1. Introduction
Sphingomyelinases are enzymes classified according to the optimal pH for their activity: acid, neutral, or alkaline [
1]. Sphingomyelinases catalyze the hydrolysis of sphingomyelin to generate ceramide [
2], which is a central molecule in modulating membrane biophysical properties and is involved in various cellular process, such as apoptosis and inflammation, as well as several pathologies and diseases [
3]. Acid sphingomyelinase generates ceramide in the outer leaflet of the plasma membrane and in lysosomes. Ceramide molecules reorganize the cell membrane, resulting in the formation of large, distinct ceramide-enriched membrane domains that serve to cluster and aggregate activated receptor molecules. Receptor clustering allows the amplification of receptor signaling and thereby efficient signal transduction into the cells [
4]. Acid sphingomyelinase and ceramide have been shown to be crucially involved in the host response to various bacteria, including pathogenic mycobacteria, several viruses, and some parasites [
5,
6,
7,
8,
9,
10,
11,
12,
13,
14]. Deficiency of acid sphingomyelinase often leads to increased susceptibility of the host to pathogens, such as
Pseudomonas aeruginosa,
Listeria monocytogenes,
Salmonella typhimurium, and
Staphylococcus aureus via distinct mechanisms including failure of internalization, loss of fusion of bacteria-containing phagosomes with lysosomes, or a defect in the nicotinamide adenine dinucleotide phosphate (NADPH)-mediated release of reactive oxygen species (ROS) [
7,
8,
9,
11,
15]. In order to effectively kill phagocytosed bacteria within phagolysosomes, various degradative enzymes are required, such as cathepsins, proteases, lysozymes, and lipases. Previous studies provide evidence for a specific interaction of acid-sphingomyelinase-derived ceramide with cathepsin D (CTSD), leading to enhanced enzymatic activity and proteolytic activation of proteins to be secreted [
16].
Diseases caused by infections with pathogenic mycobacteria such as
Mycobacterium tuberculosis and
Mycobacterium avium are among the most common severe infections worldwide. In all countries,
Mycobacterium tuberculosis is the primary cause of tuberculosis (TB), with more than 8 million new cases and approximately 1.6 million casualties annually [
17]. Most people infected with
Mycobacterium tuberculosis are clinically asymptomatic, a state that is referred as latent TB; about 5–10% of them can develop a severe systemic infection with high lethality [
17].
The first indications that sphingomyelinases and ceramide may be involved in mycobacterial infections were reported in 2003. Anes et al. found that ceramide, sphingomyelin, sphingosine, and sphingosine 1-phosphate were involved in actin nucleation on phagosomes, thereby triggering the fusion of phagosomes with lysosomes to release antimicrobial factors, which killed mycobacteria [
18]. Acid sphingomyelinase also plays a central role in phagolysosomal fusion upon infection of macrophages with
Mycobacterium avium by modifying the steric conformation of cellular membranes [
11]. A common survival strategy used by mycobacteria is to interfere with phagosome maturation and block phagosomal acidification. A recent study indicated that acid-sphingomyelinase-mediated maturation of phagosomes is regulated by neurotensin receptor 3, sortilin, and is important for controlling mycobacterial infection [
19,
20]. Studies using a zebrafish model described that
Mycobacterium marinum infection in the host resulted in rapid production of mitochondrial reactive oxygen species (ROS) in infected macrophages, which was controlled by a combined interaction between mitochondrial cyclophilin D and acid sphingomyelinase [
21]. These findings indicate that acid-sphingomyelinase-mediated maturation of phagosomes is important for controlling mycobacterial infection [
11,
19,
20]. Therefore, we characterized the role of acid sphingomyelinase in
Mycobacterium bovis Bacillus Calmette-Guérin (BCG) killing and infection in vivo and in vitro. Based on our results, we suggest a model in which acid sphingomyelinase controls mycobacterial infection by activating nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and releasing reactive oxygen species (ROS). ROS promote BCG degradation via the bactericidal enzyme cathepsin D. Acid-sphingomyelinase-deficient mice failed to activate this cascade, leading to a high susceptibility to BCG infection.
