Metformin: Metabolic Rewiring Faces Tumor Heterogeneity

 ,
,  ,
,  ,
, {kind=link}
{kind=link}
Abstract
1. A Brief Intro to Tumor Heterogeneity
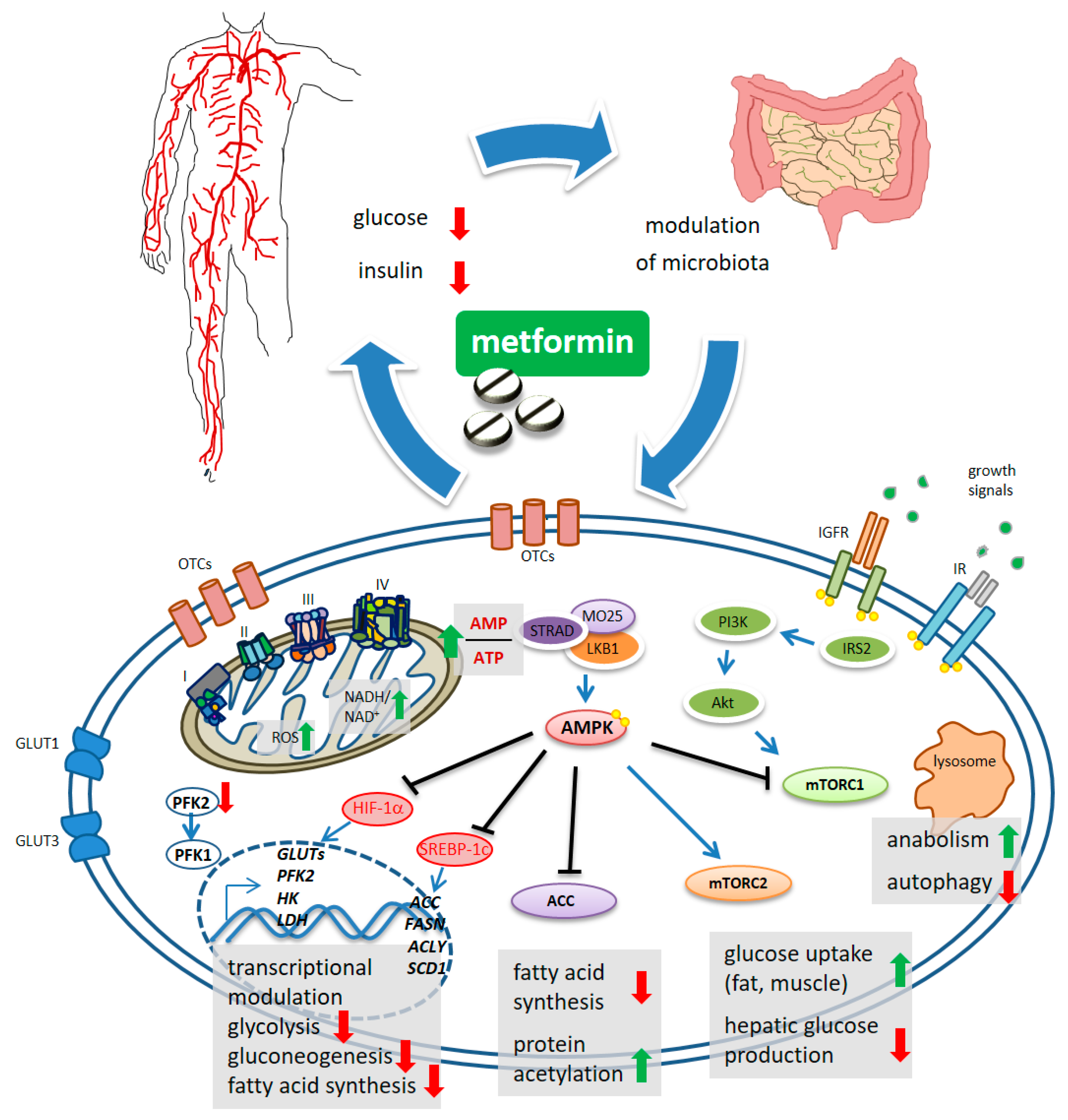
2. Metformin Elicited Signaling Disturbance
2.1. Metformin Modulates the Activity of mTORC1 and mTORC2 Complexes
2.2. Metformin Inhibits Fatty Acid Synthesis
2.3. Metformin Modulates Gut Microbiota
3. Metformin in Cancer Patients
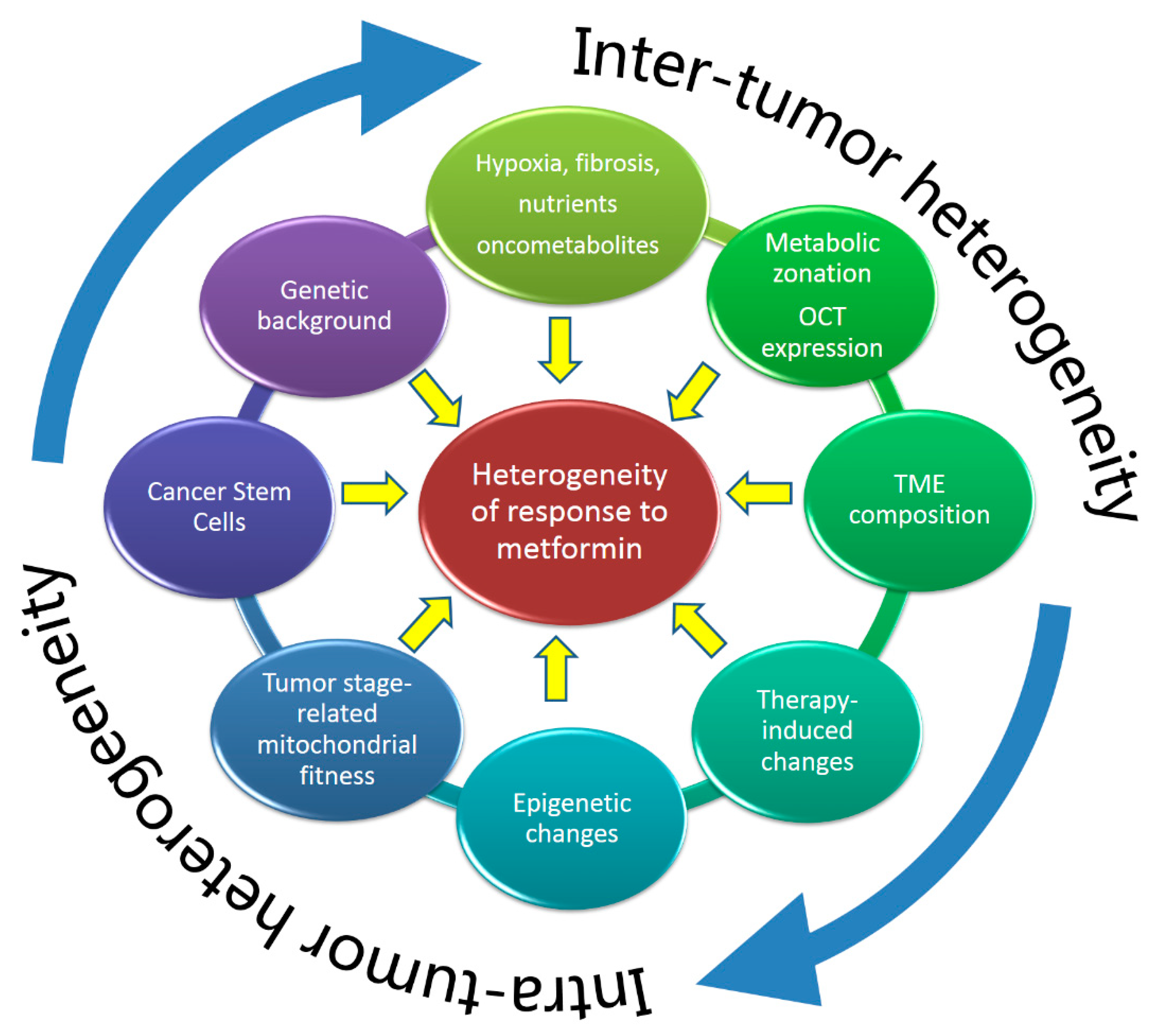
3.1. Metformin in Cancer Cells: Influence of Intra- and Inter-Tumor Heterogeneity
3.2. Molecular Mechanisms for Anticancer Effects: It May All Start from Mitochondria
3.2.1. Dose of Metformin in Cancer Studies, a Long Debate with a Recent Twist
3.2.2. Mitochondria Targeting Is Central to Metformin Anticancer Effects
4. Metformin May Impair the Tumor Response to Stress
4.1. Metformin, p53, STAT3, NFkB, ER-Stress and Other Stress Adaptive Processses
4.1.1. Metformin and p53
4.1.2. Metformin and STAT3
4.1.3. Metformin and NFkB
4.2. Metformin and Autophagy
4.3. Metformin and Mitophagy
4.4. Metformin and the Unfolded Protein Response (UPR)
4.5. Metformin and Modulation of microRNA Expression
4.6. Attenuation of Stress Adaptation by Metformin Is Complex, Integrated and Tumor Context-Specific
5. Metformin and Cancer Stem Cells: Another Layer of Complexity
Metformin Attenuates Cancer Stem Cell Features
6. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Heppner, G.H. Tumor heterogeneity. Cancer Res. 1984, 44, 2259–2265. [Google Scholar] [PubMed]
- Kim, J.; DeBerardinis, R.J. Mechanisms and Implications of Metabolic Heterogeneity in Cancer. Cell Metab. 2019, 30, 434–446. [Google Scholar] [CrossRef]
- Seth, S.; Li, C.Y.; Ho, I.L.; Corti, D.; Loponte, S.; Sapio, L.; Del Poggetto, E.; Yen, E.Y.; Robinson, F.S.; Peoples, M.; et al. Pre-existing Functional Heterogeneity of Tumorigenic Compartment as the Origin of Chemoresistance in Pancreatic Tumors. Cell Rep. 2019, 26, 1518–1532 e1519. [Google Scholar] [CrossRef]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic reprogramming and cancer progression. Science 2020, 368. [Google Scholar] [CrossRef]
- Fan, Y.; Mao, R.; Yang, J. NF-kappaB and STAT3 signaling pathways collaboratively link inflammation to cancer. Protein Cell 2013, 4, 176–185. [Google Scholar] [CrossRef]
- Loponte, S.; Lovisa, S.; Deem, A.K.; Carugo, A.; Viale, A. The Many Facets of Tumor Heterogeneity: Is Metabolism Lagging Behind? Cancers (Basel) 2019, 11, 1574. [Google Scholar] [CrossRef]
- Frattaruolo, L.; Brindisi, M.; Curcio, R.; Marra, F.; Dolce, V.; Cappello, A.R. Targeting the Mitochondrial Metabolic Network: A Promising Strategy in Cancer Treatment. Int. J. Mol. Sci. 2020, 21, 6014. [Google Scholar] [CrossRef]
- Sica, V.; Bravo-San Pedro, J.M.; Stoll, G.; Kroemer, G. Oxidative phosphorylation as a potential therapeutic target for cancer therapy. Int. J. Cancer 2020, 146, 10–17. [Google Scholar] [CrossRef]
- Gallaher, J.A.; Enriquez-Navas, P.M.; Luddy, K.A.; Gatenby, R.A.; Anderson, A.R.A. Spatial Heterogeneity and Evolutionary Dynamics Modulate Time to Recurrence in Continuous and Adaptive Cancer Therapies. Cancer Res. 2018, 78, 2127–2139. [Google Scholar] [CrossRef]
- Jia, D.; Lu, M.; Jung, K.H.; Park, J.H.; Yu, L.; Onuchic, J.N.; Kaipparettu, B.A.; Levine, H. Elucidating cancer metabolic plasticity by coupling gene regulation with metabolic pathways. Proc. Natl. Acad. Sci. USA 2019, 116, 3909–3918. [Google Scholar] [CrossRef]
- Cassim, S.; Vucetic, M.; Zdralevic, M.; Pouyssegur, J. Warburg and Beyond: The Power of Mitochondrial Metabolism to Collaborate or Replace Fermentative Glycolysis in Cancer. Cancers (Basel) 2020, 12, 1119. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Heredia, J.M.; Carnero, A. Role of Mitochondria in Cancer Stem Cell Resistance. Cells 2020, 9, 1693. [Google Scholar] [CrossRef]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and Cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef]
- Zhang, L.; Yao, Y.; Zhang, S.; Liu, Y.; Guo, H.; Ahmed, M.; Bell, T.; Zhang, H.; Han, G.; Lorence, E.; et al. Metabolic reprogramming toward oxidative phosphorylation identifies a therapeutic target for mantle cell lymphoma. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Moldogazieva, N.T.; Mokhosoev, I.M.; Terentiev, A.A. Metabolic Heterogeneity of Cancer Cells: An Interplay between HIF-1, GLUTs, and AMPK. Cancers (Basel) 2020, 12, 862. [Google Scholar] [CrossRef]
- Ngoi, N.Y.L.; Eu, J.Q.; Hirpara, J.; Wang, L.; Lim, J.S.J.; Lee, S.C.; Lim, Y.C.; Pervaiz, S.; Goh, B.C.; Wong, A.L.A. Targeting Cell Metabolism as Cancer Therapy. Antioxid Redox Signal. 2020, 32, 285–308. [Google Scholar] [CrossRef]
- Alusik, S.; Paluch, Z. Metformin: The past, presence, and future. Minerva Med. 2015, 106, 233–238. [Google Scholar]
- Bailey, C.J. Metformin: Historical overview. Diabetologia 2017, 60, 1566–1576. [Google Scholar] [CrossRef] [PubMed]
- Rojas, L.B.; Gomes, M.B. Metformin: An old but still the best treatment for type 2 diabetes. Diabetol. Metab. Syndr. 2013, 5, 6. [Google Scholar] [CrossRef]
- Sterne, J. Treatment of diabetes mellitus with N,N-dimethylguanylguanidine (LA. 6023, glucophage). Therapie 1959, 14, 625–630. [Google Scholar]
- Agius, L.; Ford, B.E.; Chachra, S.S. The Metformin Mechanism on Gluconeogenesis and AMPK Activation: The Metabolite Perspective. Int. J. Mol. Sci. 2020, 21, 3240. [Google Scholar] [CrossRef]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef]
- Shaw, R.J.; Lamia, K.A.; Vasquez, D.; Koo, S.H.; Bardeesy, N.; Depinho, R.A.; Montminy, M.; Cantley, L.C. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 2005, 310, 1642–1646. [Google Scholar] [CrossRef]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef] [PubMed]
- Spiering, M.J. The mystery of metformin. J. Biol. Chem. 2019, 294, 6689–6691. [Google Scholar] [CrossRef]
- Cameron, A.R.; Logie, L.; Patel, K.; Erhardt, S.; Bacon, S.; Middleton, P.; Harthill, J.; Forteath, C.; Coats, J.T.; Kerr, C.; et al. Metformin selectively targets redox control of complex I energy transduction. Redox Biol. 2018, 14, 187–197. [Google Scholar] [CrossRef]
- Bridges, H.R.; Jones, A.J.; Pollak, M.N.; Hirst, J. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem. J. 2014, 462, 475–487. [Google Scholar] [CrossRef]
- Andrzejewski, S.; Gravel, S.P.; Pollak, M.; St-Pierre, J. Metformin directly acts on mitochondria to alter cellular bioenergetics. Cancer Metab. 2014, 2, 12. [Google Scholar] [CrossRef] [PubMed]
- Logie, L.; Harthill, J.; Patel, K.; Bacon, S.; Hamilton, D.L.; Macrae, K.; McDougall, G.; Wang, H.H.; Xue, L.; Jiang, H.; et al. Cellular responses to the metal-binding properties of metformin. Diabetes 2012, 61, 1423–1433. [Google Scholar] [CrossRef] [PubMed]
- Bridges, H.R.; Sirvio, V.A.; Agip, A.N.; Hirst, J. Molecular features of biguanides required for targeting of mitochondrial respiratory complex I and activation of AMP-kinase. BMC. Biol. 2016, 14, 65. [Google Scholar] [CrossRef]
- Hardie, D.G. Keeping the home fires burning: AMP-activated protein kinase. J. R. Soc. Interface 2018, 15. [Google Scholar] [CrossRef]
