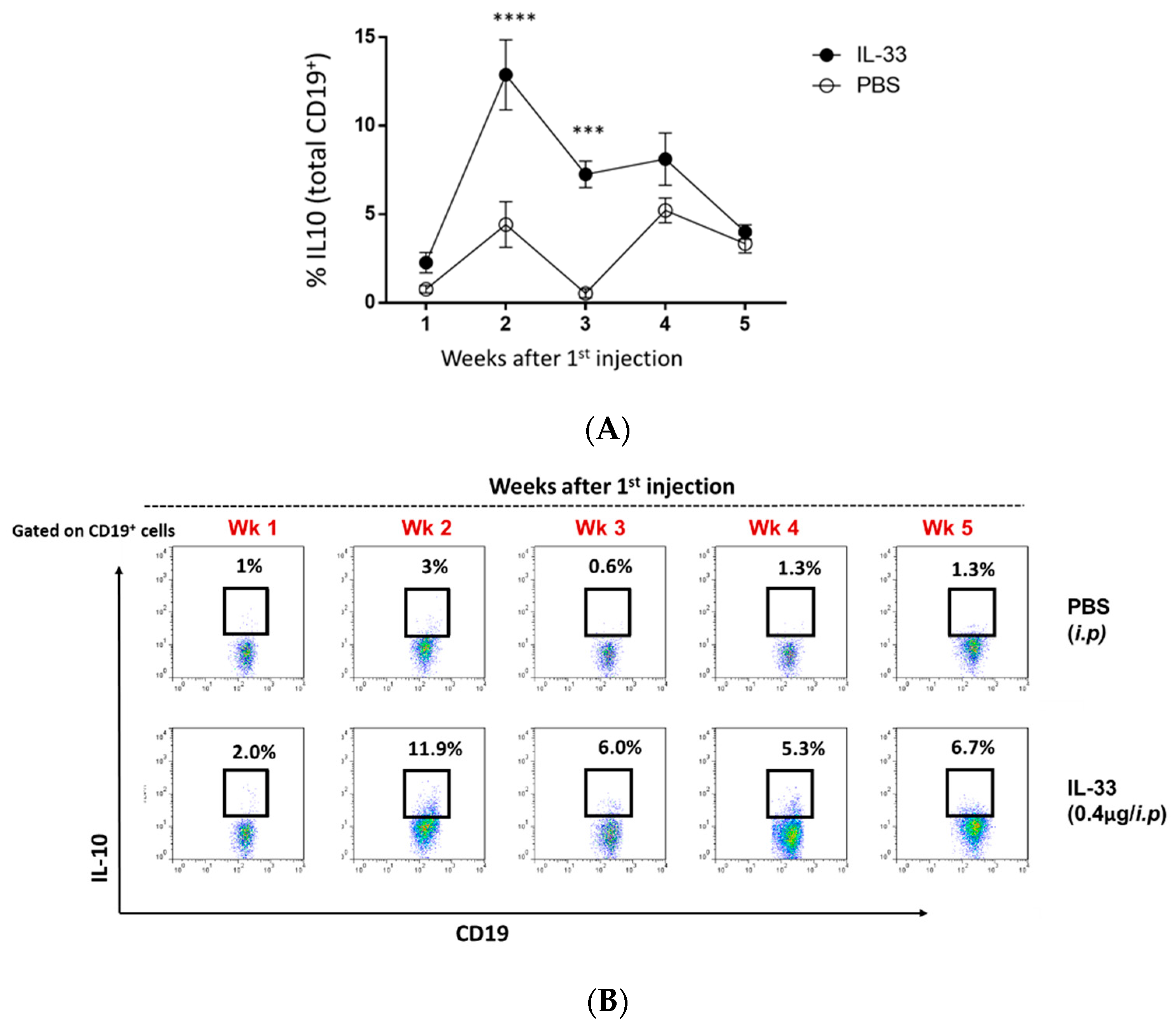
3.4. IL-33 Treatments Increased the Peripheral Breg Cells in NZB/W F1 Mice
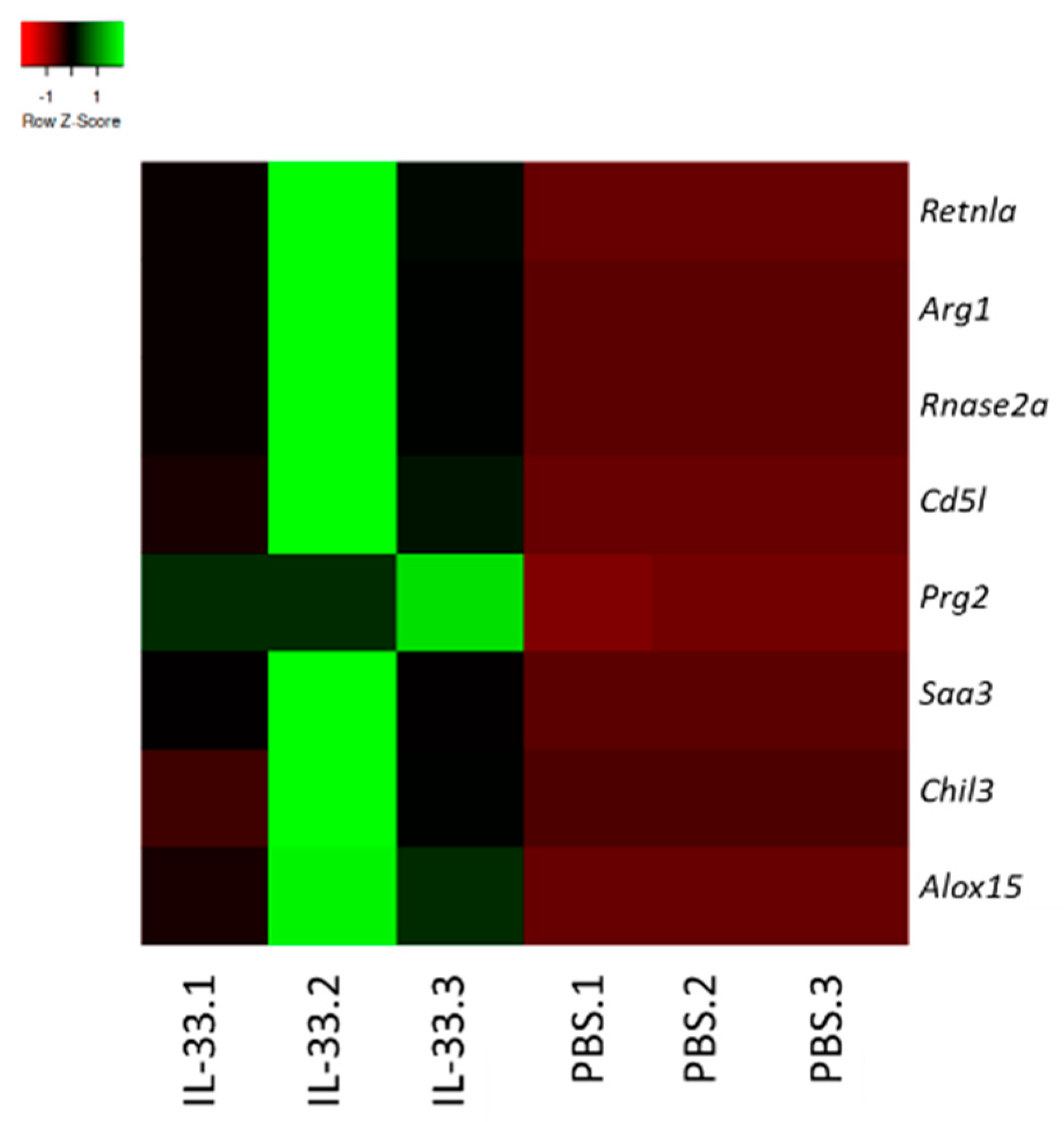
3.5. IL-33 Treatments Induced M2-Macrophage-Related Genes in NZB/W F1 Mice
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schmitz, J.; Owyang, A.; Oldham, E.; Song, Y.; Murphy, E.; McClanahan, T.K.; Zurawski, G.; Moshrefi, M.; Qin, J.; Li, X.; et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 2005, 23, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Molofsky, A.B.; Savage, A.K.; Locksley, R.M. Interleukin-33 in Tissue Homeostasis, Injury, and Inflammation. Immunity 2015, 42, 1005–1019. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Hammel, M.; He, Y.; Tainer, J.A.; Jeng, U.S.; Zhang, L.; Wang, S.; Wang, X. Structural insights into the interaction of IL-33 with its receptors. Proc. Natl. Acad. Sci. USA 2013, 110, 14918–14923. [Google Scholar] [CrossRef] [PubMed]
- Andrade, M.V.; Iwaki, S.; Ropert, C.; Gazzinelli, R.T.; Cunha-Melo, J.R.; Beaven, M.A. Amplification of cytokine production through synergistic activation of NFAT and AP-1 following stimulation of mast cells with antigen and IL-33. Eur. J. Immunol. 2011, 41, 760–772. [Google Scholar] [CrossRef]
- Kurowska-Stolarska, M.; Kewin, P.; Murphy, G.; Russo, R.C.; Stolarski, B.; Garcia, C.C.; Komai-Koma, M.; Pitman, N.; Li, Y.; McKenzie, A.N.J.; et al. IL-33 Induces Antigen-Specific IL-5 + T Cells and Promotes Allergic-Induced Airway Inflammation Independent of IL-4. J. Immunol. 2008, 181, 4780–4790. [Google Scholar] [CrossRef]
- Frisbee, A.L.; Saleh, M.M.; Young, M.K.; Leslie, J.L.; Simpson, M.E.; Abhyankar, M.M.; Cowardin, C.A.; Ma, J.Z.; Pramoonjago, P.; Turner, S.D.; et al. IL-33 drives group 2 innate lymphoid cell-mediated protection during Clostridium difficile infection. Nat. Commun. 2019, 10, 2712. [Google Scholar] [CrossRef]
- Lu, J.; Kang, J.; Zhang, C.; Zhang, X. The role of IL-33/ST2L signals in the immune cells. Immunol. Lett. 2015, 164, 11–17. [Google Scholar] [CrossRef]
- De la Fuente, M.; MacDonald, T.T.; Hermoso, M.A. The IL-33/ST2 axis: Role in health and disease. Cytokine Growth Factor Rev. 2015, 26, 615–623. [Google Scholar] [CrossRef]