2. Materials and Methods
2.1. Mice, Cells, Inhibitors
Acid-sphingomyelinase-deficient (Asm
−/−, sphingomyelin phosphodiesterase 1 knockout; Smpd1
−/−) mice and syngenic wild-type (wt) littermates were maintained on a C57BL/6J background [
22]. We used Asm
−/− mice and their wt littermates aged only 6 to 8 weeks in order to avoid an accumulation of sphingomyelin [
23]. The respective genotypes were verified by polymerase chain reaction. All mice were kept under pathogen-free conditions in the animal facility of the University of Duisburg-Essen in accordance with the criteria of the Association of Laboratory Animal Sciences. In vivo infections were approved by the Landesamt für Natur, Umwelt und Verbraucherschutz (LANUV); animal grants G 903/0, No. 887-501034.09, and G 1691/18, No. 81-02.04.2018.A192.
The in vitro experiments were carried out with freshly isolated bone-marrow-derived macrophages (BMDMs) from wt or Asm
−/− mice. To obtain BMDMs, mice were sacrificed, and femurs and tibias were rinsed with minimum essential medium (MEM; Thermo Fisher Scientific, Waltham, MA, USA) enriched with 10% fetal bovine serum (Thermo Fisher Scientific, Waltham, MA, USA), 10 mM HEPES (pH 7.4; Roth GmbH, Karlsruhe, Germany), 2 mM L-glutamine, 1 mM sodium pyruvate, 100 μM nonessential amino acids, 100 U/mL penicillin, and 100 μg/mL streptomycin (Thermo Fisher Scientific, Waltham, MA, USA). Isolated cells were passed through a 23-G needle to obtain single cells, which were cultured for 24 h in small tissue-culture flasks. Culturing of BMDMs has been previously described in detail [
15]. Briefly, cells were washed, and 3 × 10
4 or 1.2 × 10
5 non-adherent cells were cultured in 24- or 6-well plates in MEM with 20% L-cell supernatant as a source of macrophage colony-stimulating factor (M-CSF). Fresh MEM/L-cell supernatant medium was applied after 4 days of culture. Macrophages matured within the next 6 days and were used on day 10 of culture.
In the present study, we used the cathepsin D inhibitor Pepstatin A (Sigma-Aldrich, Steinheim, Germany) or the ROS inhibitor Apocynin (Abcam, Cambridge, UK), both at a concentration of 10 mM. Cells were pre-incubated with inhibitor for 1 h and then infected as described.
2.2. Infection Experiments
The in vivo infections of Asm
−/− and wt mice and the in vitro infections of bone-marrow-derived macrophages (BMDMs) were performed with green fluorescent protein-expressing BCG (GFP-BCG) [
24,
25]. To construct the GFP-BCG strain, BCG were transformed with the dual reporter plasmid pSMT3L × EGFP [
25]. For infection experiments, bacteria were shaken at 120 rpm at 37 °C in Erlenmeyer flasks with 10 mL Middlebrook 7H9 Broth supplemented with glycerol (BD Biosciences, Heidelberg, Germany) and 50 µg/mL Hygromycin B to maintain GFP plasmids. After 5 to 7 days of culture, the bacteria were used for infection experiments. To this end, the suspended bacteria were collected by centrifugation at 880×
g for 10 min. In order to separate clumped BCG, the bacterial pellet was resuspended in HEPES/saline buffer (H/S) consisting of 132 mM NaCl, 1 mM CaCl
2, 0.7 mM MgCl
2, 20 mM HEPES (pH 7.3), 5 mM KCl, and 0.8 mM MgSO
4 and vortexed for 5 min at high speed. Samples were bath-sonicated for 5 min at 4 °C and passed 10 times through a syringe with an 0.8 mm diameter needle. Unseparated clumps of bacteria were removed by centrifugation at 220×
g for 2 min. The supernatant containing single GFP-BCG was carefully collected. The bacterial number was calculated with a 100× oil lens of an inverted fluorescence microscope (DMIRE2; Leica Microsystems, Wetzlar, Germany).