- Zhang, C.S.; Li, M.; Zong, Y.; Lin, S.C. Determining AMPK Activation via the Lysosomal v-ATPase-Ragulator-AXIN/LKB1 Axis. Methods Mol. Biol. 2018, 1732, 393–411. [Google Scholar] [CrossRef]
- Jia, J.; Bissa, B.; Brecht, L.; Allers, L.; Choi, S.W.; Gu, Y.; Zbinden, M.; Burge, M.R.; Timmins, G.; Hallows, K.; et al. AMPK is activated during lysosomal damage via a galectin-ubiquitin signal transduction system. Autophagy 2020, 16, 1550–1552. [Google Scholar] [CrossRef] [PubMed]
- Carroll, B.; Dunlop, E.A. The lysosome: A crucial hub for AMPK and mTORC1 signalling. Biochem. J. 2017, 474, 1453–1466. [Google Scholar] [CrossRef]
- Jia, J.; Bissa, B.; Brecht, L.; Allers, L.; Choi, S.W.; Gu, Y.; Zbinden, M.; Burge, M.R.; Timmins, G.; Hallows, K.; et al. AMPK, a Regulator of Metabolism and Autophagy, Is Activated by Lysosomal Damage via a Novel Galectin-Directed Ubiquitin Signal Transduction System. Mol. Cell 2020, 77, 951–969 e959. [Google Scholar] [CrossRef] [PubMed]
- Zong, Y.; Zhang, C.S.; Li, M.; Wang, W.; Wang, Z.; Hawley, S.A.; Ma, T.; Feng, J.W.; Tian, X.; Qi, Q.; et al. Hierarchical activation of compartmentalized pools of AMPK depends on severity of nutrient or energy stress. Cell Res. 2019, 29, 460–473. [Google Scholar] [CrossRef]
- Madiraju, A.K.; Qiu, Y.; Perry, R.J.; Rahimi, Y.; Zhang, X.M.; Zhang, D.; Camporez, J.G.; Cline, G.W.; Butrico, G.M.; Kemp, B.E.; et al. Metformin inhibits gluconeogenesis via a redox-dependent mechanism in vivo. Nat. Med. 2018, 24, 1384–1394. [Google Scholar] [CrossRef]
- Madiraju, A.K.; Erion, D.M.; Rahimi, Y.; Zhang, X.M.; Braddock, D.T.; Albright, R.A.; Prigaro, B.J.; Wood, J.L.; Bhanot, S.; MacDonald, M.J.; et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature 2014, 510, 542–546. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Sabet, A.; Djedjos, S.; Miller, R.; Sun, X.; Hussain, M.A.; Radovick, S.; Wondisford, F.E. Metformin and insulin suppress hepatic gluconeogenesis through phosphorylation of CREB binding protein. Cell 2009, 137, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Rines, A.K.; Sharabi, K.; Tavares, C.D.; Puigserver, P. Targeting hepatic glucose metabolism in the treatment of type 2 diabetes. Nat. Rev. Drug Discov. 2016, 15, 786–804. [Google Scholar] [CrossRef]
- Kim, Y.D.; Park, K.G.; Lee, Y.S.; Park, Y.Y.; Kim, D.K.; Nedumaran, B.; Jang, W.G.; Cho, W.J.; Ha, J.; Lee, I.K.; et al. Metformin inhibits hepatic gluconeogenesis through AMP-activated protein kinase-dependent regulation of the orphan nuclear receptor SHP. Diabetes 2008, 57, 306–314. [Google Scholar] [CrossRef]
- Lee, J.M.; Seo, W.Y.; Song, K.H.; Chanda, D.; Kim, Y.D.; Kim, D.K.; Lee, M.W.; Ryu, D.; Kim, Y.H.; Noh, J.R.; et al. AMPK-dependent repression of hepatic gluconeogenesis via disruption of CREB.CRTC2 complex by orphan nuclear receptor small heterodimer partner. J. Biol. Chem. 2010, 285, 32182–32191. [Google Scholar] [CrossRef]
- Hardie, D.G. AMP-activated protein kinase: Maintaining energy homeostasis at the cellular and whole-body levels. Annu. Rev. Nutr. 2014, 34, 31–55. [Google Scholar] [CrossRef]
- Ruderman, N.B.; Cacicedo, J.M.; Itani, S.; Yagihashi, N.; Saha, A.K.; Ye, J.M.; Chen, K.; Zou, M.; Carling, D.; Boden, G.; et al. Malonyl-CoA and AMP-activated protein kinase (AMPK): Possible links between insulin resistance in muscle and early endothelial cell damage in diabetes. Biochem. Soc. Trans. 2003, 31, 202–206. [Google Scholar] [CrossRef]
- Tyszka-Czochara, M.; Bukowska-Strakova, K.; Kocemba-Pilarczyk, K.A.; Majka, M. Caffeic Acid Targets AMPK Signaling and Regulates Tricarboxylic Acid Cycle Anaplerosis while Metformin Downregulates HIF-1alpha-Induced Glycolytic Enzymes in Human Cervical Squamous Cell Carcinoma Lines. Nutrients 2018, 10, 841. [Google Scholar] [CrossRef]
- Hu, L.; Zeng, Z.; Xia, Q.; Liu, Z.; Feng, X.; Chen, J.; Huang, M.; Chen, L.; Fang, Z.; Liu, Q.; et al. Metformin attenuates hepatoma cell proliferation by decreasing glycolytic flux through the HIF-1alpha/PFKFB3/PFK1 pathway. Life Sci. 2019, 239, 116966. [Google Scholar] [CrossRef]
- Semenza, G.L. Transcriptional regulation by hypoxia-inducible factor 1 molecular mechanisms of oxygen homeostasis. Trends Cardiovasc. Med. 1996, 6, 151–157. [Google Scholar] [CrossRef]
- Sabatini, D.M. Twenty-five years of mTOR: Uncovering the link from nutrients to growth. Proc. Natl. Acad. Sci. USA 2017, 114, 11818–11825. [Google Scholar] [CrossRef]
- Howell, J.J.; Hellberg, K.; Turner, M.; Talbott, G.; Kolar, M.J.; Ross, D.S.; Hoxhaj, G.; Saghatelian, A.; Shaw, R.J.; Manning, B.D. Metformin Inhibits Hepatic mTORC1 Signaling via Dose-Dependent Mechanisms Involving AMPK and the TSC Complex. Cell Metab. 2017, 25, 463–471. [Google Scholar] [CrossRef]
- Kazyken, D.; Magnuson, B.; Bodur, C.; Acosta-Jaquez, H.A.; Zhang, D.; Tong, X.; Barnes, T.M.; Steinl, G.K.; Patterson, N.E.; Altheim, C.H.; et al. AMPK directly activates mTORC2 to promote cell survival during acute energetic stress. Sci. Signal. 2019, 12. [Google Scholar] [CrossRef]
- Kalender, A.; Selvaraj, A.; Kim, S.Y.; Gulati, P.; Brule, S.; Viollet, B.; Kemp, B.E.; Bardeesy, N.; Dennis, P.; Schlager, J.J.; et al. Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab. 2010, 11, 390–401. [Google Scholar] [CrossRef]
- Fu, W.; Hall, M.N. Regulation of mTORC2 Signaling. Genes (Basel) 2020, 11, 1045. [Google Scholar] [CrossRef]
- Angin, Y.; Beauloye, C.; Horman, S.; Bertrand, L. Regulation of Carbohydrate Metabolism, Lipid Metabolism, and Protein Metabolism by AMPK. Exp. Suppl. 2016, 107, 23–43. [Google Scholar] [CrossRef]
- Abu-Elheiga, L.; Matzuk, M.M.; Abo-Hashema, K.A.; Wakil, S.J. Continuous fatty acid oxidation and reduced fat storage in mice lacking acetyl-CoA carboxylase 2. Science 2001, 291, 2613–2616. [Google Scholar] [CrossRef]
- Han, Y.; Hu, Z.; Cui, A.; Liu, Z.; Ma, F.; Xue, Y.; Liu, Y.; Zhang, F.; Zhao, Z.; Yu, Y.; et al. Post-translational regulation of lipogenesis via AMPK-dependent phosphorylation of insulin-induced gene. Nat. Commun. 2019, 10, 623. [Google Scholar] [CrossRef]
- Bhalla, K.; Hwang, B.J.; Dewi, R.E.; Twaddel, W.; Goloubeva, O.G.; Wong, K.K.; Saxena, N.K.; Biswal, S.; Girnun, G.D. Metformin prevents liver tumorigenesis by inhibiting pathways driving hepatic lipogenesis. Cancer Prev. Res. (Phila) 2012, 5, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Liu, N.C.; Yu, I.C.; Lin, H.Y.; Lee, Y.F.; Sparks, J.D.; Chen, L.M.; Chang, C. Metformin inhibits nuclear receptor TR4-mediated hepatic stearoyl-CoA desaturase 1 gene expression with altered insulin sensitivity. Diabetes 2011, 60, 1493–1503. [Google Scholar] [CrossRef]
- Bryrup, T.; Thomsen, C.W.; Kern, T.; Allin, K.H.; Brandslund, I.; Jorgensen, N.R.; Vestergaard, H.; Hansen, T.; Hansen, T.H.; Pedersen, O.; et al. Metformin-induced changes of the gut microbiota in healthy young men: Results of a non-blinded, one-armed intervention study. Diabetologia 2019, 62, 1024–1035. [Google Scholar] [CrossRef]
- Wu, T.; Horowitz, M.; Rayner, C.K. New insights into the anti-diabetic actions of metformin: From the liver to the gut. Expert Rev. Gastroenterol. Hepatol. 2017, 11, 157–166. [Google Scholar] [CrossRef]
- Sum, C.F.; Webster, J.M.; Johnson, A.B.; Catalano, C.; Cooper, B.G.; Taylor, R. The effect of intravenous metformin on glucose metabolism during hyperglycaemia in type 2 diabetes. Diabet Med. 1992, 9, 61–65. [Google Scholar] [CrossRef]