- Liew, F.Y.; Girard, J.P.; Turnquist, H.R. Interleukin-33 in health and disease. Nat. Rev. Immunol. 2016, 16, 676–689. [Google Scholar] [CrossRef]
- Mok, C.C.; Lau, C.S. Pathogenesis of systemic lupus erythematosus. J. Clin. Pathol. 2003, 56, 481–490. [Google Scholar] [CrossRef]
- Yang, Z.; Liang, Y.; Xi, W.; Li, C.; Zhong, R. Association of increased serum IL-33 levels with clinical and laboratory characteristics of systemic lupus erythematosus in Chinese population. Clin. Exp. Med. 2011, 11, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Mok, M.Y.; Huang, F.P.; Ip, W.K.; Lo, Y.; Wong, F.Y.; Chan, E.Y.T.; Lam, K.F.; Xu, D. Serum levels of IL-33 and soluble ST2 and their association with disease activity in systemic lupus erythematosus. Rheumatology 2009, 49, 520–527. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Lin, W.; Zheng, X. IL-33 neutralization suppresses lupus disease in lupus-prone mice. Inflammation 2014, 37, 824–832. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Xie, L.; Qin, H.; Liang, J.; Yang, Y.; Xu, J.; Zhang, T. Interaction between IL-33 gene polymorphisms and current smoking with susceptibility to systemic lupus erythematosus. J. Immunol. Res. 2019, 2019, 1547578. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Liu, Y.; Ye, D. Association between IL-33 Gene Polymorphisms (rs1929992, rs7044343) and Systemic Lupus Erythematosus in a Chinese Han Population. Immunol. Investig. 2016, 45, 575–583. [Google Scholar] [CrossRef]
- Guo, J.; Xiang, Y.; Peng, Y.F.; Huang, H.T.; Lan, Y.; Wei, Y.S. The association of novel IL-33 polymorphisms with sIL-33 and risk of systemic lupus erythematosus. Mol. Immunol. 2016, 77, 1–7. [Google Scholar] [CrossRef]
- Sattler, S.; Ling, G.S.; Xu, D.; Hussaarts, L.; Romaine, A.; Zhao, H.; Fossati-Jimack, L.; Malik, T.; Cook, H.T.; Botto, M.; et al. IL-10-producing regulatory B cells induced by IL-33 (BregIL-33) effectively attenuate mucosal inflammatory responses in the gut. J. Autoimmun. 2014, 50, 107–122. [Google Scholar] [CrossRef]
