An Overview of Astrocyte Responses in Genetically Induced Alzheimer’s Disease Mouse Models


Abstract
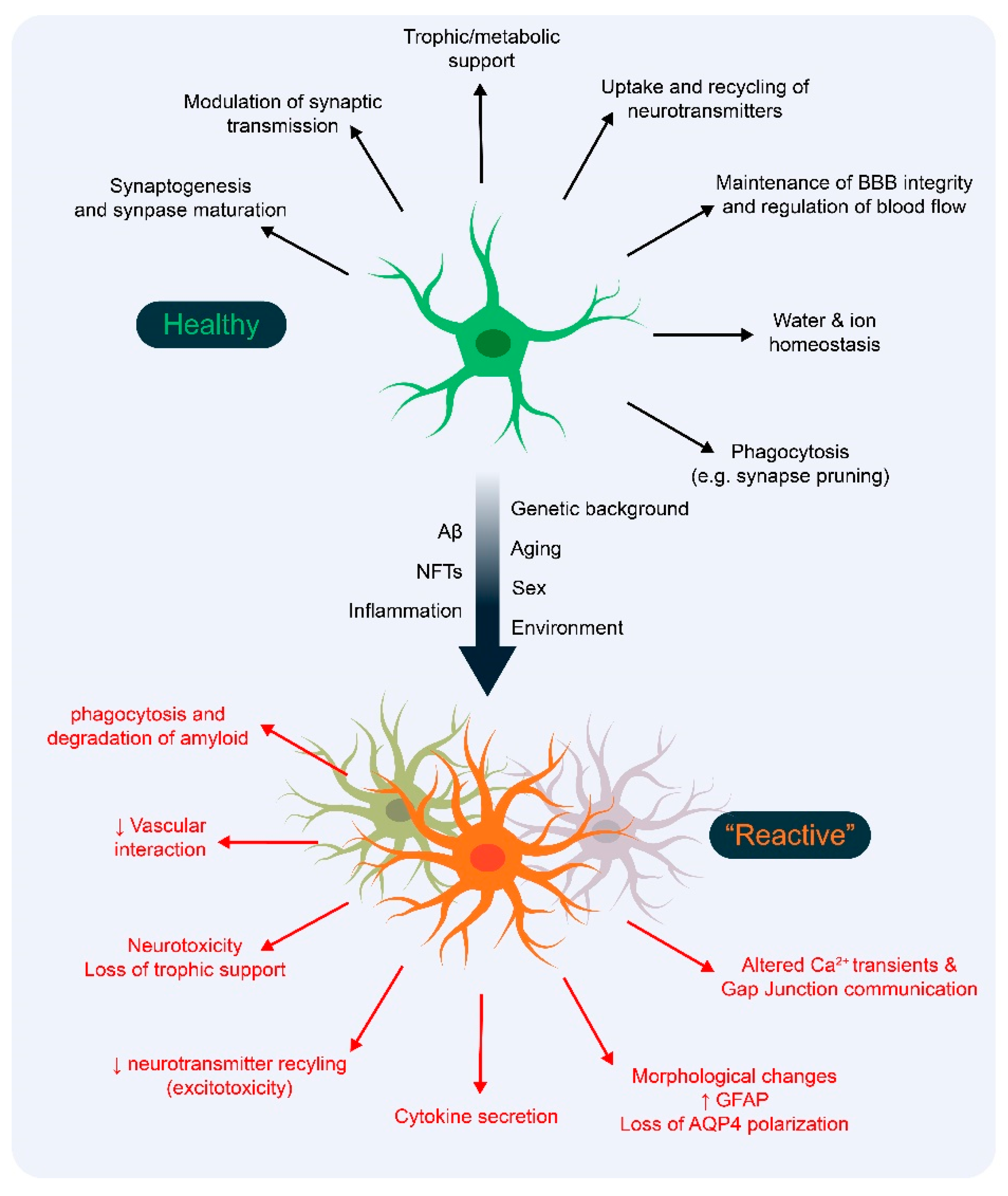
1. Introduction
2. Astrocyte Responses in Genetically Induced AD Mouse Models
2.1. Familial Alzheimer’s Disease Mouse Models
2.1.1. Models Involving the APP Pathway
2.1.2. Models Involving the MAPT Gene
2.2. Sporadic or Late-Onset Alzheimer’s Disease Mouse Models
2.2.1. Apolipoprotein-E Models in Alzheimer’s Disease
2.2.2. TREM2 Models in Alzheimer’s Disease
3. Limitations of Studying Astrocytes in Mouse Models
3.1. Limitations Restricted to Astrocytes
3.2. Other Limitations of Studying AD in Mice
4. Recent Studies and Future Perspectives for Studying Astrocytes in AD
4.1. Transcriptomic Studies
4.2. The Rise of Proteomics
4.3. Novel Mouse Models
4.4. Chimeric Mice
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jahn, H. Memory loss in Alzheimer’s disease. Clin. Res. 2013, 15, 445–454. [Google Scholar]
- Haj, M. El Memory suppression in Alzheimer’s disease. Neurol. Sci. 2016, 37, 337–343. [Google Scholar] [CrossRef]
- Bondi, M.W.; Edmonds, E.C.; Salmon, D.P. Alzheimer’s Disease: Past, Present, and Future. J. Int. Neuropsychol. Soc. 2017, 23, 818–831. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological Alterations in Alzheimer Disease. Cold Sping Harb. Perspect. Med. 2011, 1, 1–23. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, R.J.; Wong, P.C. Amyloid Precursor Protein Processing in Alzheimer’s Disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef]
- Cummings, J.L.; Morstorf, T.; Zhong, K. Alzheimer’s disease drug-development pipeline: Few candidates, frequent failures. Alzheimers Res. Ther. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, S.; Albert, M.; Fox, N.; Goedert, M.; Kivipelto, M.; Mestre-Ferrandiz, J.; Middleton, L.T. Why has therapy development for dementia failed in the last two decades? Alzheimers Dement. 2016, 12, 60–64. [Google Scholar] [CrossRef]
- Gatz, M.; Reynolds, C.A.; Fratiglioni, L.; Johansson, B.; Mortimer, J.A.; Berg, S.; Fiske, A.; Pedersen, N.L. Role of genes and environments for explaining Alzheimer disease. Arch. Gen. Psychiatry 2006, 63, 168–174. [Google Scholar] [CrossRef]
- Arranz, A.M.; De Strooper, B. The role of astroglia in Alzheimer’s disease: Pathophysiology and clinical implications. Lancet Glob. Health 2019, 4422, 25–27. [Google Scholar] [CrossRef]
- Sadick, J.S.; Liddelow, S.A. Don’t forget astrocytes when targeting Alzheimer’s disease. Br. J. Pharmacol. 2019, 176, 3585–3598. [Google Scholar] [CrossRef] [PubMed]
- Bennett, F.C.; Liddelow, S.A. Microglia Metabolic Breakdown Drives Alzheimer’ s Pathology. Cell Metab. 2019, 30, 405–406. [Google Scholar] [CrossRef] [PubMed]
- Nortley, R.; Korte, N.; Izquierdo, P.; Hirunpattarasilp, C.; Mishra, A.; Jaunmuktane, Z.; Kyrargyri, V.; Pfeiffer, T.; Khennouf, L.; Madry, C.; et al. Amyloid β oligomers constrict human capillaries in Alzheimer’s disease via signaling to pericytes. Science 2019, 365. [Google Scholar] [CrossRef] [PubMed]
- Von Bartheld, C.S.; Bahney, J.; Herculano-Houzel, S. The Search for True Numbers of Neurons and Glial Cells in the Human Brain: A Review of 150 Years of Cell Counting. J. Comp. Neurol. 2016, 524, 3865–3895. [Google Scholar] [CrossRef] [PubMed]
- Erö, C.; Gewaltig, M.O.; Keller, D.; Markram, H. A cell atlas for the mouse brain. Front. Neuroinform. 2018, 12, 1–16. [Google Scholar] [CrossRef]
- Keller, D.; Erö, C.; Markram, H. Cell densities in the mouse brain: A systematic review. Front. Neuroanat. 2018, 12. [Google Scholar] [CrossRef] [PubMed]
- Pelvig, D.P.; Pakkenberg, H.; Stark, A.K.; Pakkenberg, B. Neocortical glial cell numbers in human brains. Neurobiol. Aging 2008, 29, 1754–1762. [Google Scholar] [CrossRef]
- Allen, N.J. Astrocyte Regulation of Synaptic Behavior. Annu. Rev. Cell Dev. Biol. 2014, 30, 439–463. [Google Scholar] [CrossRef]
- Mahmoud, S.; Gharagozloo, M.; Simard, C.; Gris, D. Astrocytes Maintain Glutamate Homeostasis in the CNS by Controlling the Balance between Glutamate Uptake and Release. Cells 2019, 8, 184. [Google Scholar] [CrossRef]
- Barreto, G.E.; Gonzalez, J.; Torres, Y.; Morales, L. Astrocytic-neuronal crosstalk: Implications for neuroprotection from brain injury. Neurosci. Res. 2011, 71, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Yang, Y.; Ju, W.; Wang, X.; Zhang, H. Emerging Roles of Astrocytes in Neuro-Vascular Unit and the Tripartite Synapse with Emphasis on Reactive Gliosis in the Context of Alzheimer’s Disease. Front. Cell. Neurosci. 2018, 12, 1–12. [Google Scholar] [CrossRef]
- Macvicar, B.A.; Newman, E.A. Astrocyte Regulation of Blood Flow in the Brain. Cold Sping Harb. Perspect. Med. 2015, 7, 1–14. [Google Scholar] [CrossRef]
- Sofroniew, M.V. Astrogliosis. Cold Spring Harb. Perspect. Biol. 2015, 7, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Bignami, A.; Eng, L.F.; Dahl, D.; Uyeda, C.T. Localization of the glial fibrillary acidic protein in astrocytes by immunofluorescence. Brain Res. 1972, 43, 429–435. [Google Scholar] [CrossRef]
- Alberdi, E.; Wyssenbach, A.; Alberdi, M.; Sánchez-Gómez, M.V.; Cavaliere, F.; Rodríguez, J.J.; Verkhratsky, A.; Matute, C. Calcium-dependent endoplasmic reticulum stress correlates with astrogliosis in oligomeric amyloid β-treated astrocytes and in a model of Alzheimer’s disease. Aging Cell 2013, 12, 292–302. [Google Scholar] [CrossRef]
- Doméné, A.; Cavanagh, C.; Page, G.; Bodard, S.; Klein, C.; Delarasse, C.; Chalon, S.; Krantic, S. Expression of Phenotypic Astrocyte Marker Is Increased in a Transgenic Mouse Model of Alzheimer’s Disease versus Age-Matched Controls: A Presymptomatic Stage Study. Int. J. Alzheimers Dis. 2016. [Google Scholar] [CrossRef]
- Li, K.; Li, J.; Zheng, J.; Qin, S. Reactive Astrocytes in Neurodegenerative Diseases. Aging Dis. 2019, 10, 664–675. [Google Scholar] [CrossRef]
- Porchet, R.; Probst, A.; Bouras, C.; Dráberová, E.; Dráber, P.; Riederer, B.M. Analysis of gial acidic fibrillary protein in the human entorhinal cortex during aging and in Alzheimer’s disease. Proteomics 2003, 3, 1476–1485. [Google Scholar] [CrossRef]
- Zamanian, J.L.; Xu, L.; Foo, L.C.; Nouri, N.; Zhou, L.; Giffard, R.G.; Barres, B.A. Genomic analysis of reactive astrogliosis. J. Neurosci. 2012, 32, 6391–6410. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Habib, N.; McCabe, C.; Medina, S.; Varshavsky, M.; Kitsberg, D.; Dvir-Szternfeld, R.; Green, G.; Dionne, D.; Nguyen, L.; Marshall, J.L.; et al. Disease-associated astrocytes in Alzheimer’s disease and aging. Nat. Neurosci. 2020, 23, 701–706. [Google Scholar] [CrossRef]
- Chen, W.T.; Lu, A.; Craessaerts, K.; Pavie, B.; Sala Frigerio, C.; Corthout, N.; Qian, X.; Laláková, J.; Kühnemund, M.; Voytyuk, I.; et al. Spatial Transcriptomics and In Situ Sequencing to Study Alzheimer’s Disease. Cell 2020, 182, 976–991.e19. [Google Scholar] [CrossRef]
- Wu, T.; Dejanovic, B.; Gandham, V.D.; Gogineni, A.; Edmonds, R.; Schauer, S.; Srinivasan, K.; Huntley, M.A.; Wang, Y.; Wang, T.M.; et al. Complement C3 Is Activated in Human AD Brain and Is Required for Neurodegeneration in Mouse Models of Amyloidosis and Tauopathy. Cell Rep. 2019, 28, 2111–2123.e6. [Google Scholar] [CrossRef]
- Chen, W.T.; Lu, A.; Craessaerts, K.; Pavie, B.; Frigerio, C.S.; Mancuso, R.; Qian, X.; Lalakova, J.; Kühnemund, M.; Voytyuk, I.; et al. Spatial and temporal transcriptomics reveal microglia-astroglia crosstalk in the amyloid-β plaque cell niche of Alzheimer’s disease. bioRxiv 2019, 719930. [Google Scholar] [CrossRef]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef]