- Shin, N.R.; Lee, J.C.; Lee, H.Y.; Kim, M.S.; Whon, T.W.; Lee, M.S.; Bae, J.W. An increase in the Akkermansia spp. population induced by metformin treatment improves glucose homeostasis in diet-induced obese mice. Gut 2014, 63, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.J.; Wilcock, C.; Scarpello, J.H. Metformin and the intestine. Diabetologia 2008, 51, 1552–1553. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.Y.; Wu, C.Y.; Yu, J. The role of gut microbiota in cancer treatment: Friend or foe? Gut 2020, 69, 1867–1876. [Google Scholar] [CrossRef] [PubMed]
- de la Cuesta-Zuluaga, J.; Mueller, N.T.; Corrales-Agudelo, V.; Velasquez-Mejia, E.P.; Carmona, J.A.; Abad, J.M.; Escobar, J.S. Metformin Is Associated With Higher Relative Abundance of Mucin-Degrading Akkermansia muciniphila and Several Short-Chain Fatty Acid-Producing Microbiota in the Gut. Diabetes Care 2017, 40, 54–62. [Google Scholar] [CrossRef]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillere, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef]
- Xu, X.; Lv, J.; Guo, F.; Li, J.; Jia, Y.; Jiang, D.; Wang, N.; Zhang, C.; Kong, L.; Liu, Y.; et al. Gut Microbiome Influences the Efficacy of PD-1 Antibody Immunotherapy on MSS-Type Colorectal Cancer via Metabolic Pathway. Front. Microbiol 2020, 11, 814. [Google Scholar] [CrossRef]
- Morales, D.R.; Morris, A.D. Metformin in cancer treatment and prevention. Annu. Rev. Med. 2015, 66, 17–29. [Google Scholar] [CrossRef]
- Evans, J.M.; Donnelly, L.A.; Emslie-Smith, A.M.; Alessi, D.R.; Morris, A.D. Metformin and reduced risk of cancer in diabetic patients. BMJ 2005, 330, 1304–1305. [Google Scholar] [CrossRef]
- Franciosi, M.; Lucisano, G.; Lapice, E.; Strippoli, G.F.; Pellegrini, F.; Nicolucci, A. Metformin therapy and risk of cancer in patients with type 2 diabetes: Systematic review. PLoS ONE 2013, 8, e71583. [Google Scholar] [CrossRef]
- Jiralerspong, S.; Palla, S.L.; Giordano, S.H.; Meric-Bernstam, F.; Liedtke, C.; Barnett, C.M.; Hsu, L.; Hung, M.C.; Hortobagyi, G.N.; Gonzalez-Angulo, A.M. Metformin and pathologic complete responses to neoadjuvant chemotherapy in diabetic patients with breast cancer. J. Clin. Oncol. 2009, 27, 3297–3302. [Google Scholar] [CrossRef]
- Joshua, A.M.; Zannella, V.E.; Downes, M.R.; Bowes, B.; Hersey, K.; Koritzinsky, M.; Schwab, M.; Hofmann, U.; Evans, A.; van der Kwast, T.; et al. A pilot ‘window of opportunity’ neoadjuvant study of metformin in localised prostate cancer. Prostate Cancer Prostatic Dis. 2014, 17, 252–258. [Google Scholar] [CrossRef]
- Hadad, S.; Iwamoto, T.; Jordan, L.; Purdie, C.; Bray, S.; Baker, L.; Jellema, G.; Deharo, S.; Hardie, D.G.; Pusztai, L.; et al. Evidence for biological effects of metformin in operable breast cancer: A pre-operative, window-of-opportunity, randomized trial. Breast Cancer Res. Treat. 2011, 128, 783–794. [Google Scholar] [CrossRef]
- Hosono, K.; Endo, H.; Takahashi, H.; Sugiyama, M.; Sakai, E.; Uchiyama, T.; Suzuki, K.; Iida, H.; Sakamoto, Y.; Yoneda, K.; et al. Metformin suppresses colorectal aberrant crypt foci in a short-term clinical trial. Cancer Prev. Res. (Phila) 2010, 3, 1077–1083. [Google Scholar] [CrossRef]
- Dankner, R.; Agay, N.; Olmer, L.; Murad, H.; Keinan Boker, L.; Balicer, R.D.; Freedman, L.S. Metformin Treatment and Cancer Risk: Cox Regression Analysis, With Time-Dependent Covariates, of 320,000 Persons With Incident Diabetes Mellitus. Am. J. Epidemiol. 2019, 188, 1794–1800. [Google Scholar] [CrossRef]
- Dilman, V.M.; Berstein, L.M.; Ostroumova, M.N.; Fedorov, S.N.; Poroshina, T.E.; Tsyrlina, E.V.; Buslaeva, V.P.; Semiglazov, V.F.; Seleznev, I.K.; Bobrov Yu, F.; et al. Metabolic immunodepression and metabolic immunotherapy: An attempt of improvement in immunologic response in breast cancer patients by correction of metabolic disturbances. Oncology 1982, 39, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Dilman, V.M.; Berstein, L.M.; Zabezhinski, M.A.; Alexandrov, V.A.; Bobrov, J.F.; Pliss, G.B. Inhibition of DMBA-induced carcinogenesis by phenformin in the mammary gland of rats. Arch. Geschwulstforsch 1978, 48, 1–8. [Google Scholar]
- Pernicova, I.; Korbonits, M. Metformin--mode of action and clinical implications for diabetes and cancer. Nat. Rev. Endocrinol. 2014, 10, 143–156. [Google Scholar] [CrossRef]
- Pollak, M. The insulin receptor/insulin-like growth factor receptor family as a therapeutic target in oncology. Clin. Cancer Res. 2012, 18, 40–50. [Google Scholar] [CrossRef]
- Giannarelli, R.; Aragona, M.; Coppelli, A.; Del Prato, S. Reducing insulin resistance with metformin: The evidence today. Diabetes Metab. 2003, 29, 6S28–6S35. [Google Scholar] [CrossRef]
- Kumar, S.; Sharife, H.; Kreisel, T.; Mogilevsky, M.; Bar-Lev, L.; Grunewald, M.; Aizenshtein, E.; Karni, R.; Paldor, I.; Shlomi, T.; et al. Intra-Tumoral Metabolic Zonation and Resultant Phenotypic Diversification Are Dictated by Blood Vessel Proximity. Cell Metab. 2019, 30, 201–211 e206. [Google Scholar] [CrossRef]
- Morral, C.; Stanisavljevic, J.; Hernando-Momblona, X.; Mereu, E.; Alvarez-Varela, A.; Cortina, C.; Stork, D.; Slebe, F.; Turon, G.; Whissell, G.; et al. Zonation of Ribosomal DNA Transcription Defines a Stem Cell Hierarchy in Colorectal Cancer. Cell Stem. Cell 2020, 26, 845–861 e812. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Everett, R.S.; Thakker, D.R. Efficacious dose of metformin for breast cancer therapy is determined by cation transporter expression in tumours. Br. J. Pharm. 2019, 176, 2724–2735. [Google Scholar] [CrossRef]
- Cervenkova, L.; Vycital, O.; Bruha, J.; Rosendorf, J.; Palek, R.; Liska, V.; Daum, O.; Mohelnikova-Duchonova, B.; Soucek, P. Protein expression of ABCC2 and SLC22A3 associates with prognosis of pancreatic adenocarcinoma. Sci. Rep. 2019, 9, 19782. [Google Scholar] [CrossRef]
- Marinello, P.C.; Panis, C.; Silva, T.N.X.; Binato, R.; Abdelhay, E.; Rodrigues, J.A.; Mencalha, A.L.; Lopes, N.M.D.; Luiz, R.C.; Cecchini, R.; et al. Metformin prevention of doxorubicin resistance in MCF-7 and MDA-MB-231 involves oxidative stress generation and modulation of cell adaptation genes. Sci. Rep. 2019, 9, 5864. [Google Scholar] [CrossRef]
- Berndt, N.; Eckstein, J.; Heucke, N.; Wuensch, T.; Gajowski, R.; Stockmann, M.; Meierhofer, D.; Holzhutter, H.G. Metabolic heterogeneity of human hepatocellular carcinoma: Implications for personalized pharmacological treatment. Febs. J. 2020. [Google Scholar] [CrossRef]
- Asiedu, M.K.; Barron, M.; Aubry, M.C.; Wigle, D.A. Patient- and Cell Type-Specific Heterogeneity of Metformin Response. Basic Clin. Pharm. Toxicol. 2018, 122, 214–222. [Google Scholar] [CrossRef]
- Sanegre, S.; Lucantoni, F.; Burgos-Panadero, R.; de La Cruz-Merino, L.; Noguera, R.; Alvaro Naranjo, T. Integrating the Tumor Microenvironment into Cancer Therapy. Cancers (Basel) 2020, 12, 1677. [Google Scholar] [CrossRef]
- Shao, S.; Zhao, L.; An, G.; Zhang, L.; Jing, X.; Luo, M.; Li, W.; Meng, D.; Ning, Q.; Zhao, X.; et al. Metformin suppresses HIF-1alpha expression in cancer-associated fibroblasts to prevent tumor-stromal cross talk in breast cancer. Faseb J. 2020. [Google Scholar] [CrossRef]