- Ma, L.; Chan, K.W.; Trendell-Smith, N.J.; Wu, A.; Tian, L.; Lam, A.C.; Chan, A.K.; Lo, C.K.; Chik, S.; Ko, K.H.; et al. Systemic autoimmune disease induced by dendritic cells that have captured necrotic but not apoptotic cells in susceptible mouse strains. Eur. J. Immunol. 2005, 35, 3364–3375. [Google Scholar] [CrossRef]
- Lee, S.W.; Kim, B.S. Comparison of therapeutic efficacy between bortezomib and combination treatment of prednisolone and mycophenolate mofetil on nephritis in NZB/WF1 mice. Clin. Exp. Rheumatol. 2010, 28, 393–396. [Google Scholar]
- Chen, H.H.; Lu, P.J.; Chen, B.R.; Hsiao, M.; Ho, W.Y.; Tseng, C.J. Heme oxygenase-1 ameliorates kidney ischemia-reperfusion injury in mice through extracellular signal-regulated kinase 1/2-enhanced tubular epithelium proliferation. Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 2195–2201. [Google Scholar] [CrossRef]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Frazee, A.C.; Pertea, G.; Jaffe, A.E.; Langmead, B.; Salzberg, S.L.; Leek, J.T. Ballgown bridges the gap between transcriptome assembly and expression analysis. Nat. Biotechnol. 2015, 33, 243–246. [Google Scholar] [CrossRef]
- Macanovic, M.; Sinicropi, D.; Shak, S.; Baughman, S.; Thiru, S.; Lachmann, P.J. The treatment of systemic lupus erythematosus (SLE) in NZB/W F1 hybrid mice; studies with recombinant murine DNase and with dexamethasone. Clin. Exp. Immunol. 1996, 106, 243–252. [Google Scholar] [CrossRef]
- Jiang, C.; Zhao, M.L.; Scearce, R.M.; Diaz, M. Activation-induced deaminase-deficient mrl/lpr mice secrete high levels of protective antibodies against lupus nephritis. Arthritis Rheum. 2011, 63, 1086–1096. [Google Scholar] [CrossRef]
- Werwitzke, S.; Trick, D.; Kamino, K.; Matthias, T.; Kniesch, K.; Schlegelberger, B.; Schmidt, R.E.; Witte, T. Inhibition of lupus disease by anti-double-stranded DNA antibodies of the IgM isotype in the (NZB × NZW)F1 mouse. Arthritis Rheum. 2005, 52, 3629–3638. [Google Scholar] [CrossRef]
- Szymczak, W.A.; Deepe, G.S. The CCL7-CCL2-CCR2 Axis Regulates IL-4 Production in Lungs and Fungal Immunity. J. Immunol. 2009, 183, 1964–1974. [Google Scholar] [CrossRef]