- Zhou, Y.; Song, W.M.; Andhey, P.S.; Swain, A.; Levy, T.; Miller, K.R.; Poliani, P.L.; Cominelli, M.; Grover, S.; Gilfillan, S.; et al. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat. Med. 2020, 26, 131–142. [Google Scholar] [CrossRef]
- Grubman, A.; Chew, G.; Ouyang, J.F.; Sun, G.; Choo, X.Y.; McLean, C.; Simmons, R.K.; Buckberry, S.; Vargas-Landin, D.B.; Poppe, D.; et al. A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. Nat. Neurosci. 2019, 22, 2087–2097. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Marsh, S.E.; Stevens, B. Microglia and astrocyte interactions in health and disease: Dynamic Duo or Partners in Crime? Trends Immunol. 2020, 41, 820–835. [Google Scholar] [CrossRef]
- Delekate, A.; Füchtemeier, M.; Schumacher, T.; Ulbrich, C.; Foddis, M.; Petzold, G.C. Metabotropic P2Y1 receptor signalling mediates astrocytic hyperactivity in vivo in an Alzheimer’s disease mouse model. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhang, R.; Shi, C.; Mao, C.; Yang, Z.; Suo, Z.; Torp, R.; Xu, Y. AQP4 Association with Amyloid Deposition and Astrocyte Pathology in the Tg-ArcSwe Mouse Model of Alzheimer’s Disease. J. Alzheimers Dis. 2011, 27, 622–711. [Google Scholar] [CrossRef]
- Balducci, C.; Forloni, G. APP Transgenic Mice: Their Use and Limitations. Neuromol. Med. 2011, 13, 117–137. [Google Scholar] [CrossRef] [PubMed]
- Herrup, K.; Carrillo, M.C.; Schenk, D.; Cacace, A.; Desanti, S.; Fremeau, R.; Bhat, R.; Glicksman, M.; May, P.; Swerdlow, R.; et al. Beyond amyloid: Getting real about nonamyloid targets in Alzheimer’s disease. Alzheimers Dement. 2013, 9, 452–458. [Google Scholar] [CrossRef]
- Kitazawa, M.; Medeiros, R.; Laferla, F.M. Transgenic Mouse Models of Alzheimer Disease: Developing a Better Model as a Tool for Therapeutic Interventions. Curr. Pharm. Des. 2012, 18, 1131–1147. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Govindpani, K.; McNamara, L.G.; Smith, N.R.; Vinnakota, C.; Waldvogel, H.J.; Faull, R.L.; Kwakowsky, A. Vascular Dysfunction in Alzheimer’s Disease: A Prelude to the Pathological Process or a Consequence of It? J. Clin. Med. 2019, 8, 651. [Google Scholar] [CrossRef] [PubMed]
- Onos, K.D.; Uyar, A.; Keezer, K.J.; Jackson, H.M.; Preuss, C.; Acklin, C.J.; O’Rourke, R.; Buchanan, R.; Cossette, T.L.; Sukoff Rizzo, S.J.; et al. Enhancing face validity of mouse models of Alzheimer’s disease with natural genetic variation. PLoS Genet. 2019, 15, e1008155. [Google Scholar] [CrossRef]
- Neuner, S.M.; Heuer, S.E.; Huentelman, M.J.; O’Connell, K.M.S.; Kaczorowski, C.C. Harnessing Genetic Complexity to Enhance Translatability of Alzheimer’s Disease Mouse Models: A Path toward Precision Medicine. Neuron 2019, 101, 399–411.e5. [Google Scholar] [CrossRef]
- Dunn, A.R.; O’Connell, K.M.S.; Kaczorowski, C.C. Gene-by-environment interactions in Alzheimer’s disease and Parkinson’s disease. Neurosci. Biobehav. Rev. 2019, 103, 73–80. [Google Scholar] [CrossRef]
- Wang, H.; Li, Y.; Ryder, J.W.; Hole, J.T.; Ebert, P.J.; Airey, D.C.; Qian, H.R.; Logsdon, B.; Fisher, A.; Ahmed, Z.; et al. Genome-wide RNAseq study of the molecular mechanisms underlying microglia activation in response to pathological tau perturbation in the rTg4510 tau transgenic animal model. Mol. Neurodegener. 2018, 13, 1–19. [Google Scholar] [CrossRef]
- Mukherjee, S.; Perumal, T.M.; Daily, K.; Sieberts, S.K.; Omberg, L.; Preuss, C.; Carter, G.W.; Mangravite, L.M.; Logsdon, B.A. Identifying and ranking potential driver genes of Alzheimer’s disease using multiview evidence aggregation. Bioinformatics 2019, 35, i568–i576. [Google Scholar] [CrossRef]
- Wan, Y.W.; Al-Ouran, R.; Mangleburg, C.G.; Perumal, T.M.; Lee, T.V.; Allison, K.; Swarup, V.; Funk, C.C.; Gaiteri, C.; Allen, M.; et al. Meta-Analysis of the Alzheimer’s Disease Human Brain Transcriptome and Functional Dissection in Mouse Models. Cell Rep. 2020, 32. [Google Scholar] [CrossRef] [PubMed]
- Elder, G.A.; Gama Sosa, M.A.; De Gasperi, R. Transgenic Mouse Models of Alzheimer’s Disease. Mt. Sinai J. Med. 2010, 77, 68–81. [Google Scholar] [CrossRef]
- Hall, A.M.; Roberson, E.D. Mouse models of Alzheimer’s disease. Brain Res. Bull. 2012, 88, 3–12. [Google Scholar] [CrossRef]
- Sturchler-Pierrat, C.; Abramowski, D.; Duke, M.; Wiederhold, K.H.; Mistl, C.; Rothacher, S.; Ledermann, B.; Bürki, K.; Frey, P.; Paganetti, P.A.; et al. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc. Natl. Acad. Sci. USA 1997, 94, 13287–13292. [Google Scholar] [CrossRef] [PubMed]
- Dujardin, S.; Colin, M.; Buée, L. Invited review: Animal models of tauopathies and their implications for research/translation into the clinic. Neuropathol. Appl. Neurobiol. 2015, 41, 59–80. [Google Scholar] [CrossRef] [PubMed]
- Jackson, R.J.; Rudinskiy, N.; Herrmann, A.G.; Croft, S.; Kim, J.S.M.; Petrova, V.; Ramos-Rodriguez, J.J.; Pitstick, R.; Wegmann, S.; Garcia-Alloza, M.; et al. Human tau increases amyloid β plaque size but not amyloid β-mediated synapse loss in a novel mouse model of Alzheimer’s disease. Eur. J. Neurosci. 2016, 44, 3056–3066. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic model of Alzheimer’s Disease with plaques and tangles: Intracellular Aβ and synaptic dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef]
- Tesseur, I.; Van Dorpe, J.; Spittaels, K.; Van den Haute, C.; Moechars, D.; Van Leuven, F. Expression of human apolipoprotein E4 in neurons causes hyperphosphorylation of protein tau in the brains of transgenic mice. Am. J. Pathol. 2000, 156, 951–964. [Google Scholar] [CrossRef]
- Saito, T.; Matsuba, Y.; Mihira, N.; Takano, J.; Nilsson, P.; Itohara, S.; Iwata, N.; Saido, T.C. Single App knock-in mouse models of Alzheimer’s disease. Nat. Neurosci. 2014, 17, 661–663. [Google Scholar] [CrossRef]
- Genin, E.; Hannequin, D.; Wallon, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Bullido, M.J.; Engelborghs, S.; De Deyn, P.; Berr, C.; et al. APOE and Alzheimer disease: A major gene with semi-dominant inheritance. Mol. Psychiatry 2011, 16, 903–907. [Google Scholar] [CrossRef]
- Sims, R.; Van Der Lee, S.J.; Naj, A.C.; Bellenguez, C.; Badarinarayan, N.; Jakobsdottir, J.; Kunkle, B.W.; Boland, A.; Raybould, R.; Bis, J.C.; et al. Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat. Genet. 2017, 49, 1373–1384. [Google Scholar] [CrossRef]
- Waring, S.C.; Rosenberg, R.N. Genome-wide association studies in Alzheimer disease. Arch. Neurol. 2008, 65, 329–334. [Google Scholar] [CrossRef]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; Jun, G.; DeStefano, A.L.; Bis, J.C.; Beecham, G.W.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef]
- Saunders, A.; Macosko, E.Z.; Wysoker, A.; Goldman, M.; Krienen, F.M.; de Rivera, H.; Bien, E.; Baum, M.; Bortolin, L.; Wang, S.; et al. Molecular Diversity and Specializations among the Cells of the Adult Mouse Brain. Cell 2018, 174, 1015–1030. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef]
- Cai, Y.; An, S.S.A.; Kim, S. Mutations in presenilin 2 and its implications in Alzheimer’s disease and other dementia-associated disorders. Clin. Interv. Aging 2015, 10, 1163–1172. [Google Scholar] [CrossRef]
- Kelleher, R.J.; Shen, J. Presenilin-1 mutations and Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, 629–631. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Benzing, W.C.; Wujek, J.R.; Ward, E.K.; Shaffer, D.; Ashe, K.H.; Younkin, S.G.; Brunden, K.R. Evidence for glial-mediated inflammation in aged APP(SW) transgenic mice. Neurobiol. Aging 1999, 20, 581–589. [Google Scholar] [CrossRef]