- Kim, H.S.; Ren, G.; Kim, T.; Bhatnagar, S.; Yang, Q.; Bahk, Y.Y.; Kim, J.A. Metformin reduces saturated fatty acid-induced lipid accumulation and inflammatory response by restoration of autophagic flux in endothelial cells. Sci. Rep. 2020, 10, 13523. [Google Scholar] [CrossRef]
- Kurelac, I.; Umesh Ganesh, N.; Iorio, M.; Porcelli, A.M.; Gasparre, G. The multifaceted effects of metformin on tumor microenvironment. Semin. Cell Dev. Biol 2020, 98, 90–97. [Google Scholar] [CrossRef]
- Dowling, R.J.; Niraula, S.; Stambolic, V.; Goodwin, P.J. Metformin in cancer: Translational challenges. J. Mol. Endocrinol. 2012, 48, R31–R43. [Google Scholar] [CrossRef]
- He, L.; Wondisford, F.E. Metformin action: Concentrations matter. Cell Metab. 2015, 21, 159–162. [Google Scholar] [CrossRef] [PubMed]
- Kajbaf, F.; De Broe, M.E.; Lalau, J.D. Therapeutic Concentrations of Metformin: A Systematic Review. Clin. Pharm. 2016, 55, 439–459. [Google Scholar] [CrossRef]
- Fontaine, E. Metformin-Induced Mitochondrial Complex I Inhibition: Facts, Uncertainties, and Consequences. Front. Endocrinol. (Lausanne) 2018, 9, 753. [Google Scholar] [CrossRef]
- Liu, X.; Romero, I.L.; Litchfield, L.M.; Lengyel, E.; Locasale, J.W. Metformin Targets Central Carbon Metabolism and Reveals Mitochondrial Requirements in Human Cancers. Cell Metab. 2016, 24, 728–739. [Google Scholar] [CrossRef] [PubMed]
- Boukalova, S.; Stursa, J.; Werner, L.; Ezrova, Z.; Cerny, J.; Bezawork-Geleta, A.; Pecinova, A.; Dong, L.; Drahota, Z.; Neuzil, J. Mitochondrial Targeting of Metformin Enhances Its Activity against Pancreatic Cancer. Mol. Cancer 2016, 15, 2875–2886. [Google Scholar] [CrossRef]
- Boyle, K.A.; Van Wickle, J.; Hill, R.B.; Marchese, A.; Kalyanaraman, B.; Dwinell, M.B. Mitochondria-targeted drugs stimulate mitophagy and abrogate colon cancer cell proliferation. J. Biol. Chem. 2018, 293, 14891–14904. [Google Scholar] [CrossRef]
- Cheng, G.; Zielonka, J.; Ouari, O.; Lopez, M.; McAllister, D.; Boyle, K.; Barrios, C.S.; Weber, J.J.; Johnson, B.D.; Hardy, M.; et al. Mitochondria-Targeted Analogues of Metformin Exhibit Enhanced Antiproliferative and Radiosensitizing Effects in Pancreatic Cancer Cells. Cancer Res. 2016, 76, 3904–3915. [Google Scholar] [CrossRef]
- Muller, S.; Versini, A.; Sindikubwabo, F.; Belthier, G.; Niyomchon, S.; Pannequin, J.; Grimaud, L.; Caneque, T.; Rodriguez, R. Metformin reveals a mitochondrial copper addiction of mesenchymal cancer cells. PLoS ONE 2018, 13, e0206764. [Google Scholar] [CrossRef]
- Repiscak, P.; Erhardt, S.; Rena, G.; Paterson, M.J. Biomolecular mode of action of metformin in relation to its copper binding properties. Biochemistry 2014, 53, 787–795. [Google Scholar] [CrossRef]
- Wilcock, C.; Bailey, C.J. Accumulation of metformin by tissues of the normal and diabetic mouse. Xenobiotica 1994, 24, 49–57. [Google Scholar] [CrossRef]
- Wilcock, C.; Wyre, N.D.; Bailey, C.J. Subcellular distribution of metformin in rat liver. J. Pharm. Pharm. 1991, 43, 442–444. [Google Scholar] [CrossRef]
- Lai, X.; Li, Q.; Wu, F.; Lin, J.; Chen, J.; Zheng, H.; Guo, L. Epithelial-Mesenchymal Transition and Metabolic Switching in Cancer: Lessons From Somatic Cell Reprogramming. Front. Cell Dev. Biol. 2020, 8, 760. [Google Scholar] [CrossRef] [PubMed]
- Dowling, R.J.; Lam, S.; Bassi, C.; Mouaaz, S.; Aman, A.; Kiyota, T.; Al-Awar, R.; Goodwin, P.J.; Stambolic, V. Metformin Pharmacokinetics in Mouse Tumors: Implications for Human Therapy. Cell Metab. 2016, 23, 567–568. [Google Scholar] [CrossRef]
- Janzer, A.; German, N.J.; Gonzalez-Herrera, K.N.; Asara, J.M.; Haigis, M.C.; Struhl, K. Metformin and phenformin deplete tricarboxylic acid cycle and glycolytic intermediates during cell transformation and NTPs in cancer stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, 10574–10579. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, H.H.; Cao, Y.T.; Zhang, L.L.; Huang, F.; Yi, C. The Role of Mitochondrial Dynamics and Mitophagy in Carcinogenesis, Metastasis and Therapy. Front. Cell Dev. Biol. 2020, 8, 413. [Google Scholar] [CrossRef]
- Boroughs, L.K.; DeBerardinis, R.J. Metabolic pathways promoting cancer cell survival and growth. Nat. Cell Biol. 2015, 17, 351–359. [Google Scholar] [CrossRef]
- Vasan, K.; Werner, M.; Chandel, N.S. Mitochondrial Metabolism as a Target for Cancer Therapy. Cell Metab. 2020, 32, 341–352. [Google Scholar] [CrossRef]
- O’Malley, J.; Kumar, R.; Inigo, J.; Yadava, N.; Chandra, D. Mitochondrial Stress Response and Cancer. Trends Cancer 2020, 6, 688–701. [Google Scholar] [CrossRef]
- Zhou, Z.; Ibekwe, E.; Chornenkyy, Y. Metabolic Alterations in Cancer Cells and the Emerging Role of Oncometabolites as Drivers of Neoplastic Change. Antioxidants (Basel) 2018, 7, 16. [Google Scholar] [CrossRef] [PubMed]
- Grasso, D.; Zampieri, L.X.; Capeloa, T.; Van de Velde, J.A.; Sonveaux, P. Mitochondria in cancer. Cell Stress 2020, 4, 114–146. [Google Scholar] [CrossRef]
- Roth, K.G.; Mambetsariev, I.; Kulkarni, P.; Salgia, R. The Mitochondrion as an Emerging Therapeutic Target in Cancer. Trends Mol. Med. 2020, 26, 119–134. [Google Scholar] [CrossRef]
- Gui, D.Y.; Sullivan, L.B.; Luengo, A.; Hosios, A.M.; Bush, L.N.; Gitego, N.; Davidson, S.M.; Freinkman, E.; Thomas, C.J.; Vander Heiden, M.G. Environment Dictates Dependence on Mitochondrial Complex I for NAD+ and Aspartate Production and Determines Cancer Cell Sensitivity to Metformin. Cell Metab. 2016, 24, 716–727. [Google Scholar] [CrossRef] [PubMed]
- Griss, T.; Vincent, E.E.; Egnatchik, R.; Chen, J.; Ma, E.H.; Faubert, B.; Viollet, B.; DeBerardinis, R.J.; Jones, R.G. Metformin Antagonizes Cancer Cell Proliferation by Suppressing Mitochondrial-Dependent Biosynthesis. PLoS Biol. 2015, 13, e1002309. [Google Scholar] [CrossRef] [PubMed]
- Guimaraes, T.A.; Farias, L.C.; Santos, E.S.; de Carvalho Fraga, C.A.; Orsini, L.A.; de Freitas Teles, L.; Feltenberger, J.D.; de Jesus, S.F.; de Souza, M.G.; Santos, S.H.; et al. Metformin increases PDH and suppresses HIF-1alpha under hypoxic conditions and induces cell death in oral squamous cell carcinoma. Oncotarget 2016, 7, 55057–55068. [Google Scholar] [CrossRef]
- Kruiswijk, F.; Labuschagne, C.F.; Vousden, K.H. p53 in survival, death and metabolic health: A lifeguard with a licence to kill. Nat. Rev. Mol. Cell Biol. 2015, 16, 393–405. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, C.; Hu, W.; Feng, Z. Tumor suppressor p53 and metabolism. J. Mol. Cell Biol. 2019, 11, 284–292. [Google Scholar] [CrossRef]
- Liu, Y.; He, Y.; Jin, A.; Tikunov, A.P.; Zhou, L.; Tollini, L.A.; Leslie, P.; Kim, T.H.; Li, L.O.; Coleman, R.A.; et al. Ribosomal protein-Mdm2-p53 pathway coordinates nutrient stress with lipid metabolism by regulating MCD and promoting fatty acid oxidation. Proc. Natl. Acad. Sci. USA 2014, 111, E2414–E2422. [Google Scholar] [CrossRef]
- Simabuco, F.M.; Morale, M.G.; Pavan, I.C.B.; Morelli, A.P.; Silva, F.R.; Tamura, R.E. p53 and metabolism: From mechanism to therapeutics. Oncotarget 2018, 9, 23780–23823. [Google Scholar] [CrossRef]