- Sanjurjo, L.; Aran, G.; Téllez, É.; Amézaga, N.; Armengol, C.; López, D.; Prats, C.; Sarrias, M.R. CD5L promotes M2 macrophage polarization through autophagy-mediated upregulation of ID3. Front. Immunol. 2018, 9, 480. [Google Scholar] [CrossRef]
- Yamada, K.J.; Barker, T.; Dyer, K.D.; Rice, T.A.; Percopo, C.M.; Garcia-Crespo, K.E.; Cho, S.; Lee, J.J.; Druey, K.M.; Rosenberg, H.F. Eosinophil-associated ribonuclease 11 is a macrophage chemoattractant. J. Biol. Chem. 2015, 290, 8863–8875. [Google Scholar] [CrossRef]
- Snelgrove, R.J.; Gregory, L.G.; Peiró, T.; Akthar, S.; Campbell, G.A.; Walker, S.A.; Lloyd, C.M. Alternaria-derived serine protease activity drives IL-33-mediated asthma exacerbations. J. Allergy Clin. Immunol. 2014, 134, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Ye, R.D.; Sun, L. Emerging functions of serum amyloid A in inflammation. J. Leukoc. Biol. 2015, 98, 923–929. [Google Scholar] [CrossRef] [PubMed]
- Witte, T.; Hartung, K.; Sachse, C.; Matthias, T.; Fricke, M.; Deicher, H.; Kalden, J.R.; Lakomek, H.J.; Peter, H.H.; Schmidt, R.E. IgM anti-dsDNA antibodies in systemic lupus erythematosus: Negative association with nephritis. Rheumatol. Int. 1998, 18, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Villalta, D.; Bizzaro, N.; Bassi, N.; Zen, M.; Gatto, M.; Ghirardello, A.; Iaccarino, L.; Punzi, L.; Doria, A. Anti-dsDNA Antibody Isotypes in Systemic Lupus Erythematosus: IgA in Addition to IgG Anti-dsDNA Help to Identify Glomerulonephritis and Active Disease. PLoS ONE 2013, 8, e71458. [Google Scholar] [CrossRef]
- Zhu, J.F.; Xu, Y.; Zhao, J.; Li, X.; Meng, X.; Wang, T.Q.; Zou, B.Y.; Zhao, P.Y.; Liu, Q.; Lu, C.L.; et al. IL-33 protects mice against dss-induced chronic colitis by increasing both regulatory B cell and regulatory T cell responses as well as decreasing Th17 Cell response. J. Immunol. Res. 2018, 2018, 1827901. [Google Scholar] [CrossRef]
- Sokolov, A.V.; Shmidt, A.A.; Lomakin, Y.A. B cell regulation in autoimmune diseases. Acta Nat. 2018, 10, 11–22. [Google Scholar] [CrossRef]