- Matsuoka, Y.; Picciano, M.; Maleste, B.; LaFrancois, J.; Zehr, C.; Daeschner, J.A.M.; Olschowka, J.A.; Fonseca, M.I.; O’Banion, M.K.; Tenner, A.J.; et al. Inflammatory responses to amyloidosis in a transgenic mouse model of Alzheimer’s disease. Am. J. Pathol. 2001, 158, 1345–1354. [Google Scholar] [CrossRef]
- Manaye, K.F.; Wang, P.C.; O’Neil, J.N.; Huang, S.Y.; Xu, T.; Lei, D.L.; Tizabi, Y.; Ottinger, M.A.; Ingram, D.K.; Mouton, P.R. Neuropathological quantification of dtg APP/PS1: Neuroimaging, stereology, and biochemistry. Age 2007, 29, 87–96. [Google Scholar] [CrossRef]
- Jorda, A.; Cauli, O.; Santonja, J.M.; Aldasoro, M.; Aldasoro, C.; Obrador, E.; Vila, J.M.; Mauricio, M.D.; Iradi, A.; Guerra-Ojeda, S.; et al. Changes in chemokines and chemokine receptors expression in a mouse model of alzheimer’s disease. Int. J. Biol. Sci. 2019, 15, 453–463. [Google Scholar] [CrossRef]
- Liu, L.; Liu, Y.; Li, N.; Huang, R.; Zheng, X.; Huang, L.; Hou, S.; Yuan, Q. Multiple inflammatory profiles of microglia and altered neuroimages in APP/PS1 transgenic AD mice. Brain Res. Bull. 2020, 156, 86–104. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Yarishkin, O.; Hwang, Y.J.; Chun, Y.E.; Park, M.; Woo, D.H.; Bae, J.Y.; Kim, T.; Lee, J.; Chun, H.; et al. GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat. Med. 2014, 20, 886–896. [Google Scholar] [CrossRef]
- Lian, H.; Litvinchuk, A.; Chiang, A.C.A.; Aithmitti, N.; Jankowsky, J.L.; Zheng, H. Astrocyte-microglia cross talk through complement activation modulates amyloid pathology in mouse models of alzheimer’s disease. J. Neurosci. 2016, 36, 577–589. [Google Scholar] [CrossRef]
- Sérrière, S.; Tauber, C.; Vercouillie, J.; Mothes, C.; Pruckner, C.; Guilloteau, D.; Kassiou, M.; Doméné, A.; Garreau, L.; Page, G.; et al. Amyloid load and translocator protein 18kDa in APPswePS1-dE9 mice: A longitudinal study. Neurobiol. Aging 2015, 36, 1639–1652. [Google Scholar] [CrossRef]
- Ruan, L.; Kang, Z.; Pei, G.; Le, Y. Amyloid Deposition and Inflammation in APPswe/PS1dE9 Mouse Model of Alzheimer’s Disease. Curr. Alzheimer Res. 2009, 6, 531–540. [Google Scholar] [CrossRef]
- Kamphuis, W.; Orre, M.; Kooijman, L.; Dahmen, M.; Hol, E.M. Differential cell proliferation in the cortex of the APPswePS1dE9 Alzheimer’s disease mouse model. Glia 2012, 60, 615–629. [Google Scholar] [CrossRef]
- Janota, C.S.; Brites, D.; Lemere, C.A.; Brito, M.A. Glio-vascular changes during ageing in wild-type and Alzheimer’s disease-like APP/PS1 mice. Brain Res. 2015, 1620, 153–168. [Google Scholar] [CrossRef][Green Version]
- Huang, H.; Nie, S.; Cao, M.; Marshall, C.; Gao, J.; Xiao, N.; Hu, G.; Xiao, M. Characterization of AD-like phenotype in aged APPSwe/PS1dE9 mice. Age 2016, 38, 303–322. [Google Scholar] [CrossRef]
- Galea, E.; Morrison, W.; Hudry, E.; Arbel-Ornath, M.; Bacskai, B.J.; Gómez-Isla, T.; Stanley, H.E.; Hyman, B.T. Topological analyses in APP/PS1 mice reveal that astrocytes do not migrate to amyloid-β plaques. Proc. Natl. Acad. Sci. USA 2015, 112, 15556–15561. [Google Scholar] [CrossRef]
- Minkeviciene, R.; Ihalainen, J.; Malm, T.; Matilainen, O.; Keksa-Goldsteine, V.; Goldsteins, G.; Iivonen, H.; Leguit, N.; Glennon, J.; Koistinaho, J.; et al. Age-related decrease in stimulated glutamate release and vesicular glutamate transporters in APP/PS1 transgenic and wild-type mice. J. Neurochem. 2008, 105, 584–594. [Google Scholar] [CrossRef]
- Jackson, H.M.; Soto, I.; Graham, L.C.; Carter, G.W.; Howell, G.R. Clustering of transcriptional profiles identifies changes to insulin signaling as an early event in a mouse model of Alzheimer’s disease. BMC Genom. 2013, 14. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef]
- Shi, Q.; Chowdhury, S.; Ma, R.; Le, K.X.; Hong, S.; Caldarone, B.J.; Stevens, B.; Lemere, C.A. Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Viña, J.; Lloret, A. Why women have more Alzheimer’s disease than men: Gender and mitochondrial toxicity of amyloid-β peptide. J. Alzheimers Dis. 2010, 20, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Turner, R.S. Alzheimer’s disease in man and transgenic mice: Females at higher risk. Am. J. Pathol. 2001, 158, 797–801. [Google Scholar] [CrossRef]
- Yang, J.T.; Wang, Z.J.; Cai, H.Y.; Yuan, L.; Hu, M.M.; Wu, M.N.; Qi, J.S. Sex Differences in Neuropathology and Cognitive Behavior in APP/PS1/tau Triple-Transgenic Mouse Model of Alzheimer’s Disease. Neurosci. Bull. 2018, 34, 736–746. [Google Scholar] [CrossRef]
- Carroll, J.C.; Rosario, E.R.; Kreimer, S.; Villamagna, A.; Gentzschein, E.; Stanczyk, F.Z.; Pike, C.J. Sex differences in β-amyloid accumulation in 3xTg-AD mice: Role of neonatal sex steroid hormone exposure. Brain Res. 2010, 1366, 233–245. [Google Scholar] [CrossRef]
- Roy, U.; Stute, L.; Höfling, C.; Hartlage-Rübsamen, M.; Matysik, J.; Roβner, S.; Alia, A. Sex- and age-specific modulation of brain GABA levels in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2018, 62, 168–179. [Google Scholar] [CrossRef]
- Kowalski, K.; Mulak, A. Brain-gut-microbiota axis in Alzheimer’s disease. J. Neurogastroenterol. Motil. 2019, 25, 48–60. [Google Scholar] [CrossRef]
- Richetin, K.; Petsophonsakul, P.; Roybon, L.; Guiard, B.P.; Rampon, C. Differential alteration of hippocampal function and plasticity in females and males of the APPxPS1 mouse model of Alzheimer’s disease. Neurobiol. Aging 2017, 57, 220–231. [Google Scholar] [CrossRef]
- Kamphuis, W.; Mamber, C.; Moeton, M.; Kooijman, L.; Sluijs, J.A.; Jansen, A.H.P.; Verveer, M.; de Groot, L.R.; Smith, V.D.; Rangarajan, S.; et al. GFAP isoforms in adult mouse brain with a focus on neurogenic astrocytes and reactive astrogliosis in mouse models of Alzheimer disease. PLoS ONE 2012, 7, e42823. [Google Scholar] [CrossRef]
- Ceyzériat, K.; Ben Haim, L.; Denizot, A.; Pommier, D.; Matos, M.; Guillemaud, O.; Palomares, M.A.; Abjean, L.; Petit, F.; Gipchtein, P.; et al. Modulation of astrocyte reactivity improves functional deficits in mouse models of Alzheimer’s disease. Acta Neuropathol. Commun. 2018, 6, 104. [Google Scholar] [CrossRef]
- Gomez-Arboledas, A.; Davila, J.C.; Sanchez-Mejias, E.; Navarro, V.; Nuñez-Diaz, C.; Sanchez-Varo, R.; Sanchez-Mico, M.V.; Trujillo-Estrada, L.; Fernandez-Valenzuela, J.J.; Vizuete, M.; et al. Phagocytic clearance of presynaptic dystrophies by reactive astrocytes in Alzheimer’s disease. Glia 2018, 66, 637–653. [Google Scholar] [CrossRef]
- Kimbrough, I.F.; Robel, S.; Roberson, E.D.; Sontheimer, H. Vascular amyloidosis impairs the gliovascular unit in a mouse model of Alzheimer’s disease. Brain 2015, 138, 3716–3733. [Google Scholar] [CrossRef]
- Beauquis, J.; Vinuesa, A.; Pomilio, C.; Pavia, P.; Galvan, V.; Saravia, F. Neuronal and Glial Alterations, Increased Anxiety, and Cognitive Impairment Before Hippocampal Amyloid Deposition in PDAPP Mice, Model of Alzheimer’s Disease Juan. Hippocampus 2014, 24, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Lunde, L.K.; Nuntagij, P.; Oguchi, T.; Camassa, L.M.A.; Nilsson, L.N.G.; Lannfelt, L.; Xu, Y.; Amiry-Moghaddam, M.; Ottersen, O.P.; et al. Loss of astrocyte polarization in the Tg-ArcSwe mouse model of Alzheimer’s disease. J. Alzheimers Dis. 2011, 27, 711–722. [Google Scholar] [CrossRef] [PubMed]
- Heiland, T.; Zeitschel, U.; Puchades, M.A.; Kuhn, P.H.; Lichtenthaler, S.F.; Bjaalie, J.G.; Hartlage-Rübsamen, M.; Roßner, S.; Höfling, C. Defined astrocytic expression of human amyloid precursor protein in Tg2576 mouse brain. Glia 2019, 67, 393–403. [Google Scholar] [CrossRef]