For in vitro assays, BMDMs were kept in MEM/10 mM HEPES (pH 7.4) and either left uninfected or infected with GFP-BCG for the indicated time at a ratio of bacteria-to-host cells (multiplicity of infection, MOI) of 5:1 to 10:1. In order to achieve increased interaction between bacteria and host cells and to obtain synchronous infection conditions, the bacteria were centrifuged onto the cells for 8 min at 55× g. The end of centrifugation was defined as the starting point of infection. The infection was terminated either by fixation or lysis, as described below. For in vivo infections, bacteria were prepared as described above and then pelleted at 2.240× g for 10 min. BCG were resuspended in 0.9% NaCl and 1 × 107 colony-forming units (CFU) of bacteria in 100 μL were injected intravenously into mice.
2.3. Discrimination between Intra- and Extracellular GFP-BCG
To discriminate between binding and internalization of GFP-BCG, we carried out reduction in green fluorescence of Trypan blue quenched bacteria by excitation energy transfer [
26,
27]. To this end, we washed infected or uninfected bone-marrow-derived macrophages in cold phosphate buffered saline (PBS) buffer (137 mM NaCl, 2.7 mM KCl, 10 mM Na
2HPO
4, 2.0 mM KH
2PO
4; pH adjusted to 7.4) and incubated them in PBS on ice for 15 min. Cells were harvested, normalized to a concentration of 1 × 10
6 cells/50 µL in PBS, and either fixed in 4% paraformaldehyde (PFA; Sigma-Aldrich, Steinheim, Germany) for 10 min at room temperature or processed to quench adherent bacteria. To quench the fluorescence of adherent GFP-BCG, 500 µL Trypan blue (0.4%
w/
v in PBS; Corning Inc., Corning, NY, USA) was added for 1 min, followed by washing twice in PBS and fixation in 4% PFA for 10 min at room temperature. Quenching with Trypan blue reduced the GFP fluorescence of only adherent bacteria (not internalized bacteria) by excitation energy transfer [
26,
27]. Bacterial binding (without Trypan blue treatment) and internalization (with Trypan blue treatment) were analyzed by Attune NxT flow cytometry (Thermo Fisher Scientific, Waltham, MA, USA) and FlowJo software v10 (FlowJo LLC, Ashland, OR, USA).
2.4. Depletion and Transplantation of Bone-Marrow-Derived Macrophages
Liposomes were purchased from Liposoma BV, Netherlands, stored at 4 °C, and calibrated to room temperature for 2 h before injection. For bone-marrow-derived macrophage (BMDM) depletion, clodronate, or PBS liposomes (200 µL) were infused intravenously. For reconstitution, macrophages were generated by culturing BMDMs as previously described. Two days after depletion, mice were left untreated or intravenously injected with 106, 5 × 106, or 107 macrophages. Mice were sacrificed 3 days after transplantation of macrophages. Liver, spleen, and bone marrow were collected for detection of macrophages by Attune NxT flow cytometer (Thermo Fisher Scientific, Waltham, MA, USA). Briefly, cell suspension was harvested from bone marrow, liver, and spleen and adjusted to a concentration of 1 × 106 cells/50 µL in PBS. Cells were incubated for 30 min at 4 °C with anti-mouse CD16/32 antibody (Biolegend, San Diego, CA, USA) to block Fcγ receptors, followed by incubation with primary fluorochrome-conjugated antibodies specific to mouse PE-Gr-1 (Ly6G/C; clone RB6-8C5), Brilliant Violet-CD115 (clone AFS98), Pacific Blue-CD11b (clone M1/70), PerCP-CD3 (clone 145-2C11), PerCP-CD45R/B220 (clone RA3-6B2), PerCP-Ter119 (clone TER-119), APC-F4/80 (clone BM8), or PE/Cy7-NK1.1 (clone PK136), all from Biolegend, San Diego, CA, USA for 45 min at 4 °C. After washing 2 times with PBS, cells were resuspended in PBS and analyzed with an NxT flow cytometer (Thermo Fisher Scientific, Waltham, MA, USA). Stained cell suspensions were analyzed with multiparameter FlowJo software v10 (FlowJo LLC, Ashland, OR, USA).