- Menon, M.; Blair, P.A.; Isenberg, D.A.; Mauri, C. A Regulatory Feedback between Plasmacytoid Dendritic Cells and Regulatory B Cells Is Aberrant in Systemic Lupus Erythematosus. Immunity 2016, 44, 683–697. [Google Scholar] [CrossRef]
- Watanabe, R.; Ishiura, N.; Nakashima, H.; Kuwano, Y.; Okochi, H.; Tamaki, K.; Sato, S.; Tedder, T.F.; Fujimoto, M. Regulatory B Cells (B10 Cells) Have a Suppressive Role in Murine Lupus: CD19 and B10 Cell Deficiency Exacerbates Systemic Autoimmunity. J. Immunol. 2010, 184, 4801–4809. [Google Scholar] [CrossRef]
- Wang, C.; Dong, C.; Xiong, S. IL-33 enhances macrophage M2 polarization and protects mice from CVB3-induced viral myocarditis. J. Mol. Cell. Cardiol. 2017, 103, 22–30. [Google Scholar] [CrossRef]
- Tu, L.; Chen, J.; Xu, D.; Xie, Z.; Yu, B.; Tao, Y.; Shi, G.; Duan, L. IL-33-induced alternatively activated macrophage attenuates the development of TNBS-induced colitis. Oncotarget 2017, 8, 27704–27714. [Google Scholar] [CrossRef]
- Mohd Jaya, F.N.; Garcia, S.G.; Borràs, F.E.; Chan, G.C.F.; Franquesa, M. Paradoxical role of Breg-inducing cytokines in autoimmune diseases. J. Transl. Autoimmun. 2019, 2, 100011. [Google Scholar] [CrossRef] [PubMed]
- Peine, M.; Marek, R.M.; Löhning, M. IL-33 in T Cell Differentiation, Function, and Immune Homeostasis. Trends Immunol. 2016, 37, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, T.; Horikawa, M.; Iwata, Y.; Tedder, T.F. Regulatory B Cells (B10 Cells) and Regulatory T Cells Have Independent Roles in Controlling Experimental Autoimmune Encephalomyelitis Initiation and Late-Phase Immunopathogenesis. J. Immunol. 2010, 185, 2240–2252. [Google Scholar] [CrossRef] [PubMed]
- Yanaba, K.; Hamaguchi, Y.; Venturi, G.M.; Steeber, D.A.; Clair, E.W.S.; Tedder, T.F. B Cell Depletion Delays Collagen-Induced Arthritis in Mice: Arthritis Induction Requires Synergy between Humoral and Cell-Mediated Immunity. J. Immunol. 2007, 179, 1369–1380. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.R.; Milovanović, M.; Allan, D.; Niedbala, W.; Besnard, A.G.; Fukada, S.Y.; Alves-Filho, J.C.; Togbe, D.; Goodyear, C.S.; Linington, C.; et al. IL-33 attenuates EAE by suppressing IL-17 and IFN-γ production and inducing alternatively activated macrophages. Eur. J. Immunol. 2012, 42, 1804–1814. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Li, Y.; Liu, X.; Gao, X.; Wang, Y. IL-33 blockade suppresses the development of experimental autoimmune encephalomyelitis in C57BL/6 mice. J. Neuroimmunol. 2012, 247, 25–31. [Google Scholar] [CrossRef]