- Waniek, A.; Hartlage-Rübsamen, M.; Höfling, C.; Kehlen, A.; Schilling, S.; Demuth, H.U.; Roßner, S. Identification of thyrotropin-releasing hormone as hippocampal glutaminyl cyclase substrate in neurons and reactive astrocytes. Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 146–155. [Google Scholar] [CrossRef]
- Wilcock, D.M.; Vitek, M.P.; Colton, C.A. Vascular amyloid alters astrocytic water and potassium channels in mouse models and humans with Alzheimer’s disease. Neuroscience 2009, 159, 1055–1069. [Google Scholar] [CrossRef] [PubMed]
- Merlini, M.; Meyer, E.P.; Nitsch, A.U.R.M. Vascular β-amyloid and early astrocyte alterations impair cerebrovascular function and cerebral metabolism in transgenic arcAβ mice. Acta Neuropathol. 2011, 293–311. [Google Scholar] [CrossRef]
- Ugolini, F.; Lana, D.; Nardiello, P.; Nosi, D.; Pantano, D.; Casamenti, F.; Giovannini, M.G. Different Patterns of Neurodegeneration and Glia Activation in CA1 and CA3 Hippocampal Regions of TgCRND8 Mice. Front. Aging Neurosci. 2018, 10, 1–23. [Google Scholar] [CrossRef]
- Freeman, M.R. Specification and morphogenesis of astrocytes. Science 2010, 330, 774–778. [Google Scholar] [CrossRef]
- Lanjakornsiripan, D.; Pior, B.J.; Kawaguchi, D.; Furutachi, S.; Tahara, T.; Katsuyama, Y.; Suzuki, Y.; Fukazawa, Y.; Gotoh, Y. Layer-specific morphological and molecular differences in neocortical astrocytes and their dependence on neuronal layers. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Rockenstein, E.; Mallory, M.; Mante, M.; Sisk, A.; Masliaha, E. Early formation of mature amyloid-β protein deposits in a mutant APP transgenic model depends on levels of Aβ1-42. J. Neurosci. Res. 2001, 66, 573–582. [Google Scholar] [CrossRef]
- Hsiao, K.; Chapman, P.; Nilsen, S.; Eckman, C.; Harigaya, Y.; Younkin, S.; Yang, F.; Cole, G. Correlative Memory Deficits, Aβ Elevation, and Amyloid Plaques in Transgenic Mice. Science 1996, 274, 99–102. [Google Scholar] [CrossRef]
- Rönnbäck, A.; Sagelius, H.; Bergstedt, K.D.; Näslund, J.; Westermark, G.T.; Winblad, B.; Graff, C. Amyloid neuropathology in the single Arctic APP transgenic model affects interconnected brain regions. Neurobiol. Aging 2012, 33, 11–19. [Google Scholar] [CrossRef]
- Lord, A.; Kalimo, H.; Eckman, C.; Zhang, X.Q.; Lannfelt, L.; Nilsson, L.N.G. The Arctic Alzheimer mutation facilitates early intraneuronal Aβ aggregation and senile plaque formation in transgenic mice. Neurobiol. Aging 2006, 27, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.; Xu, F.; Deane, R.; Romanov, G.; Previti, M.L.; Zeigler, K.; Zlokovic, B.V.; Van Nostrand, W.E. Early-onset and Robust Cerebral Microvascular Accumulation of Amyloid β-Protein in Transgenic Mice Expressing Low Levels of a Vasculotropic Dutch/Iowa Mutant Form of Amyloid β-Protein Precursor. J. Biol. Chem. 2004, 279, 20296–20306. [Google Scholar] [CrossRef]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal β-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef]
- Radde, R.; Bolmont, T.; Kaeser, S.A.; Coomaraswamy, J.; Lindau, D.; Stoltze, L.; Calhoun, M.E.; Jäggi, F.; Wolburg, H.; Gengler, S.; et al. Aβ42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep. 2006, 7, 940–946. [Google Scholar] [CrossRef]
- Jankowsky, J.L.; Fadale, D.J.; Anderson, J.; Xu, G.M.; Gonzales, V.; Jenkins, N.A.; Copeland, N.G.; Lee, M.K.; Younkin, L.H.; Wagner, S.L.; et al. Mutant presenilins specifically elevate the levels of the 42 residue β-amyloid peptide in vivo: Evidence for augmentation of a 42-specific γ secretase. Hum. Mol. Genet. 2004, 13, 159–170. [Google Scholar] [CrossRef]
- Jankowsky, J.L.; Slunt, H.H.; Ratovitski, T.; Jenkins, N.A.; Copeland, N.G.; Borchelt, D.R. Co-expression of multiple transgenes in mouse CNS: A comparison of strategies. Biomol. Eng. 2001, 17, 157–165. [Google Scholar] [CrossRef]
- Blanchard, V.; Moussaoui, S.; Czech, C.; Touchet, N.; Bonici, B.; Planche, M.; Canton, T.; Jedidi, I.; Gohin, M.; Wirths, O.; et al. Time sequence of maturation of dystrophic neurites associated with Aβ deposits in APP/PS1 transgenic mice. Exp. Neurol. 2003, 184, 247–263. [Google Scholar] [CrossRef]
- Richards, J.G.; Higgins, G.A.; Ouagazzal, A.M.; Ozmen, L.; Kew, J.N.C.; Bohrmann, B.; Malherbe, P.; Brockhaus, M.; Loetscher, H.; Czech, C.; et al. PS2APP transgenic mice, coexpressing hPS2mut and hAPPswe, show age-related cognitive deficits associated with discrete brain amyloid deposition and inflammation. J. Neurosci. 2003, 23, 8989–9003. [Google Scholar] [CrossRef] [PubMed]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.M.; Iwata, N.; Saido, T.C.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M.Y. Synapse Loss and Microglial Activation Precede Tangles in a P301S Tauopathy Mouse Model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef]
- Tatebayashi, Y.; Miyasaka, T.; Chui, D.H.; Akagi, T.; Mishima, K.I.; Iwasaki, K.; Fujiwara, M.; Tanemura, K.; Murayama, M.; Ishiguro, K.; et al. Tau filament formation and associative memory deficit in aged mice expressing mutant (R406W) human tau. Proc. Natl. Acad. Sci. USA 2002, 99, 13896–13901. [Google Scholar] [CrossRef]
- Ramsden, M.; Kotilinek, L.; Forster, C.; Paulson, J.; Mcgowan, E.; Santacruz, K.; Guimaraes, A.; Yue, M.; Lewis, J.; Carlson, G.; et al. Age-Dependent Neurofibrillary Tangle Formation, Neuron Loss, and Memory Impairment in a Mouse Model of Human Tauopathy (P301L). Neurobiol. Dis. 2005, 25, 10637–10647. [Google Scholar] [CrossRef]
- SantaCruz, K.; Lewis, J.; Spires, T.; Paulson, J.; Kotilinek, L.; Ingelsson, M.; Guimaraes, A.; DeTure, M.; Ramsden, M.; McGowan, E.; et al. Tau Suppression in a Neurodegenerative Mouse Model Improves Memory Function. Science 2005, 309. [Google Scholar] [CrossRef]
- Liu, L.; Drouet, V.; Wu, J.W.; Witter, M.P.; Small, S.A.; Clelland, C.; Duff, K. Trans-synaptic spread of tau pathology in vivo. PLoS ONE 2012, 7, e31302. [Google Scholar] [CrossRef]
- De Calignon, A.; Polydoro, M.; Suárez-Calvet, M.; William, C.; Adamowicz, D.H.; Kopeikina, K.J.; Pitstick, R.; Sahara, N.; Ashe, K.H.; Carlson, G.A.; et al. Propagation of Tau Pathology in a Model of Early Alzheimer’s Disease. Neuron 2012, 73, 685–697. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Gómez-Isla, T.; Growdon, J.H.; Frosch, M.P.; Hyman, B.T. A phenotypic change but not proliferation underlies glial responses in Alzheimer disease. Am. J. Pathol. 2013, 182, 2332–2344. [Google Scholar] [CrossRef]
- Perez-Nievas, B.G.; Serrano-Pozo, A. Deciphering the astrocyte reaction in Alzheimer’s disease. Front. Aging Neurosci. 2018, 10, 1–23. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, X.; Wang, M.; Zhou, D.; Pan, H.; Shu, Q.; Sun, B. Early Activation of Astrocytes does not Affect Amyloid Plaque Load in an Animal Model of Alzheimer’s Disease. Neurosci. Bull. 2018, 34, 912–920. [Google Scholar] [CrossRef] [PubMed]
- Chai, H.; Diaz-Castro, B.; Shigetomi, E.; Monte, E.; Octeau, J.C.; Yu, X.; Cohn, W.; Rajendran, P.S.; Vondriska, T.M.; Whitelegge, J.P.; et al. Neural Circuit-Specialized Astrocytes: Transcriptomic, Proteomic, Morphological, and Functional Evidence. Neuron 2017, 95, 531–549. [Google Scholar] [CrossRef]
- Bayraktar, O.A.; Bartels, T.; Holmqvist, S.; Kleshchevnikov, V.; Martirosyan, A.; Polioudakis, D.; Ben Haim, L.; Young, A.M.H.; Batiuk, M.Y.; Prakash, K.; et al. Astrocyte layers in the mammalian cerebral cortex revealed by a single-cell in situ transcriptomic map. Nat. Neurosci. 2020, 23, 500–509. [Google Scholar] [CrossRef]