- Budanov, A.V. Stress-responsive sestrins link p53 with redox regulation and mammalian target of rapamycin signaling. Antioxid Redox Signal. 2011, 15, 1679–1690. [Google Scholar] [CrossRef]
- Moyer, S.M.; Wasylishen, A.R.; Qi, Y.; Fowlkes, N.; Su, X.; Lozano, G. p53 drives a transcriptional program that elicits a non-cell-autonomous response and alters cell state in vivo. Proc. Natl. Acad. Sci. USA 2020. [Google Scholar] [CrossRef]
- Gomes, A.S.; Ramos, H.; Soares, J.; Saraiva, L. p53 and glucose metabolism: An orchestra to be directed in cancer therapy. Pharm. Res. 2018, 131, 75–86. [Google Scholar] [CrossRef]
- Itahana, Y.; Itahana, K. Emerging Roles of p53 Family Members in Glucose Metabolism. Int. J. Mol. Sci. 2018, 19, 776. [Google Scholar] [CrossRef]
- Jones, R.G.; Plas, D.R.; Kubek, S.; Buzzai, M.; Mu, J.; Xu, Y.; Birnbaum, M.J.; Thompson, C.B. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 2005, 18, 283–293. [Google Scholar] [CrossRef]
- He, G.; Zhang, Y.W.; Lee, J.H.; Zeng, S.X.; Wang, Y.V.; Luo, Z.; Dong, X.C.; Viollet, B.; Wahl, G.M.; Lu, H. AMP-activated protein kinase induces p53 by phosphorylating MDMX and inhibiting its activity. Mol. Cell Biol. 2014, 34, 148–157. [Google Scholar] [CrossRef]
- Cerezo, M.; Tichet, M.; Abbe, P.; Ohanna, M.; Lehraiki, A.; Rouaud, F.; Allegra, M.; Giacchero, D.; Bahadoran, P.; Bertolotto, C.; et al. Metformin blocks melanoma invasion and metastasis development in AMPK/p53-dependent manner. Mol. Cancer 2013, 12, 1605–1615. [Google Scholar] [CrossRef]
- Drakos, E.; Atsaves, V.; Li, J.; Leventaki, V.; Andreeff, M.; Medeiros, L.J.; Rassidakis, G.Z. Stabilization and activation of p53 downregulates mTOR signaling through AMPK in mantle cell lymphoma. Leukemia 2009, 23, 784–790. [Google Scholar] [CrossRef]
- Lee, M.; Hirpara, J.L.; Eu, J.Q.; Sethi, G.; Wang, L.; Goh, B.C.; Wong, A.L. Targeting STAT3 and oxidative phosphorylation in oncogene-addicted tumors. Redox Biol. 2019, 25, 101073. [Google Scholar] [CrossRef]
- Darnell, J.E., Jr. STAT3, HIF-1, glucose addiction and Warburg effect. Aging (Albany Ny) 2010, 2, 890–891. [Google Scholar] [CrossRef][Green Version]
- Valle-Mendiola, A.; Soto-Cruz, I. Energy Metabolism in Cancer: The Roles of STAT3 and STAT5 in the Regulation of Metabolism-Related Genes. Cancers (Basel) 2020, 12, 124. [Google Scholar] [CrossRef]
- Wegrzyn, J.; Potla, R.; Chwae, Y.J.; Sepuri, N.B.; Zhang, Q.; Koeck, T.; Derecka, M.; Szczepanek, K.; Szelag, M.; Gornicka, A.; et al. Function of mitochondrial Stat3 in cellular respiration. Science 2009, 323, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Avalle, L.; Poli, V. Nucleus, Mitochondrion, or Reticulum? STAT3 a La Carte. Int. J. Mol. Sci. 2018, 19, 2820. [Google Scholar] [CrossRef]
- Deng, X.S.; Wang, S.; Deng, A.; Liu, B.; Edgerton, S.M.; Lind, S.E.; Wahdan-Alaswad, R.; Thor, A.D. Metformin targets Stat3 to inhibit cell growth and induce apoptosis in triple-negative breast cancers. Cell Cycle 2012, 11, 367–376. [Google Scholar] [CrossRef]
- Esparza-Lopez, J.; Alvarado-Munoz, J.F.; Escobar-Arriaga, E.; Ulloa-Aguirre, A.; de Jesus Ibarra-Sanchez, M. Metformin reverses mesenchymal phenotype of primary breast cancer cells through STAT3/NF-kappaB pathways. BMC Cancer 2019, 19, 728. [Google Scholar] [CrossRef]
- Leidgens, V.; Proske, J.; Rauer, L.; Moeckel, S.; Renner, K.; Bogdahn, U.; Riemenschneider, M.J.; Proescholdt, M.; Vollmann-Zwerenz, A.; Hau, P.; et al. Stattic and metformin inhibit brain tumor initiating cells by reducing STAT3-phosphorylation. Oncotarget 2017, 8, 8250–8263. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Yang, G.L.; Yang, J.H.; Lin, S.L.; Liu, N.; Liu, S.S.; Liu, M.Y.; Zhang, L.H.; Huang, Y.R.; Shen, R.L.; et al. Metformin can block precancerous progression to invasive tumors of bladder through inhibiting STAT3-mediated signaling pathways. J. Exp. Clin. Cancer Res. 2015, 34, 77. [Google Scholar] [CrossRef] [PubMed]
- Tong, D.; Liu, Q.; Liu, G.; Xu, J.; Lan, W.; Jiang, Y.; Xiao, H.; Zhang, D.; Jiang, J. Metformin inhibits castration-induced EMT in prostate cancer by repressing COX2/PGE2/STAT3 axis. Cancer Lett. 2017, 389, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Ba, W.; Xu, Y.; Yin, G.; Yang, J.; Wang, R.; Chi, S.; Wang, Y.; Li, C. Metformin inhibits pro-inflammatory responses via targeting nuclear factor-kappaB in HaCaT cells. Cell Biochem. Funct. 2019, 37, 4–10. [Google Scholar] [CrossRef]
- Saengboonmee, C.; Seubwai, W.; Cha’on, U.; Sawanyawisuth, K.; Wongkham, S.; Wongkham, C. Metformin Exerts Antiproliferative and Anti-metastatic Effects Against Cholangiocarcinoma Cells by Targeting STAT3 and NF-kB. Anticancer Res. 2017, 37, 115–123. [Google Scholar] [CrossRef]
- Mauro, C.; Leow, S.C.; Anso, E.; Rocha, S.; Thotakura, A.K.; Tornatore, L.; Moretti, M.; De Smaele, E.; Beg, A.A.; Tergaonkar, V.; et al. NF-kappaB controls energy homeostasis and metabolic adaptation by upregulating mitochondrial respiration. Nat. Cell Biol. 2011, 13, 1272–1279. [Google Scholar] [CrossRef]
- Capece, D.; Verzella, D.; Di Francesco, B.; Alesse, E.; Franzoso, G.; Zazzeroni, F. NF-kappaB and mitochondria cross paths in cancer: Mitochondrial metabolism and beyond. Semin. Cell Dev. Biol. 2020, 98, 118–128. [Google Scholar] [CrossRef]
- Vaisitti, T.; Gaudino, F.; Ouk, S.; Moscvin, M.; Vitale, N.; Serra, S.; Arruga, F.; Zakrzewski, J.L.; Liou, H.C.; Allan, J.N.; et al. Targeting metabolism and survival in chronic lymphocytic leukemia and Richter syndrome cells by a novel NF-kappaB inhibitor. Haematologica 2017, 102, 1878–1889. [Google Scholar] [CrossRef]
- Ferro, F.; Servais, S.; Besson, P.; Roger, S.; Dumas, J.F.; Brisson, L. Autophagy and mitophagy in cancer metabolic remodelling. Semin. Cell Dev. Biol. 2020, 98, 129–138. [Google Scholar] [CrossRef]
- Gozuacik, D.; Kimchi, A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene 2004, 23, 2891–2906. [Google Scholar] [CrossRef] [PubMed]
- Folkerts, H.; Hilgendorf, S.; Vellenga, E.; Bremer, E.; Wiersma, V.R. The multifaceted role of autophagy in cancer and the microenvironment. Med. Res. Rev. 2019, 39, 517–560. [Google Scholar] [CrossRef]
- Zhou, W.; Xu, G.; Wang, Y.; Xu, Z.; Liu, X.; Xu, X.; Ren, G.; Tian, K. Oxidative stress induced autophagy in cancer associated fibroblast enhances proliferation and metabolism of colorectal cancer cells. Cell Cycle 2017, 16, 73–81. [Google Scholar] [CrossRef]
- New, J.; Arnold, L.; Ananth, M.; Alvi, S.; Thornton, M.; Werner, L.; Tawfik, O.; Dai, H.; Shnayder, Y.; Kakarala, K.; et al. Secretory Autophagy in Cancer-Associated Fibroblasts Promotes Head and Neck Cancer Progression and Offers a Novel Therapeutic Target. Cancer Res. 2017, 77, 6679–6691. [Google Scholar] [CrossRef]
- Liu, S.; Yue, C.; Chen, H.; Chen, Y.; Li, G. Metformin Promotes Beclin1-Dependent Autophagy to Inhibit the Progression of Gastric Cancer. Onco Targets 2020, 13, 4445–4455. [Google Scholar] [CrossRef]
- Gao, C.; Fang, L.; Zhang, H.; Zhang, W.S.; Li, X.O.; Du, S.Y. Metformin Induces Autophagy via the AMPK-mTOR Signaling Pathway in Human Hepatocellular Carcinoma Cells. Cancer Manag. Res. 2020, 12, 5803–5811. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, W.; Yan, Z.; Zhao, W.; Mi, J.; Li, J.; Yan, H. Metformin induces autophagy and G0/G1 phase cell cycle arrest in myeloma by targeting the AMPK/mTORC1 and mTORC2 pathways. J. Exp. Clin. Cancer Res. 2018, 37, 63. [Google Scholar] [CrossRef]