- Chen, H.; Sun, Y.; Lai, L.; Wu, H.; Xiao, Y.; Ming, B.; Gao, M.; Zou, H.; Xiong, P.; Xu, Y.; et al. Interleukin-33 is released in spinal cord and suppresses experimental autoimmune encephalomyelitis in mice. Neuroscience 2015, 308, 157–168. [Google Scholar] [CrossRef]
- Altara, R.; Ghali, R.; Mallat, Z.; Cataliotti, A.; Booz, G.W.; Zouein, F.A. Conflicting vascular and metabolic impact of the IL-33/sST2 axis. Cardiovasc. Res. 2018, 114, 1578–1594. [Google Scholar] [CrossRef]
- Miller, A.M.; Xu, D.; Asquith, D.L.; Denby, L.; Li, Y.; Sattar, N.; Baker, A.H.; McInnes, I.B.; Liew, F.Y. IL-33 reduces the development of atherosclerosis. J. Exp. Med. 2008, 205, 339–346. [Google Scholar] [CrossRef]
- Ngkelo, A.; Richart, A.; Kirk, J.A.; Bonnin, P.; Vilar, J.; Lemitre, M.; Marck, P.; Branchereau, M.; Le Gall, S.; Renault, N.; et al. Mast cells regulate myofilament calcium sensitization and heart function after myocardial infarction. J. Exp. Med. 2016, 213, 1353–1374. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohd Jaya, F.N.; Liu, Z.; Chan, G.C.-F. Early Treatment of Interleukin-33 can Attenuate Lupus Development in Young NZB/W F1 Mice. Cells 2020, 9, 2448. https://doi.org/10.3390/cells9112448
Mohd Jaya FN, Liu Z, Chan GC-F. Early Treatment of Interleukin-33 can Attenuate Lupus Development in Young NZB/W F1 Mice. Cells. 2020; 9(11):2448. https://doi.org/10.3390/cells9112448
Chicago/Turabian StyleMohd Jaya, Fatin Nurizzati, Zhongyi Liu, and Godfrey Chi-Fung Chan. 2020. "Early Treatment of Interleukin-33 can Attenuate Lupus Development in Young NZB/W F1 Mice" Cells 9, no. 11: 2448. https://doi.org/10.3390/cells9112448
APA StyleMohd Jaya, F. N., Liu, Z., & Chan, G. C.-F. (2020). Early Treatment of Interleukin-33 can Attenuate Lupus Development in Young NZB/W F1 Mice. Cells, 9(11), 2448. https://doi.org/10.3390/cells9112448