- Xiao, Q.; Yan, P.; Ma, X.; Liu, H.; Perez, R.; Zhu, A.; Gonzales, E.; Burchett, J.M.; Schuler, D.R.; Cirrito, J.R.; et al. Enhancing astrocytic lysosome biogenesis facilitates Aβ clearance and attenuates amyloid plaque pathogenesis. J. Neurosci. 2014, 34, 9607–9620. [Google Scholar] [CrossRef]
- Jones, R.S.; Minogue, A.M.; Connor, T.J.; Lynch, M.A. Amyloid-β-induced astrocytic phagocytosis is mediated by CD36, CD47 and RAGE. J. Neuroimmune Pharmacol. 2013, 8, 301–311. [Google Scholar] [CrossRef]
- Wyss-Coray, T.; Loike, J.D.; Brionne, T.C.; Lu, E.; Anankov, R.; Yan, F.; Silverstein, S.C.; Husemann, J. Adult mouse astrocytes degrade amyloid-β in vitro and in situ. Nat. Med. 2003, 9, 453–457. [Google Scholar] [CrossRef]
- Lee, Y.; Messing, A.; Su, M.; Brenner, M. GFAP promoter elements required for region-specific and astrocyte-specific expression. Glia 2008, 56, 481–493. [Google Scholar] [CrossRef]
- Mader, S.; Brimberg, L. Aquaporin-4 Water Channel in the Brain and Its Implication for Health and Disease. Cells 2019, 8, 90. [Google Scholar] [CrossRef]
- Chandra, A.; Farrell, C.; Wilson, H.; Dervenoulas, G.; De Natale, E.R.; Politis, M. Aquaporin-4 polymorphisms predict amyloid burden and clinical outcome in the Alzheimer’s disease spectrum. Neurobiol. Aging 2020. [Google Scholar] [CrossRef]
- Xu, Z.; Xiao, N.; Chen, Y.; Huang, H.; Marshall, C.; Gao, J.; Cai, Z.; Wu, T.; Hu, G.; Xiao, M. Deletion of aquaporin-4 in APP/PS1 mice exacerbates brain Aβ accumulation and memory deficits. Mol. Neurodegener. 2015, 10, 1–16. [Google Scholar] [CrossRef]
- Sloan, S.A.; Darmanis, S.; Huber, N.; Khan, T.; Birey, F.; Caneda, C.; Reimer, R.; Quake, S.R.; Barres, B.A.; Sergiu, P. Human Astrocyte Maturation Captured in 3D Cerebral Cortical Spheroids Derived from Pluripotent Stem Cells. Neuron 2017, 95, 779–790. [Google Scholar] [CrossRef]
- Marton, R.M.; Pașca, S.P. Organoid and Assembloid Technologies for Investigating Cellular Crosstalk in Human Brain Development and Disease. Trends Cell Biol. 2020, 30, 133–143. [Google Scholar] [CrossRef]
- Barbar, L.; Jain, T.; Zimmer, M.; Kruglikov, I.; Sadick, J.S.; Wang, M.; Kalpana, K.; Rose, I.V.L.; Burstein, S.R.; Rusielewicz, T.; et al. CD49f Is a Novel Marker of Functional and Reactive Human iPSC-Derived Astrocytes. Neuron 2020, 107, 436–453. [Google Scholar] [CrossRef]
- Johnson, G.V.W.; Stoothoff, W.H. Tau phosphorylation in neuronal cell function and dysfunction. J. Cell Sci. 2004, 117, 5721–5729. [Google Scholar] [CrossRef]
- Noble, W.; Hanger, D.P.; Miller, C.C.J.; Lovestone, S. The importance of tau phosphorylation for neurodegenerative diseases. Front. Neurol. 2013, 4, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hampton, D.W.; Webber, D.J.; Bilican, B.; Goedert, M.; Spillantini, M.G.; Chandran, S. Cell-mediated neuroprotection in a mouse model of human tauopathy. J. Neurosci. 2010, 30, 9973–9983. [Google Scholar] [CrossRef]
- Crescenzi, R.; DeBrosse, C.; Nanga, R.P.R.; Byrne, M.D.; Krishnamoorthy, G.; D’aquilla, K.; Al, E. Longitudinal imaging reveals sub-hippocampal dynamics in glutamate levels associated with histopathologic events in a mouse model of tauopathy and healthy mice. Hippocampus 2017, 27, 285–302. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Hussaini, S.A.; Wegmann, S.; Profaci, C.; Daniels, J.D.; Herman, M.; Emrani, S.; Figueroa, H.Y.; Hyman, B.T.; Davies, P.; et al. 3D Visualization of the temporal and spatial spread of tau pathology reveals extensive sites of tau accumulation associated with neuronal loss and recognition memory deficit in aged tau transgenic mice. PLoS ONE 2016, 11, e159463. [Google Scholar] [CrossRef]
- Nilson, A.N.; English, K.C.; Gerson, J.E.; Whittle, T.B. Tau Oligomers Associate with Inflammation in the Brain and Retina of Tauopathy Mice and in Neurodegenerative Diseases. J. Alzheimers Dis. 2017, 55, 1083–1099. [Google Scholar] [CrossRef]
- Helboe, L.; Egebjerg, J.; Barkholt, P.; Volbracht, C. Early depletion of CA1 neurons and late neurodegeneration in a mouse tauopathy model. Brain Res. 2017, 1665, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Spires, T.L.; Orne, J.D.; SantaCruz, K.; Pitstick, R.; Carlson, G.A.; Ashe, K.H.; Hyman, B.T. Region-specific dissociation of neuronal loss and neurofibrillary pathology in a mouse model of tauopathy. Am. J. Pathol. 2006, 168, 1598–1607. [Google Scholar] [CrossRef]
- Ricklin, D.; Reis, Edimara, S.; Mastellos, D.S.; Gros, P.; Lambris, J.D. Complement component C3—The “Swiss Army Knife” of innate immunity and host defense. Immunol. Rev. 2016, 274, 33–58. [Google Scholar] [CrossRef]
- Litvinchuk, A.; Wan, Y.W.; Swartzlander, D.B.; Chen, F.; Cole, A.; Propson, N.E.; Wang, Q.; Zhang, B.; Liu, Z.; Zheng, H. Complement C3aR Inactivation Attenuates Tau Pathology and Reverses an Immune Network Deregulated in Tauopathy Models and Alzheimer’s Disease. Neuron 2018, 100, 1337–1353. [Google Scholar] [CrossRef]
- Shiratori-Hayashi, M.; Koga, K.; Tozaki-Saitoh, H.; Kohro, Y.; Toyonaga, H.; Yamaguchi, C.; Hasegawa, A.; Nakahara, T.; Hachisuka, J.; Akira, S.; et al. STAT3-dependent reactive astrogliosis in the spinal dorsal horn underlies chronic itch. Nat. Med. 2015, 21, 927–931. [Google Scholar] [CrossRef]
- Clarke, L.E.; Liddelow, S.A.; Chakraborty, C.; Münch, A.E.; Heiman, M. Normal aging induces A1-like astrocyte reactivity. Proc. Natl. Acad. Sci. USA 2018, 115, E1896–E1905. [Google Scholar] [CrossRef]
- Brunello, C.A.; Merezhko, M.; Uronen, R.L.; Huttunen, H.J. Mechanisms of secretion and spreading of pathological tau protein. Cell. Mol. Life Sci. 2019, 77, 1721–1744. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, G.S.; Lee, V.M.Y.; Trojanowski, J.Q. Mechanisms of Cell-to-Cell Transmission of Pathological Tau: A Review. JAMA Neurol. 2019, 76, 101–108. [Google Scholar] [CrossRef]
- Franzmeier, N.; Rubinski, A.; Neitzel, J.; Kim, Y.; Damm, A.; Na, D.L.; Kim, H.J.; Lyoo, C.H.; Cho, H.; Finsterwalder, S.; et al. Functional connectivity associated with tau levels in ageing, Alzheimer’s, and small vessel disease. Brain 2019, 142, 1093–1107. [Google Scholar] [CrossRef]
- Wu, J.W.; Hussaini, S.A.; Bastille, I.M.; Rodriguez, G.A.; Mrejeru, A.; Rilett, K.; Sanders, D.W.; Cook, C.; Fu, H.; Boonen, R.A.C.M.; et al. Neuronal activity enhances tau propagation and tau pathology in vivo. Nat. Neurosci. 2016, 19, 1085–1092. [Google Scholar] [CrossRef]
- Perea, G.; Navarrete, M.; Araque, A. Tripartite synapses: Astrocytes process and control synaptic information. Trends Neurosci. 2009, 32, 421–431. [Google Scholar] [CrossRef]
- Perea, J.R.; López, E.; Diéz-Ballesteros, J.C.; Ávila, J.; Hernández, F.; Bolós, M. Extracellular monomeric tau is internalized by astrocytes. Front. Neurosci. 2019, 13, 1–7. [Google Scholar] [CrossRef]
- Otero-Garcia, M.; Xue, Y.-Q.; Shakouri, T.; Deng, Y.; Morabito, S.; Allison, T.; Lowry, W.E.; Kawaguchi, R.; Swarup, V.; Cobos, I. Single-soma transcriptomics of tangle-bearing neurons in Alzheimer’s disease reveals the signatures of tau-associated synaptic dysfunction. BioRxiv 2020. [Google Scholar] [CrossRef]
- Ikeda, M.; Shoji, M.; Kawarai, T.; Kawarabayashi, T.; Matsubara, E.; Murakami, T.; Sasaki, A.; Tomidokoro, Y.; Ikarashi, Y.; Kuribara, H.; et al. Accumulation of filamentous tau in the cerebral cortex of human tau R406W transgenic mice. Am. J. Pathol. 2005, 166, 521–531. [Google Scholar] [CrossRef]
- Narasimhan, S.; Guo, J.L.; Changolkar, L.; Stieber, A.; McBride, J.D.; Silva, L.V.; He, Z.; Zhang, B.; Gathagan, R.J.; Trojanowski, J.Q.; et al. Pathological tau strains from human brains recapitulate the diversity of tauopathies in nontransgenic mouse brain. J. Neurosci. 2017, 37, 11406–11423. [Google Scholar] [CrossRef]