- Li, K.; Zhang, T.T.; Hua, F.; Hu, Z.W. Metformin reduces TRIB3 expression and restores autophagy flux: An alternative antitumor action. Autophagy 2018, 14, 1278–1279. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.R.; Kim, H.; Kim, K.I.; Baek, S.H. Epigenetic and transcriptional regulation of autophagy. Autophagy 2016, 12, 2248–2249. [Google Scholar] [CrossRef]
- Coomans de Brachene, A.; Demoulin, J.B. FOXO transcription factors in cancer development and therapy. Cell Mol. Life Sci. 2016, 73, 1159–1172. [Google Scholar] [CrossRef]
- Cheng, Z. The FoxO-Autophagy Axis in Health and Disease. Trends Endocrinol. Metab. 2019, 30, 658–671. [Google Scholar] [CrossRef]
- Hu, T.; Chung, Y.M.; Guan, M.; Ma, M.; Ma, J.; Berek, J.S.; Hu, M.C. Reprogramming ovarian and breast cancer cells into non-cancerous cells by low-dose metformin or SN-38 through FOXO3 activation. Sci. Rep. 2014, 4, 5810. [Google Scholar] [CrossRef]
- Queiroz, E.A.; Puukila, S.; Eichler, R.; Sampaio, S.C.; Forsyth, H.L.; Lees, S.J.; Barbosa, A.M.; Dekker, R.F.; Fortes, Z.B.; Khaper, N. Metformin induces apoptosis and cell cycle arrest mediated by oxidative stress, AMPK and FOXO3a in MCF-7 breast cancer cells. PLoS ONE 2014, 9, e98207. [Google Scholar] [CrossRef]
- Zhou, X.; Chen, J.; Yi, G.; Deng, M.; Liu, H.; Liang, M.; Shi, B.; Fu, X.; Chen, Y.; Chen, L.; et al. Metformin suppresses hypoxia-induced stabilization of HIF-1alpha through reprogramming of oxygen metabolism in hepatocellular carcinoma. Oncotarget 2016, 7, 873–884. [Google Scholar] [CrossRef]
- Saladini, S.; Aventaggiato, M.; Barreca, F.; Morgante, E.; Sansone, L.; Russo, M.A.; Tafani, M. Metformin Impairs Glutamine Metabolism and Autophagy in Tumour Cells. Cells 2019, 8, 49. [Google Scholar] [CrossRef] [PubMed]
- Mates, J.M.; Campos-Sandoval, J.A.; Santos-Jimenez, J.L.; Marquez, J. Dysregulation of glutaminase and glutamine synthetase in cancer. Cancer Lett. 2019, 467, 29–39. [Google Scholar] [CrossRef]
- Li, T.; Le, A. Glutamine Metabolism in Cancer. Adv. Exp. Med. Biol. 2018, 1063, 13–32. [Google Scholar] [CrossRef]
- De Vitto, H.; Perez-Valencia, J.; Radosevich, J.A. Glutamine at focus: Versatile roles in cancer. Tumour Biol. 2016, 37, 1541–1558. [Google Scholar] [CrossRef]
- Tyszka-Czochara, M.; Bukowska-Strakova, K.; Majka, M. Metformin and caffeic acid regulate metabolic reprogramming in human cervical carcinoma SiHa/HTB-35 cells and augment anticancer activity of Cisplatin via cell cycle regulation. Food Chem. Toxicol. 2017, 106, 260–272. [Google Scholar] [CrossRef]
- Eng, C.H.; Yu, K.; Lucas, J.; White, E.; Abraham, R.T. Ammonia derived from glutaminolysis is a diffusible regulator of autophagy. Sci. Signal. 2010, 3, ra31. [Google Scholar] [CrossRef]
- Vara-Perez, M.; Felipe-Abrio, B.; Agostinis, P. Mitophagy in Cancer: A Tale of Adaptation. Cells 2019, 8, 493. [Google Scholar] [CrossRef]
- Min, X.; Zhang, T.; Lin, Y.; Wang, B.; Zhu, K. Metformin inhibits the growth of ovarian cancer cells by promoting the Parkin-induced p53 ubiquitination. Biosci. Rep. 2020. [Google Scholar] [CrossRef]
- Zhao, Y.; Sun, M. Metformin rescues Parkin protein expression and mitophagy in high glucose-challenged human renal epithelial cells by inhibiting NF-kappaB via PP2A activation. Life Sci. 2020, 246, 117382. [Google Scholar] [CrossRef]
- Bhansali, S.; Bhansali, A.; Dutta, P.; Walia, R.; Dhawan, V. Metformin upregulates mitophagy in patients with T2DM: A randomized placebo-controlled study. J. Cell Mol. Med. 2020, 24, 2832–2846. [Google Scholar] [CrossRef]
- Santamaria, P.G.; Mazon, M.J.; Eraso, P.; Portillo, F. UPR: An Upstream Signal to EMT Induction in Cancer. J. Clin. Med. 2019, 8, 624. [Google Scholar] [CrossRef]
- Shapiro, D.J.; Livezey, M.; Yu, L.; Zheng, X.; Andruska, N. Anticipatory UPR Activation: A Protective Pathway and Target in Cancer. Trends Endocrinol. Metab. 2016, 27, 731–741. [Google Scholar] [CrossRef]
- Leclerc, G.M.; Leclerc, G.J.; Kuznetsov, J.N.; DeSalvo, J.; Barredo, J.C. Metformin induces apoptosis through AMPK-dependent inhibition of UPR signaling in ALL lymphoblasts. PLoS ONE 2013, 8, e74420. [Google Scholar] [CrossRef]
- Tong, X.; Yu, N.; Han, R.; Wang, T. Function of Dicer with regard to Energy Homeostasis Regulation, Structural Modification, and Cellular Distribution. Int. J. Endocrinol. 2020, 2020, 6420816. [Google Scholar] [CrossRef]
- Hua, Q.; Mi, B.; Huang, G. The emerging co-regulatory role of long noncoding RNAs in epithelial-mesenchymal transition and the Warburg effect in aggressive tumors. Crit. Rev. Oncol. Hematol. 2018, 126, 112–120. [Google Scholar] [CrossRef]
- Bernstein, E.; Caudy, A.A.; Hammond, S.M.; Hannon, G.J. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 2001, 409, 363–366. [Google Scholar] [CrossRef]
- Blandino, G.; Valerio, M.; Cioce, M.; Mori, F.; Casadei, L.; Pulito, C.; Sacconi, A.; Biagioni, F.; Cortese, G.; Galanti, S.; et al. Metformin elicits anticancer effects through the sequential modulation of DICER and c-MYC. Nat. Commun. 2012, 3, 865. [Google Scholar] [CrossRef] [PubMed]
- Malinowski, B.; Musiala, N.; Wicinski, M. Metformin’s Modulatory Effects on miRNAs Function in Cancer Stem Cells-A Systematic Review. Cells 2020, 9, 1401. [Google Scholar] [CrossRef]
- Pulito, C.; Donzelli, S.; Muti, P.; Puzzo, L.; Strano, S.; Blandino, G. microRNAs and cancer metabolism reprogramming: The paradigm of metformin. Ann. Transl. Med. 2014, 2, 58. [Google Scholar] [CrossRef]
- Pulito, C.; Mori, F.; Sacconi, A.; Goeman, F.; Ferraiuolo, M.; Pasanisi, P.; Campagnoli, C.; Berrino, F.; Fanciulli, M.; Ford, R.J.; et al. Metformin-induced ablation of microRNA 21-5p releases Sestrin-1 and CAB39L antitumoral activities. Cell Discov. 2017, 3, 17022. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Yang, W.; Wu, F.; Wang, C.; Yu, L.; Tang, L.; Qiu, B.; Li, Y.; Guo, L.; Wu, M.; et al. Prognostic significance of AMPK activation and therapeutic effects of metformin in hepatocellular carcinoma. Clin. Cancer Res. 2013, 19, 5372–5380. [Google Scholar] [CrossRef] [PubMed]
- DeWaal, D.; Nogueira, V.; Terry, A.R.; Patra, K.C.; Jeon, S.M.; Guzman, G.; Au, J.; Long, C.P.; Antoniewicz, M.R.; Hay, N. Hexokinase-2 depletion inhibits glycolysis and induces oxidative phosphorylation in hepatocellular carcinoma and sensitizes to metformin. Nat. Commun. 2018, 9, 446. [Google Scholar] [CrossRef]
- Wandee, J.; Prawan, A.; Senggunprai, L.; Kongpetch, S.; Kukongviriyapan, V. Metformin sensitizes cholangiocarcinoma cell to cisplatin-induced cytotoxicity through oxidative stress mediated mitochondrial pathway. Life Sci. 2019, 217, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Ke, C.; Tang, Q.; Dong, H.; Zheng, X.; Lin, W.; Ke, J.; Huang, J.; Yeung, S.C.; Zhang, H. Metformin promotes autophagy and apoptosis in esophageal squamous cell carcinoma by downregulating Stat3 signaling. Cell Death Dis. 2014, 5, e1088. [Google Scholar] [CrossRef]