2.5. Transfection of Bone-Marrow-Derived Macrophages
The all-in-one mouse CTSD gRNA/CRISPR-cas9 plasmid and CRISPR-cas9 control plasmid (without CTSD-gRNA) were purchased from GenScript (Piscataway, NJ, USA). Plasmids were transformed to DH5α-competent cells and amplified in LB medium (Roth GmbH, Karlsruhe, Germany) containing ampicillin (Sigma-Aldrich, Steinheim, Germany) overnight at 37 °C with shaking at 225 rpm. The bacteria were harvested, and plasmids were extracted and purified using a Qiagen plasmid extraction and Midi purification kit (Qiagen, Hilden, Germany) following the manufacturer’s instructions. The purified CTSD gRNA/CRISPR-Cas9 plasmid and CRISPR-Cas9 control plasmid (without CTSD gRNA) were transfected with Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA) into mature bone-marrow-derived macrophages according to the procedure provided by the company. Transfected cells with segments containing either control or CTSD gRNA were selected by adding puromycin (6 μg/mL; Thermo Fisher Scientific, Waltham, MA, USA) and were used 2–3 days after transfection for infection in puromycin-free medium.
2.6. Determination of Acid Sphingomyelinase Activity
For determination of Asm activity in macrophages, we performed a recently described assay that makes use of green fluorescent BODIPY FL C
12-sphingomyelin (Thermo Fisher Scientific, Waltham, MA, USA) as a substrate for the acid sphingomyelinase [
28]. For this purpose, cells were left uninfected or infected for the indicated time, harvested, and lysed in 250 mM sodium acetate (Sigma-Aldrich, Steinheim, Germany) with 1% Nonidet P-40 (pH 5.0; Sigma-Aldrich, Steinheim, Germany) for 5 min on ice. Cells were further disrupted by sonification for 10 min in an ice bath sonicator (Bandelin Electronic, Berlin, Germany). Aliquots were taken for protein measurement by a Bradford protein assay (BioRad, Feldkirchen, Germany), and 5 µg of protein in 20 μL lysis buffer was added to 250 mM sodium acetate (pH 5.0) containing 100 pmol green fluorescent BODIPY-FL
C12-sphingomyelin. The samples were shaken for 1 h at 300 U and 37 °C, and the reaction was stopped by adding 1 mL of chloroform/methanol (2:1,
v/
v), followed by centrifugation at 14,000 rpm for 5 min. The lower phase was dried in a SpeedVac Concentrator (Thermo Fisher Scientific, Waltham, MA, USA), resuspended in chloroform/methanol (2:1,
v/
v), spotted on a thin-layer chromatography (TLC) plate (Merck, Darmstadt, Germany), and separated with chloroform/methanol (80:20,
v/
v). For analysis, the samples were scanned with a Typhoon FLA 9500 laser scanner (GE Healthcare Life Sciences, Freiburg, Germany) and quantified with ImageQuant software (GE Healthcare Life Sciences, Freiburg, Germany).