- Lin, W.L.; Lewis, J.; Yen, S.H.; Hutton, M.; Dickson, D.W. Filamentous tau in oligodendrocytes and astrocytes of transgenic mice expressing the human tau isoform with the P301L mutation. Am. J. Pathol. 2003, 162, 213–218. [Google Scholar] [CrossRef]
- Bronzuoli, M.R.; Facchinetti, R.; Valenza, M.; Cassano, T.; Steardo, L.; Scuderi, C. Astrocyte function is affected by aging and not Alzheimer’s disease: A preliminary investigation in hippocampi of 3xTg-AD mice. Front. Pharmacol. 2019, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Griffith, C.M.; Xie, M.X.; Qiu, W.Y.; Sharp, A.A.; Ma, C.; Pan, A.; Yan, X.X.; Patrylo, P.R. Aberrant expression of the pore-forming KATP channel subunit Kir6.2 in hippocampal reactive astrocytes in the 3xTg-AD mouse model and human Alzheimer’s disease. Neuroscience 2016, 336, 81–101. [Google Scholar] [CrossRef]
- Wyssenbach, A.; Quintela, T.; Llavero, F.; Zugaza, J.L.; Matute, C.; Alberdi, E. Amyloid β-induced astrogliosis is mediated by β1-integrin via NADPH oxidase 2 in Alzheimer’s disease. Aging Cell 2016, 15, 1140–1152. [Google Scholar] [CrossRef]
- Grimaldi, A.; Brighi, C.; Peruzzi, G.; Ragozzino, D.; Bonanni, V.; Limatola, C.; Ruocco, G.; Di Angelantonio, S. Inflammation, neurodegeneration and protein aggregation in the retina as ocular biomarkers for Alzheimer’s disease in the 3xTg-AD mouse model. Cell Death Dis. 2018, 9. [Google Scholar] [CrossRef]
- Ontiveros-Torres, M.A.; Labra-Barrios, M.L.; Diaz-Cintra, S.; Aguilar-Vazquez, R.A.; Al, E. Fibrillar Amyloid-β Accumulation Triggers an Inflammatory Mechanism Leading to Hyperphosphorylation of the Carboxyl-Terminal End of Tau Polypeptide in the Hippocampal Formation of the 3×Tg-AD Transgenic Mouse. J. Alzheimers Dis. 2016, 52, 243–269. [Google Scholar] [CrossRef]
- Guedes, J.R.; Custódia, C.M.; Silva, R.J.; de Almeida, L.P.; de Lima, M.C.P.; Cardoso, A.L. Early miR-155 upregulation contributes to neuroinflammation in Alzheimer’s disease triple transgenic mouse model. Hum. Mol. Genet. 2014, 23, 6286–6301. [Google Scholar] [CrossRef] [PubMed]
- Mckeon-O’Malley, C.; Tanzi, R. Etiology, Genetics, and Pathogenesis of Alzheimer’s Disease. Funct. Neurobiol. Aging 2001, 333–348. [Google Scholar] [CrossRef]
- Verghese, P.B.; Castellano, J.M.; Holtzman, D.M. Apolipoprotein E in Alzheimer’s disease and other neurological disorders. Lancet Neurol. 2011, 10, 241–252. [Google Scholar] [CrossRef]
- Zhu, Y.; Nwabuisi-Heath, E.; Dumanis, S.B.; Tai, L.M.; Yu, C.; Rebeck, G.W.; Ladu, M.J. APOE genotype alters glial activation and loss of synaptic markers in mice. Glia 2012, 60, 559–569. [Google Scholar] [CrossRef]
- Hussain, G.; Wang, J.; Rasul, A.; Anwar, H.; Imran, A.; Qasim, M.; Zafar, S.; Kamran, S.K.S.; Razzaq, A.; Aziz, N.; et al. Role of cholesterol and sphingolipids in brain development and neurological diseases. Lipids Health Dis. 2019, 18, 1–12. [Google Scholar] [CrossRef]
- Zheng, J.; Sun, J.; Ji, C.; Shen, L.; Chen, Z.; Xie, P.; Sun, Y.Z.; Yu, R. Selective deletion of apolipoprotein E in astrocytes ameliorates the spatial learning and memory deficits in Alzheimer’s disease (APP/PS1) mice by inhibiting TGF-β/Smad2/STAT3 signaling. Neurobiol. Aging 2017, 54, 112–132. [Google Scholar] [CrossRef]
- Bales, K.R.; Verina, T.; Cummins, D.J.; Du, Y.; Dodel, R.C.; Saura, J.; Fishman, C.E.; Delong, C.A.; Piccardo, P.; Petegnief, V.; et al. Apolipoprotein E is essential for amyloid deposition in the APP(V717F) transgenic mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 1999, 96, 15233–15238. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, G.A.; Tai, L.M.; LaDu, M.J.; Rebeck, G.W. Human APOE4 increases microglia reactivity at Aβ plaques in a mouse model of Aβ deposition. J. Neuroinflamm. 2014, 11. [Google Scholar] [CrossRef]
- Dorey, E.; Bamji-Mirza, M.; Najem, D.; Li, Y.; Liu, H.; Callaghan, D.; Walker, D.; Lue, L.F.; Stanimirovic, D.; Zhang, W. Apolipoprotein E Isoforms Differentially Regulate Alzheimer’s Disease and Amyloid-β-Induced Inflammatory Response in vivo and in vitro. J. Alzheimers Dis. 2017, 57, 1265–1279. [Google Scholar] [CrossRef]
- Zhang, R.; Wu, Y.; Xie, F.; Zhong, Y.; Wang, Y.; Xu, M.; Feng, J.; Charish, J.; Monnier, P.P.; Qin, X. RGMa mediates reactive astrogliosis and glial scar formation through TGFβ1/Smad2/3 signaling after stroke. Cell Death Differ. 2018, 25, 1503–1516. [Google Scholar] [CrossRef]
- Rolyan, H.; Feike, A.C.; Upadhaya, A.R.; Waha, A.; Van Dooren, T.; Haass, C.; Birkenmeier, G.; Pietrzik, C.U.; Van Leuven, F.; Thal, D.R. Amyloid-β protein modulates the perivascular clearance of neuronal apolipoprotein e in mouse models of Alzheimer’s disease. J. Neural Transm. 2011, 118, 699–712. [Google Scholar] [CrossRef]
- Shi, Y.; Yamada, K.; Liddelow, S.A.; Smith, S.T.; Zhao, L.; Luo, W.; Tsai, R.M.; Spina, S.; Grinberg, L.T.; Rojas, J.C.; et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 2017, 549, 523–527. [Google Scholar] [CrossRef]
- Ulland, T.K.; Colonna, M. TREM2—A key player in microglial biology and Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 667–675. [Google Scholar] [CrossRef]
- Turnbull, I.R.; Gilfillan, S.; Cella, M.; Aoshi, T.; Miller, M.; Piccio, L.; Hernandez, M.; Colonna, M. Cutting Edge: TREM-2 Attenuates Macrophage Activation. J. Immunol. 2006, 177, 3520–3524. [Google Scholar] [CrossRef] [PubMed]
- Leyns, C.E.G.; Ulrich, J.D.; Finn, M.B.; Stewart, F.R.; Koscal, L.J.; Serrano, J.R.; Robinson, G.O.; Anderson, E.; Colonna, M.; Holtzman, D.M. TREM2 deficiency attenuates neuroinflammation and protects against neurodegeneration in a mouse model of tauopathy. Proc. Natl. Acad. Sci. USA 2017, 114, 11524–11529. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sloan, S.A.; Clarke, L.E.; Caneda, C.; Plaza, C.A.; Blumenthal, P.D.; Vogel, H.; Steinberg, G.K.; Edwards, M.S.B.; Li, G.; et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 2016, 89, 37–53. [Google Scholar] [CrossRef]
- Oberheim, N.A.; Takano, T.; Han, X.; He, W.; Lin, J.H.C.; Wang, F.; Xu, Q.; Wyatt, J.D.; Pilcher, W.; Ojemann, J.G.; et al. Uniquely hominid features of adult human astrocytes. J. Neurosci. 2009, 29, 3276–3287. [Google Scholar] [CrossRef]
- Windrem, M.S.; Schanz, S.J.; Morrow, C.; Munir, J.; Chandler-Militello, D.; Wang, S.; Goldman, S.A. A competitive advantage by neonatally engrafted human glial progenitors yields mice whose brains are chimeric for human glia. J. Neurosci. 2014, 34, 16153–16161. [Google Scholar] [CrossRef]
- Hsu, E.T.; Gangolli, M.; Su, S.; Holleran, L.; Stein, T.D.; Alvarez, V.E.; McKee, A.C.; Schmidt, R.E.; Brody, D.L. Astrocytic degeneration in chronic traumatic encephalopathy. Acta Neuropathol. 2018, 136, 955–972. [Google Scholar] [CrossRef]
- Krutzik, S.R.; Tan, B.; Li, H.; Ochoa, M.T.; Liu, P.T.; Sharfstein, S.E.; Graeber, T.G.; Sieling, P.A.; Liu, Y.J.; Rea, T.H.; et al. TLR activation triggers the rapid differentiation of monocytes into macrophages and dendritic cells. Nat. Med. 2005, 11, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Underhill, D.M.; Ozinsky, A.; Hajjar, A.M.; Stevens, A.; Wilson, C.B.; Bassetti, M.; Aderem, A. The Toll-like receptor 2 is recruited to macrophage phagosomes and discriminates between pathogens. Nature 1999, 401, 811–815. [Google Scholar] [CrossRef]
- Frederiksen, H.R.; Haukedal, H.; Freude, K.; Muthuraju, S. Cell type specific expression of toll-like receptors in human brains and implications in Alzheimer’s disease. Biomed. Res. Int. 2019, 2019. [Google Scholar] [CrossRef]