- Sekino, N.; Kano, M.; Matsumoto, Y.; Sakata, H.; Akutsu, Y.; Hanari, N.; Murakami, K.; Toyozumi, T.; Takahashi, M.; Otsuka, R.; et al. Antitumor effects of metformin are a result of inhibiting nuclear factor kappa B nuclear translocation in esophageal squamous cell carcinoma. Cancer Sci. 2018, 109, 1066–1074. [Google Scholar] [CrossRef]
- Chai, X.; Chu, H.; Yang, X.; Meng, Y.; Shi, P.; Gou, S. Metformin Increases Sensitivity of Pancreatic Cancer Cells to Gemcitabine by Reducing CD133+ Cell Populations and Suppressing ERK/P70S6K Signaling. Sci. Rep. 2015, 5, 14404. [Google Scholar] [CrossRef]
- Yue, W.; Zheng, X.; Lin, Y.; Yang, C.S.; Xu, Q.; Carpizo, D.; Huang, H.; DiPaola, R.S.; Tan, X.L. Metformin combined with aspirin significantly inhibit pancreatic cancer cell growth in vitro and in vivo by suppressing anti-apoptotic proteins Mcl-1 and Bcl-2. Oncotarget 2015, 6, 21208–21224. [Google Scholar] [CrossRef]
- Pan, Y.H.; Jiao, L.; Lin, C.Y.; Lu, C.H.; Li, L.; Chen, H.Y.; Wang, Y.B.; He, Y. Combined treatment with metformin and gefitinib overcomes primary resistance to EGFR-TKIs with EGFR mutation via targeting IGF-1R signaling pathway. Biologics 2018, 12, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.H.; Lin, C.Y.; Lu, C.H.; Li, L.; Wang, Y.B.; Chen, H.Y.; He, Y. Metformin synergistically enhances the antitumor activity of the third-generation EGFR-TKI CO-1686 in lung cancer cells through suppressing NF-kappaB signaling. Clin. Respir. J. 2018, 12, 2642–2652. [Google Scholar] [CrossRef]
- Park, J.H.; Kim, Y.H.; Park, E.H.; Lee, S.J.; Kim, H.; Kim, A.; Lee, S.B.; Shim, S.; Jang, H.; Myung, J.K.; et al. Effects of metformin and phenformin on apoptosis and epithelial-mesenchymal transition in chemoresistant rectal cancer. Cancer Sci. 2019, 110, 2834–2845. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Park, D.; Lee, Y. Metformin Synergistically Potentiates the Antitumor Effects of Imatinib in Colorectal Cancer Cells. Dev. Reprod 2017, 21, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Bermudez, A.; Vicente-Blanco, R.J.; Laza-Briviesca, R.; Garcia-Grande, A.; Laine-Menendez, S.; Gutierrez, L.; Calvo, V.; Romero, A.; Martin-Acosta, P.; Garcia, J.M.; et al. PGC-1alpha levels correlate with survival in patients with stage III NSCLC and may define a new biomarker to metabolism-targeted therapy. Sci. Rep. 2017, 7, 16661. [Google Scholar] [CrossRef]
- Denise, C.; Paoli, P.; Calvani, M.; Taddei, M.L.; Giannoni, E.; Kopetz, S.; Kazmi, S.M.; Pia, M.M.; Pettazzoni, P.; Sacco, E.; et al. 5-fluorouracil resistant colon cancer cells are addicted to OXPHOS to survive and enhance stem-like traits. Oncotarget 2015, 6, 41706–41721. [Google Scholar] [CrossRef]
- Vellinga, T.T.; Borovski, T.; de Boer, V.C.; Fatrai, S.; van Schelven, S.; Trumpi, K.; Verheem, A.; Snoeren, N.; Emmink, B.L.; Koster, J.; et al. SIRT1/PGC1alpha-Dependent Increase in Oxidative Phosphorylation Supports Chemotherapy Resistance of Colon Cancer. Clin. Cancer Res. 2015, 21, 2870–2879. [Google Scholar] [CrossRef]
- Matassa, D.S.; Amoroso, M.R.; Lu, H.; Avolio, R.; Arzeni, D.; Procaccini, C.; Faicchia, D.; Maddalena, F.; Simeon, V.; Agliarulo, I.; et al. Oxidative metabolism drives inflammation-induced platinum resistance in human ovarian cancer. Cell Death Differ. 2016, 23, 1542–1554. [Google Scholar] [CrossRef]
- Thiagarajan, P.S.; Wu, X.; Zhang, W.; Shi, I.; Bagai, R.; Leahy, P.; Feng, Y.; Veigl, M.; Lindner, D.; Danielpour, D.; et al. Transcriptomic-metabolomic reprogramming in EGFR-mutant NSCLC early adaptive drug escape linking TGFbeta2-bioenergetics-mitochondrial priming. Oncotarget 2016, 7, 82013–82027. [Google Scholar] [CrossRef] [PubMed]
- Walcher, L.; Kistenmacher, A.K.; Suo, H.; Kitte, R.; Dluczek, S.; Strauss, A.; Blaudszun, A.R.; Yevsa, T.; Fricke, S.; Kossatz-Boehlert, U. Cancer Stem Cells-Origins and Biomarkers: Perspectives for Targeted Personalized Therapies. Front. Immunol 2020, 11, 1280. [Google Scholar] [CrossRef]
- Yuan, S.; Norgard, R.J.; Stanger, B.Z. Cellular Plasticity in Cancer. Cancer Discov. 2019, 9, 837–851. [Google Scholar] [CrossRef] [PubMed]
- Canino, C.; Mori, F.; Cambria, A.; Diamantini, A.; Germoni, S.; Alessandrini, G.; Borsellino, G.; Galati, R.; Battistini, L.; Blandino, R.; et al. SASP mediates chemoresistance and tumor-initiating-activity of mesothelioma cells. Oncogene 2012, 31, 3148–3163. [Google Scholar] [CrossRef]
- Tong, Y.; Gao, W.Q.; Liu, Y. Metabolic heterogeneity in cancer: An overview and therapeutic implications. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188421. [Google Scholar] [CrossRef]
- Ben Sahra, I.; Tanti, J.F.; Bost, F. The combination of metformin and 2-deoxyglucose inhibits autophagy and induces AMPK-dependent apoptosis in prostate cancer cells. Autophagy 2010, 6, 670–671. [Google Scholar] [CrossRef]
- Cheong, J.H.; Park, E.S.; Liang, J.; Dennison, J.B.; Tsavachidou, D.; Nguyen-Charles, C.; Wa Cheng, K.; Hall, H.; Zhang, D.; Lu, Y.; et al. Dual inhibition of tumor energy pathway by 2-deoxyglucose and metformin is effective against a broad spectrum of preclinical cancer models. Mol. Cancer 2011, 10, 2350–2362. [Google Scholar] [CrossRef]
- Elgendy, M.; Ciro, M.; Hosseini, A.; Weiszmann, J.; Mazzarella, L.; Ferrari, E.; Cazzoli, R.; Curigliano, G.; DeCensi, A.; Bonanni, B.; et al. Combination of Hypoglycemia and Metformin Impairs Tumor Metabolic Plasticity and Growth by Modulating the PP2A-GSK3beta-MCL-1 Axis. Cancer Cell 2019, 35, 798–815 e795. [Google Scholar] [CrossRef]
- Tyszka-Czochara, M.; Konieczny, P.; Majka, M. Caffeic Acid Expands Anti-Tumor Effect of Metformin in Human Metastatic Cervical Carcinoma HTB-34 Cells: Implications of AMPK Activation and Impairment of Fatty Acids De Novo Biosynthesis. Int. J. Mol. Sci. 2017, 18, 462. [Google Scholar] [CrossRef] [PubMed]
- Tyszka-Czochara, M.; Lasota, M.; Majka, M. Caffeic Acid and Metformin Inhibit Invasive Phenotype Induced by TGF-beta1 in C-4I and HTB-35/SiHa Human Cervical Squamous Carcinoma Cells by Acting on Different Molecular Targets. Int. J. Mol. Sci. 2018, 19, 266. [Google Scholar] [CrossRef]
- Li, J.; Condello, S.; Thomes-Pepin, J.; Ma, X.; Xia, Y.; Hurley, T.D.; Matei, D.; Cheng, J.X. Lipid Desaturation Is a Metabolic Marker and Therapeutic Target of Ovarian Cancer Stem Cells. Cell Stem. Cell 2017, 20, 303–314.e305,. [Google Scholar] [CrossRef]
- Sanchez-Martinez, R.; Cruz-Gil, S.; Gomez de Cedron, M.; Alvarez-Fernandez, M.; Vargas, T.; Molina, S.; Garcia, B.; Herranz, J.; Moreno-Rubio, J.; Reglero, G.; et al. A link between lipid metabolism and epithelial-mesenchymal transition provides a target for colon cancer therapy. Oncotarget 2015, 6, 38719–38736. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.W.; Lim, I.K. Sensitization of metformin-cytotoxicity by dichloroacetate via reprogramming glucose metabolism in cancer cells. Cancer Lett. 2014, 346, 300–308. [Google Scholar] [CrossRef]
- Kolesnik, D.L.; Pyaskovskaya, O.N.; Yurchenko, O.V.; Solyanik, G.I. Metformin enhances antitumor action of sodium dichloroacetate against glioma C6. Exp. Oncol. 2019, 41, 123–129. [Google Scholar] [CrossRef]
- Li, B.; Li, X.; Ni, Z.; Zhang, Y.; Zeng, Y.; Yan, X.; Huang, Y.; He, J.; Lyu, X.; Wu, Y.; et al. Dichloroacetate and metformin synergistically suppress the growth of ovarian cancer cells. Oncotarget 2016, 7, 59458–59470. [Google Scholar] [CrossRef]