2.7. Western Blots
Infection of bone-marrow-derived macrophages (BMDMs) was carried out for the indicated time. Both infected and uninfected cells were washed in cold H/S buffer and lysed for 5 min on ice in 125 mM NaCl, 25 mM TrisHCl (pH 7.4), 10 mM ethylenediaminetetraacetic acid (EDTA), 10 mM sodium pyrophosphate, and 3% NP-40 supplemented with 10 µg/mL aprotinin/leupeptin (A/L). Cell lysates were pelleted by centrifugation for 10 min at 10.510× g. The supernatants were collected, added to 5× sodium dodecyl sulfate (SDS) sample buffer, and boiled for 5 min at 95 °C. Proteins were separated by 8.5% to 12.5% SDS polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose membranes (Protran Premium 0.2 µm; GE Healthcare, Life Sciences, Freiburg, Germany) for 2 h at 4 °C (80 V). The blots were washed with PBS and blocked for 1 h at room temperature in Starting Block Tris-buffered saline (TBS) buffer (Thermo Fisher Scientific, Waltham, MA, USA). After 2 additional washes in PBS, they were then incubated overnight at 4 °C in blocking buffer with specific primary antibodies against mature cathepsin D (Santa Cruz Biotechnology, Dallas, TX, USA), p47phox (mouse) (Merck, Darmstadt, Germany), or actin (Santa Cruz Biotechnology, Dallas, TX, USA) at 1:1000 dilution. After being subjected to 6 washing steps in TBS/Tween, blots were incubated for 1 h at room temperature in TBS/Tween containing 10% blocking buffer with alkaline phosphatase (AP)-conjugated secondary antibodies (Santa Cruz Biotechnology, Dallas, TX, USA). Samples were washed extensively and developed with CDP-Star substrate (Perkin Elmer, Rodgau, Germany). For in vivo measurement of cathepsin D expression, mice were sacrificed and tissues were removed, homogenized, and processed, as described above.
2.8. Measurement of Superoxide Production
Superoxide production was measured by electron spin resonance (ESR), as previously described [
29]. In brief, 10
6 cells were infected with GFP-BCG for the indicated time, the medium was removed, and the cells were scraped into 20 mM HEPES (pH 7.5), 1 mM EDTA, and 255 mM sucrose then shock-frozen in liquid nitrogen. Proteins were isolated and resuspended with modified Krebs-HEPES buffer containing deferoxamine (100 μM; Sigma) and diethyldithiocarbamate (5 μM; Sigma-Aldrich, Steinheim, Germany). A spin trap with 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine (1 mM final concentration; Noxygen Science Transfer and Diagnostics, Elzach, Germany) was then added to the mixture in the presence or absence of manganese-dependent superoxide dismutase (SOD, 200 U/mL; Sigma-Aldrich, Steinheim, Germany). The mixture was loaded into glass capillaries and immediately kinetically analyzed for O
2. formation for 10 min. The SOD-inhibited fraction of the signal was used to calibrate the system. The ESR settings were as follows: biofield, 3350; field sweep, 60 G; microwave frequency, 9.78 GHz; microwave power, 20 mW; modulation amplitude, 3 G; points of resolution, 4096; receiver gain, 100; and kinetic time, 10 min. The ESR signal strength was recorded in arbitrary units, and the final results were expressed as fold change from control strength, as previously described [
30].
2.9. Immunocytochemistry
Bone-marrow-derived macrophages (BMDMs) were grown on coverslips and were infected or left uninfected. After fixation in 1% paraformaldehyde (PFA; Sigma-Aldrich, Steinheim, Germany) for 15 min at room temperature, they were washed in PBS (pH 7.4) for further staining. Cells were permeabilized with 0.1% Triton X-100 (Sigma-Aldrich, Steinheim, Germany) in PBS (pH 7.4) for 10 min at room temperature, washed once with H/S and once with H/S with 0.05% Tween-20 (Sigma-Aldrich, Steinheim, Germany), and blocked for 45 min in H/S supplemented with 5% fetal calf serum (FCS; Thermo Fisher Scientific, Waltham, MA, USA). BMDMs were washed 3 times in H/S with 0.05% Tween-20 and incubated for 45 min with cathepsin D (R&D, Minneapolis, MN, USA) or rabbit-anti-p47phox (mouse) antibodies (Merck, Darmstadt, Germany) in H/S supplemented with 1% FCS (Thermo Fisher Scientific, Waltham, MA, USA). Cells were washed 3 times in H/S with 0.05% Tween-20 and incubated with secondary antibodies corresponding to the primary antibodies for 45 min (final concentration of all antibodies, 1.5 μg/mL, diluted in 5% FCS/PBS; all antibodies from Jackson Immuno Research, Europe Ltd., Cambridgeshire, UK). To confirm the specificity of fluorescent staining, samples were incubated with secondary antibody controls. After 3 further washes in H/S with 0.05% Tween-20 and a final wash with H/S, cells were mounted on glass microscope slides with Mowiol (Kuraray Specialities Europe GmbH, Frankfurt, Germany). Cells were examined with a Leica TCS SP5 confocal microscope (Leica Microsystems, Wetzlar, Germany). For quantification of co-localization between GFP-BCG and CTSD or p47phox, randomly selected fields were chosen and at least 50 bacteria/sample were analyzed. To acquire the percentage of bacterial co-localization, the number of GFP-BCGs that co-localized with CTSD or p47phox was divided by total number of GFP-BCGs, and the results were multiplied by 100.