- Bauer, S.; Kirschning, C.J.; Häcker, H.; Redecke, V.; Hausmann, S.; Akira, S.; Wagner, H.; Lipford, G.B. Human TLR9 confers responsiveness to bacterial DNA via species-specific CpG motif recognition. Proc. Natl. Acad. Sci. USA 2001, 98, 9237–9242. [Google Scholar] [CrossRef] [PubMed]
- Jones, V.C.; Atkinson-Dell, R.; Verkhratsky, A.; Mohamet, L. Aberrant iPSC-derived human astrocytes in Alzheimer’s disease. Cell Death Dis. 2017, 8, 1–11. [Google Scholar] [CrossRef]
- Kondo, T.; Asai, M.; Tsukita, K.; Kutoku, Y.; Ohsawa, Y.; Sunada, Y.; Imamura, K.; Egawa, N.; Yahata, N.; Okita, K.; et al. Modeling Alzheimer’s disease with iPSCs reveals stress phenotypes associated with intracellular Aβ and differential drug responsiveness. Cell Stem Cell 2013, 12, 487–496. [Google Scholar] [CrossRef]
- Lin, Y.T.; Seo, J.; Gao, F.; Feldman, H.M.; Wen, H.L.; Penney, J.; Cam, H.P.; Gjoneska, E.; Raja, W.K.; Cheng, J.; et al. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron 2018, 98, 1141–1154. [Google Scholar] [CrossRef]
- Niccoli, T.; Partridge, L. Ageing as a risk factor for disease. Curr. Biol. 2012, 22, R741–R752. [Google Scholar] [CrossRef]
- Soreq, L.; Rose, J.; Soreq, E.; Hardy, J.; Trabzuni, D.; Cookson, M.R.; Smith, C.; Ryten, M.; Patani, R.; Ule, J. Major Shifts in Glial Regional Identity Are a Transcriptional Hallmark of Human Brain Aging. Cell Rep. 2017, 18, 557–570. [Google Scholar] [CrossRef]
- Boisvert, M.M.; Erikson, G.A.; Shokhirev, M.N.; Allen, N.J. The Aging Astrocyte Transcriptome from Multiple Regions of the Mouse Brain. Cell Rep. 2018, 22, 269–285. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Liu, L.; Ji, H.F. Alzheimer’s disease histological and behavioral manifestations in transgenic mice correlate with specific gut microbiome state. J. Alzheimers Dis. 2017, 56, 385–390. [Google Scholar] [CrossRef]
- Sochocka, M.; Donskow-Łysoniewska, K.; Diniz, B.S.; Kurpas, D.; Brzozowska, E.; Leszek, J. The Gut Microbiome Alterations and Inflammation-Driven Pathogenesis of Alzheimer’s Disease—A Critical Review. Mol. Neurobiol. 2019, 56, 1841–1851. [Google Scholar] [CrossRef]
- Harach, T.; Marungruang, N.; Duthilleul, N.; Cheatham, V.; Mc Coy, K.D.; Frisoni, G.; Neher, J.J.; Fåk, F.; Jucker, M.; Lasser, T.; et al. Reduction of Abeta amyloid pathology in APPPS1 transgenic mice in the absence of gut microbiota. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Rothhammer, V.; Mascanfroni, I.D.; Bunse, L.; Takenaka, M.C.; Kenison, J.E.; Mayo, L.; Chao, C.C.; Patel, B.; Yan, R.; Blain, M.; et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat. Med. 2016, 22, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Marizzoni, M.; Cattaneo, A.; Mirabello, P.; Festari, C.; Lopizzo, N.; Nicolosi, V.; Mombelli, E.; Mazzelli, M.; Luongo, D.; Naviglio, D.; et al. Short-chain fatty acids and lipopolysaccharide as mediators between gut dysbiosis and amyloid pathology in Alzheimer’s Disease. J. Alzheimers. Dis. 2020. [Google Scholar] [CrossRef]
- Moir, R.D.; Lathe, R.; Tanzi, R.E. The antimicrobial protection hypothesis of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 1602–1614. [Google Scholar] [CrossRef]
- Casellas, J. Inbred mouse strains and genetic stability: A review. Animal 2011, 5, 1–7. [Google Scholar] [CrossRef][Green Version]
- Park, R.; Kook, S.Y.; Park, J.C.; Mook-Jung, I. Aβ1-42 reduces P-glycoprotein in the blood-brain barrier through RAGE-NF-κB signaling. Cell Death Dis. 2014, 5, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Letronne, F.; Laumet, G.; Ayral, A.M.; Chapuis, J.; Demiautte, F.; Laga, M.; Vandenberghe, M.E.; Malmanche, N.; Leroux, F.; Eysert, F.; et al. ADAM30 Downregulates APP-Linked Defects Through Cathepsin D Activation in Alzheimer’s Disease. EBioMedicine 2016, 9, 278–292. [Google Scholar] [CrossRef]
- Urbanelli, L.; Emiliani, C.; Massini, C.; Persichetti, E.; Orlacchio, A.; Pelicci, G.; Sorbi, S.; Hasilik, A.; Bernardi, G.; Orlacchio, A. Cathepsin D expression is decreased in Alzheimer’s disease fibroblasts. Neurobiol. Aging 2008, 29, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Chai, Y.L.; Chong, J.R.; Weng, J.; Howlett, D.; Halsey, A.; Lee, J.H.; Attems, J.; Aarsland, D.; Francis, P.T.; Chen, C.P.; et al. Lysosomal cathepsin D is upregulated in Alzheimer’s disease neocortex and may be a marker for neurofibrillary degeneration. Brain Pathol. 2019, 29, 63–74. [Google Scholar] [CrossRef]
- Yin, C.; Ackermann, S.; Ma, Z.; Mohanta, S.K.; Zhang, C.; Li, Y.; Nietzsche, S.; Westermann, M.; Peng, L.; Hu, D.; et al. ApoE attenuates unresolvable inflammation by complex formation with activated C1q. Nat. Med. 2019, 25, 496–506. [Google Scholar] [CrossRef]
- Das, S.; Li, Z.; Noori, A.; Hyman, B.T.; Serrano-Pozo, A. Meta-analysis of mouse transcriptomic studies supports a context-dependent astrocyte reaction in acute CNS injury versus neurodegeneration. J. Neuroinflamm. 2020, 17, 227. [Google Scholar] [CrossRef]
- Wang, M.; Roussos, P.; McKenzie, A.; Zhou, X.; Kajiwara, Y.; Brennand, K.J.; De Luca, G.C.; Crary, J.F.; Casaccia, P.; Buxbaum, J.D.; et al. Integrative network analysis of nineteen brain regions identifies molecular signatures and networks underlying selective regional vulnerability to Alzheimer’s disease. Genome Med. 2016, 8, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, K.; Sepulveda-Falla, D.; Rose, I.V.L.; Madore, C.; Muth, C.; Matschke, J.; Butovsky, O.; Liddelow, S.; Glatzel, M.; Krasemann, S. Complement 3+-astrocytes are highly abundant in prion diseases, but their abolishment led to an accelerated disease course and early dysregulation of microglia. Acta Neuropathol. Commun. 2019, 7, 83. [Google Scholar] [CrossRef]
- Yun, S.P.; Kam, T.I.; Panicker, N.; Kim, S.; Oh, Y.; Park, J.S.; Kwon, S.H.; Park, Y.J.; Karuppagounder, S.S.; Park, H.; et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat. Med. 2018, 24, 931–938. [Google Scholar] [CrossRef]
- Johnson, E.C.B.; Dammer, E.B.; Duong, D.M.; Ping, L.; Zhou, M.; Yin, L.; Higginbotham, L.A.; Guajardo, A.; White, B.; Troncoso, J.C.; et al. Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat. Med. 2020, 26. [Google Scholar] [CrossRef]
- Bai, B.; Wang, X.; Li, Y.; Chen, P.C.; Yu, K.; Dey, K.K.; Yarbro, J.M.; Han, X.; Lutz, B.M.; Rao, S.; et al. Deep Multilayer Brain Proteomics Identifies Molecular Networks in Alzheimer’s Disease Progression. Neuron 2020, 105, 975–991.e7. [Google Scholar] [CrossRef]
- Böttcher, C.; Schlickeiser, S.; Sneeboer, M.A.M.; Kunkel, D.; Knop, A.; Paza, E.; Fidzinski, P.; Kraus, L.; Snijders, G.J.L.; Kahn, R.S.; et al. Human microglia regional heterogeneity and phenotypes determined by multiplexed single-cell mass cytometry. Nat. Neurosci. 2019, 22, 78–90. [Google Scholar] [CrossRef]
- Neff, E.P. Animal models of Alzheimer’s disease embrace diversity. Lab. Anim. 2019, 48, 255–259. [Google Scholar] [CrossRef]
- Vandamme, T. Use of rodents as models of human diseases. J. Pharm. Bioallied Sci. 2014, 6, 2–9. [Google Scholar] [CrossRef]
- Mariani, J.N.; Zou, L.; Goldman, S.A. Human Glial Chimeric Mice to Define the Role of Glial Pathology in Human Disease. Methods Mol. Biol. 2019, 1936, 311–331. [Google Scholar] [CrossRef] [PubMed]
- Benraiss, A.; Wang, S.; Herrlinger, S.; Li, X.; Chandler-Militello, D.; Mauceri, J.; Burm, H.B.; Toner, M.; Osipovitch, M.; Jim Xu, Q.; et al. Human glia can both induce and rescue aspects of disease phenotype in Huntington disease. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef]