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006, 3, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Gentles, A.; Nair, R.V.; Huang, M.; Lin, Y.; Lee, C.Y.; Cai, S.; Scheeren, F.A.; Kuo, A.H.; Diehn, M. Targeting unique metabolic properties of breast tumor initiating cells. Stem. Cells 2014, 32, 1734–1745. [Google Scholar] [CrossRef]
- Steg, A.D.; Bevis, K.S.; Katre, A.A.; Ziebarth, A.; Dobbin, Z.C.; Alvarez, R.D.; Zhang, K.; Conner, M.; Landen, C.N. Stem cell pathways contribute to clinical chemoresistance in ovarian cancer. Clin. Cancer Res. 2012, 18, 869–881. [Google Scholar] [CrossRef]
- Abubaker, K.; Latifi, A.; Luwor, R.; Nazaretian, S.; Zhu, H.; Quinn, M.A.; Thompson, E.W.; Findlay, J.K.; Ahmed, N. Short-term single treatment of chemotherapy results in the enrichment of ovarian cancer stem cell-like cells leading to an increased tumor burden. Mol. Cancer 2013, 12, 24. [Google Scholar] [CrossRef]
- Hirsch, H.A.; Iliopoulos, D.; Tsichlis, P.N.; Struhl, K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009, 69, 7507–7511. [Google Scholar] [CrossRef]
- Lonardo, E.; Cioffi, M.; Sancho, P.; Sanchez-Ripoll, Y.; Trabulo, S.M.; Dorado, J.; Balic, A.; Hidalgo, M.; Heeschen, C. Metformin targets the metabolic achilles heel of human pancreatic cancer stem cells. PLoS ONE 2013, 8, e76518. [Google Scholar] [CrossRef]
- Yang, S.H.; Li, S.; Lu, G.; Xue, H.; Kim, D.H.; Zhu, J.J.; Liu, Y. Metformin treatment reduces temozolomide resistance of glioblastoma cells. Oncotarget 2016, 7, 78787–78803. [Google Scholar] [CrossRef]
- Marcato, P.; Dean, C.A.; Giacomantonio, C.A.; Lee, P.W. Aldehyde dehydrogenase: Its role as a cancer stem cell marker comes down to the specific isoform. Cell Cycle 2011, 10, 1378–1384. [Google Scholar] [CrossRef] [PubMed]
- Farnie, G.; Sotgia, F.; Lisanti, M.P. High mitochondrial mass identifies a sub-population of stem-like cancer cells that are chemo-resistant. Oncotarget 2015, 6, 30472–30486. [Google Scholar] [CrossRef] [PubMed]
- Cioce, M.; Valerio, M.; Casadei, L.; Pulito, C.; Sacconi, A.; Mori, F.; Biagioni, F.; Manetti, C.; Muti, P.; Strano, S.; et al. Metformin-induced metabolic reprogramming of chemoresistant ALDHbright breast cancer cells. Oncotarget 2014, 5, 4129–4143. [Google Scholar] [CrossRef]
- Walsh, H.R.; Cruickshank, B.M.; Brown, J.M.; Marcato, P. The Flick of a Switch: Conferring Survival Advantage to Breast Cancer Stem Cells Through Metabolic Plasticity. Front. Oncol. 2019, 9, 753. [Google Scholar] [CrossRef] [PubMed]
- Shin, M.K.; Cheong, J.H. Mitochondria-centric bioenergetic characteristics in cancer stem-like cells. Arch. Pharm. Res. 2019, 42, 113–127. [Google Scholar] [CrossRef]
- Iliopoulos, D.; Hirsch, H.A.; Struhl, K. Metformin decreases the dose of chemotherapy for prolonging tumor remission in mouse xenografts involving multiple cancer cell types. Cancer Res. 2011, 71, 3196–3201. [Google Scholar] [CrossRef]
- Shank, J.J.; Yang, K.; Ghannam, J.; Cabrera, L.; Johnston, C.J.; Reynolds, R.K.; Buckanovich, R.J. Metformin targets ovarian cancer stem cells in vitro and in vivo. Gynecol. Oncol. 2012, 127, 390–397. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Suh, D.H.; Kim, M.K.; Song, Y.S. Metformin against cancer stem cells through the modulation of energy metabolism: Special considerations on ovarian cancer. Biomed. Res. Int. 2014, 2014, 132702. [Google Scholar] [CrossRef]
- Sancho, P.; Burgos-Ramos, E.; Tavera, A.; Bou Kheir, T.; Jagust, P.; Schoenhals, M.; Barneda, D.; Sellers, K.; Campos-Olivas, R.; Grana, O.; et al. MYC/PGC-1alpha Balance Determines the Metabolic Phenotype and Plasticity of Pancreatic Cancer Stem Cells. Cell Metab. 2015, 22, 590–605. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Martin, A.; Cufi, S.; Lopez-Bonet, E.; Corominas-Faja, B.; Oliveras-Ferraros, C.; Martin-Castillo, B.; Menendez, J.A. Metformin limits the tumourigenicity of iPS cells without affecting their pluripotency. Sci. Rep. 2012, 2, 964. [Google Scholar] [CrossRef]
- Wang, Y.J.; Herlyn, M. The emerging roles of Oct4 in tumor-initiating cells. Am. J. Physiol. Cell Physiol. 2015, 309, C709–C718. [Google Scholar] [CrossRef]
- Brown, J.R.; Chan, D.K.; Shank, J.J.; Griffith, K.A.; Fan, H.; Szulawski, R.; Yang, K.; Reynolds, R.K.; Johnston, C.; McLean, K.; et al. Phase II clinical trial of metformin as a cancer stem cell-targeting agent in ovarian cancer. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Chen, G.; Yu, C.; Tang, Z.; Liu, S.; An, F.; Zhu, J.; Wu, Q.; Cao, J.; Zhan, Q.; Zhang, S. Metformin suppresses gastric cancer progression through calmodulinlike protein 3 secreted from tumorassociated fibroblasts. Oncol. Rep. 2019, 41, 405–414. [Google Scholar] [CrossRef]
- Tebbe, C.; Chhina, J.; Dar, S.A.; Sarigiannis, K.; Giri, S.; Munkarah, A.R.; Rattan, R. Metformin limits the adipocyte tumor-promoting effect on ovarian cancer. Oncotarget 2014, 5, 4746–4764. [Google Scholar] [CrossRef]
- Melzer, C.; Yang, Y.; Hass, R. Interaction of MSC with tumor cells. Cell Commun. Signal. 2016, 14, 20. [Google Scholar] [CrossRef]
- Li, D.; Jiao, W.; Liang, Z.; Wang, L.; Chen, Y.; Wang, Y.; Liang, Y.; Niu, H. Variation in energy metabolism arising from the effect of the tumor microenvironment on cell biological behaviors of bladder cancer cells and endothelial cells. Biofactors 2020, 46, 64–75. [Google Scholar] [CrossRef]
- Li, I.; Nabet, B.Y. Exosomes in the tumor microenvironment as mediators of cancer therapy resistance. Mol. Cancer 2019, 18, 32. [Google Scholar] [CrossRef]
- El Hout, M.; Cosialls, E.; Mehrpour, M.; Hamai, A. Crosstalk between autophagy and metabolic regulation of cancer stem cells. Mol. Cancer 2020, 19, 27. [Google Scholar] [CrossRef]
- Mandhair, H.K.; Arambasic, M.; Novak, U.; Radpour, R. Molecular modulation of autophagy: New venture to target resistant cancer stem cells. World J. Stem Cells 2020, 12, 303–322. [Google Scholar] [CrossRef]
- Smith, A.G.; Macleod, K.F. Autophagy, cancer stem cells and drug resistance. J. Pathol. 2019, 247, 708–718. [Google Scholar] [CrossRef]
- Garcia-Calzon, S.; Perfilyev, A.; Martinell, M.; Ustinova, M.; Kalamajski, S.; Franks, P.W.; Bacos, K.; Elbere, I.; Pihlajamaki, J.; Volkov, P.; et al. Epigenetic markers associated with metformin response and intolerance in drug-naive patients with type 2 diabetes. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cioce, M.; Pulito, C.; Strano, S.; Blandino, G.; Fazio, V.M. Metformin: Metabolic Rewiring Faces Tumor Heterogeneity. Cells 2020, 9, 2439. https://doi.org/10.3390/cells9112439
Cioce M, Pulito C, Strano S, Blandino G, Fazio VM. Metformin: Metabolic Rewiring Faces Tumor Heterogeneity. Cells. 2020; 9(11):2439. https://doi.org/10.3390/cells9112439
Chicago/Turabian StyleCioce, Mario, Claudio Pulito, Sabrina Strano, Giovanni Blandino, and Vito Michele Fazio. 2020. "Metformin: Metabolic Rewiring Faces Tumor Heterogeneity" Cells 9, no. 11: 2439. https://doi.org/10.3390/cells9112439
APA StyleCioce, M., Pulito, C., Strano, S., Blandino, G., & Fazio, V. M. (2020). Metformin: Metabolic Rewiring Faces Tumor Heterogeneity. Cells, 9(11), 2439. https://doi.org/10.3390/cells9112439