- Windrem, M.S.; Osipovitch, M.; Liu, Z.; Bates, J.; Chandler-Militello, D.; Zou, L.; Munir, J.; Schanz, S.; McCoy, K.; Miller, R.H.; et al. Human iPSC Glial Mouse Chimeras Reveal Glial Contributions to Schizophrenia. Cell Stem Cell 2017, 21, 195–208. [Google Scholar] [CrossRef]
- Han, X.; Chen, M.; Wang, F.; Windrem, M.; Wang, S.; Shanz, S.; Xu, Q.; Oberheim, N.A.; Bekar, L.; Betstadt, S.; et al. Forebrain engraftment by human glial progenitor cells enhances synaptic plasticity and learning in adult mice. Cell Stem Cell 2013, 12, 342–353. [Google Scholar] [CrossRef]
- Engel, M.; Do-Ha, D.; Muñoz, S.S.; Ooi, L. Common pitfalls of stem cell differentiation: A guide to improving protocols for neurodegenerative disease models and research. Cell. Mol. Life Sci. 2016, 73, 3693–3709. [Google Scholar] [CrossRef] [PubMed]
- Atkinson-Del, R.; Mohamet, L. Induced Pluripotent Stem Cell-Derived Astroglia: A New Tool for Research Towards the Treatment of Alzheimer’s Disease; Verkhratsky, A., Ho, M.S., Zorec, R., Parpura, V., Eds.; Springer: Singapore, 2019; Volume 1175. [Google Scholar]
- Essayan-Perez, S.; Zhou, B.; Nabet, A.M.; Wernig, M.; Huang, Y.W.A. Modeling Alzheimer’s disease with human iPS cells: Advancements, lessons, and applications. Neurobiol. Dis. 2019, 130, 104503. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Finding | Method | Age | Brain Area | References |
|---|---|---|---|---|
| Tg(APPswe/PSEN1dE9) (also known as 2xTg, 2xTg-AD, APP/PS1) | ||||
| ↑ GFAP protein (including staining intensity) (for male vs. female see [71,92]) | WB, IHC | 6–19 mo 3 mo (HPC) | CTX, HPC STR, CB (19 mo) | [26,72,73,74,75,76,93] |
| ↑ GFAP+ cell density | IHC | 6, 12–14, 23–28 mo ? 6, 12–14 mo 3 mo 16 mo | CTX HPC DG CA3 | [26,73,79,85] |
| ↓ GFAP+ cell density | IHC | 24 mo | HPC | [80] |
| NC GFAP+ cell density | IHC | 5–9 mo | CTX layers II/III | [81] |
| ↑ GFAP+ area in 8–12 mo | IHC | 2–6, 8–12 mo | FtC, HPC | [77] |
| ↓ GFAP+ area | IHC | 24 mo | HPC | [80] |
| NC GFAP+ cells/blood vessel | IHC | 6, 12–14, 23–28 mo | HPC | [79] |
| NC A1, A2, pan-reactive genes 235 genes differentially expressed [↑ Cytokines/Chemokines (Il1β, Ccl2, Ccl4, Ccl6), Complement (C1qa, C1qb, C1qc, C4b)] | RNA-seq of FACS-isolated astrocytes | 9 mo | HPC | [94] |
| No uptake of Methoxy-X04+ amyloid fibrils by astrocytes | FACS | 9 mo | HPC | [94] |
| Fibrillar Aβ not engulfed by GFAP+ cells | IHC | 3, 6, 9, 12 mo | CTX, HPC | [73] |
| Observation: hypertrophic astrocytes close to plaques, atrophic distant to plaques | IHC | 24 mo | CTX, HPC | [71,73,77,79,80] |
| Tg(APPSwLon/PSEN1*M146L) | ||||
| GFAP+ cells engulf APP+ dystrophic neurites | IHC, EM | 4, 6, 12 mo | HPC | [95] |
| Tg(PDGFB-APPSwInd) (also known as hAPP-J20, APP/J20, J20) | ||||
| ↑ GFAP+ area from 12–29 mo | IHC | 3, 9, 12–16, 29 mo | CTX | [96] |
| ↓ GFAP+ surface/volume per cell | IHC | 5 mo | HPC | [97] |
| NC GFAP+ cell surface and volume | IHC | 5 mo | HPC | [97] |
| Observations: Vascular amyloidosis can partially or fully displace astrocyte endfeet from vessels | IHC, EM | 27 mo | CTX | [96] |
| Tg(Thy1-APPSw/Prnp-PSEN2*N141I) (also known as PS2APP) | ||||
| ↑GFAP+ area | IHC | 6 mo | HPC | [33] |
| ↑classical components (↑ C1q in RNA-seq, but not NC fluorescence intensity in IHC) NC C3 expression ↑A1-specific and pan-reactive genes | RNA-seq of FACS-isolated astrocytes (validated with IHC) | 7, 11.5, 13 mo | HPC | [33] |
| Observation: C3 mostly associates with astrocytes | IHC | 6 mo | HPC | [33] |
| Tg(Thy1-APPSweArc)B (also known as Tg-Arc/Swe, TgArcSwe) | ||||
| ↑ AQP4 in 9 mo, NC AQP4 at 12 mo | WB | 9, 12 mo | FtC | [98] |
| ↑ AQP4 staining intensity | IHC | 4, 16 mo | CTX | [40] |
| Observation: Loss of AQP4 polarization in astrocytes close to Aβ plaques | IHC | 8–16 mo | * | [98] |
| Observations: 3 senile plaque stages characterized: (1) GFAP+/AQP4-; (2) GFAP+/AQP4+; (3) GFAP-/AQP4- 2 types of astrocytes related to Aβ observed: (1) ↑ AQP4, (2) less ↑ AQP4, rich in mitochondria, microvesicles | IHC, EM | 8, 12, 16 mo | CTX | [40] |
| Tg(APPSwe)2576 (also known as APPSw, APPswe, Tg2576) | ||||
| GFAP colocalization with human APP | IHC | 3, 18 mo | CTX, CC | [99] |
| GFAP associates with pyroglutamate-modified Aβ peptides | IHC | * | * | [100] |
| NC Astrocyte end-feet (assessed by GFAP) | IHC | 12 mo | FtC, HPC | [101] |
| NC AQP4 associated w/vessels | IHC | 12 mo | FtC, HPC | [101] |
| Tg(Thy1-APPArc)M8 (also known as TgAPParc, Thy1.2-hAPParc) | ||||
| Observations: Loss of endfeet contact with vessels in plaques (6–13 mo). Maintained GFAP-vessel interaction at non-CAA vessels 16–22 mo | IHC | 6, 9–13, 16–22 mo | CTX | [102] |
| Tg(PRNP-APPSweInd)8 (also known as TgCRND8, Tg19959) | ||||
| ↑ GFAP+ cell density around plaques | IHC | 3, 6 mo | CA1 | [103] |
| ↑ GFAP signal intensity | IHC | 3, 6 mo | CA1 | [103] |
| ↑ GFAP+ branch length | IHC | 3, 6 mo | CA1 | [103] |
| NC GFAP signal intensity | IHC | 3, 6 mo | CA3 | [103] |
| NC GFAP+ cell density around plaques | IHC | 3, 6 mo | CA3 | [103] |
| NC GFAP+ branch length | IHC | 3, 6 mo | CA3 | [103] |
| Tg(Thy1-APPSwDutIowa) (also known as TgSwDI) | ||||
| ↓ astrocyte end feet number | IHC (using GFAP) | 12 mo | FtC, HPC | [101] |
| ↓ AQP4 vessel coverage | IHC (using GFAP) | 12 mo | FtC, HPC | [101] |
| Model | Gene(s) | Mutation(s) | Promoter | References |
|---|---|---|---|---|
| mThy1-hAPP751 | APP | APP751; Swedish (K670N/M671L) and London (V717I) | mThy1 | [106] |
| Tg2576 | APP | APP695; Swedish (K670N/M671L) | Hamster Prnp | [107] |
| TgAPParc | APP | APP695; Arctic (E693G) | mThy1.2 | [108] |
| TgArcSwe | APP | APP695; Arctic (E693G) and Swedish (KM670/671NL) | mThy1 | [109] |
| TgSwDI | APP | APP770; Swedish (K670N/M671L), Dutch (E693Q) and Iowa (D694N) | mThy1 | [110] |
| 5xFAD | APP, PSEN1 | APP695: Swedish (K670N/M671L), Florida (I716V), London (V717I) PSEN1: M146L and L286V | mThy1 | [111] |
| APPPS1 | APP, PSEN1 | APP751: Swedish (K670N/M671L) PSEN1: L166P | mThy1.2 | [112] |
| APPswe/PSEN1dE9 | APP, PSEN1 | APP695: Swedish (K670N/M671L) PSEN1: exon 9 removed | mPrnp | [113,114] |
| APPSWE/LON/PSEN1M146L | APP, PSEN1 | APP751: Swedish (K670N/M671L) and London (V717I) PSEN1: M146L | mThy1 (APP) HMG (PSEN1) | [115] |
| PSEN2APP | APP, PSEN2 | APP751: Swedish (K670N/M671L) PSEN2: N141I | mThy1.2 mPrnp | [116] |
| PS19 | MAPT | MAPT (1N4R): P301S | mPrnp | [117] |
| TauR406W | MAPT | MAPT (2N4R): R406W, contains myc and FLAG tags at N-and C-terminal ends, respectively | CamkII | [118] |
| TauP301L | MAPT | MAPT (2N4R): Tau-4R/2N isoform; P301L | mThy1 | [118] |
| rTg4510 | MAPT | MAPT (0N4R): P301L | tetO CaMKIIα-tTA | [119,120] |
| rTgTauEC | MAPT | MAPT (0N4R): P301L | tetO Neuropsin-tTA | [121,122] |
| 3xTg | APP, PSEN1, MAPT | APP751: Swedish (K670N/M671L) PSEN1: M146V MAPT (0N4R): P301L | mThy1 * | [57] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spanos, F.; Liddelow, S.A. An Overview of Astrocyte Responses in Genetically Induced Alzheimer’s Disease Mouse Models. Cells 2020, 9, 2415. https://doi.org/10.3390/cells9112415
Spanos F, Liddelow SA. An Overview of Astrocyte Responses in Genetically Induced Alzheimer’s Disease Mouse Models. Cells. 2020; 9(11):2415. https://doi.org/10.3390/cells9112415
Chicago/Turabian StyleSpanos, Fokion, and Shane A. Liddelow. 2020. "An Overview of Astrocyte Responses in Genetically Induced Alzheimer’s Disease Mouse Models" Cells 9, no. 11: 2415. https://doi.org/10.3390/cells9112415
APA StyleSpanos, F., & Liddelow, S. A. (2020). An Overview of Astrocyte Responses in Genetically Induced Alzheimer’s Disease Mouse Models. Cells, 9(11), 2415. https://doi.org/10.3390/cells9112415