2.10. Histopathologic Assessment
After the indicated infection time, mice were sacrificed by cervical dislocation, and livers and spleens were removed. Tissues were embedded in Tissue-Tec (Sakura Finetek USA, Torrance, CA, USA) and shock-frozen in liquid nitrogen, and 6 µm thick sections were cut with a cryotome (CM1850 UV, Leica Microsystems, Wetzlar, Germany). For staining, sections were thawed, air-dried for 5 min, and fixed in ice-cold acetone for 10 min. Fluorescent visualization of mycobacteria was performed with the Truant TB Fluorescent Stain Kit (BD Difco, Becton Dickinson, Franklin Lakes, NJ, USA) according to the manufacturer’s instructions. Evaluation of bacterial distribution was carried out with an inverted fluorescence microscope or a confocal microscope (DMIRE2; Leica Microsystems, Wetzlar, Germany). For hematoxylin and eosin (H&E) staining, liver and spleen sections were prepared as above and stained for 20 min with Mayer’s hemalum solution (Roth GmbH, Karlsruhe, Germany). Samples were washed in water for 15 min, stained with 1% eosin solution for an additional 2 min, and washed again with water. After dehydration in ethanol, the samples were embedded in Eukitt mounting medium (Sigma-Aldrich, Steinheim, Germany) and analyzed with a Leica DMIRE2 microscope (Leica Microsystems, Wetzlar, Germany). Granuloma formation was examined with a 20× lens and an inverted fluorescence microscope.
For visualization of cathepsin D and murine macrophages in tissue, fixed sections were stained with antibodies against cathepsin D (Santa Cruz, Dallas, TX, USA) and corresponding Cy3-coupled secondary antibodies, APC-F4/80 (Biolegend, San Diego, CA, USA), as described above.
2.11. Quantification of Bacterial Numbers
To quantify mycobacteria in tissue, we removed livers and spleens from infected mice and added 5 mg/mL saponin (Serva Electrophoresis GmbH, Heidelberg, Germany) in H/S for the release of intracellular bacteria. Tissue was homogenized in a loose Dounce homogenizer (Braun, Kronberg, Germany) and incubated for 30 min at 37 °C in a thermomixer for the release of intracellular bacteria. Samples were centrifuged for 2 min at 220× g, resuspended in PBS, and plated on Middlebrook 7H10 agar plates supplemented with oleic acid (OADC; BD Biosciences, Heidelberg, Germany) to determine colony-forming units (CFUs). To determine the CFUs of BCG in macrophages, infected cells were washed once with MEM/10 mM HEPES (pH 7.4) after the indicated infection time to remove nonadherent bacteria, and then lysed in 3 mg/mL saponin for 30 min at 37 °C. We plated 100 µL aliquots and counted bacterial CFUs after the plates had been incubated for approximately 2 weeks in a humidified 37 °C atmosphere.
2.12. Statistical Analysis
All data were obtained from independent measurements and expressed as arithmetic means ± standard deviation (SD). Data were tested with the David–Pearson–Stephens test for normal distribution. Statistical analysis was performed with Student’s t-test for single comparisons and ANOVA for multiple comparisons. GraphPad Prism statistical software 6 (GraphPad Software, La Jolla, CA, USA) was used for analysis.
4. Discussion
Although it has been reported that both acid sphingomyelinase (Asm), and ROS are involved in mycobacterial infection [
11,
19,
21], and that Asm-derived ceramide binds to and activates cathepsin D (CTSD) [
16], our work, for the first time, suggests a novel mechanistic link between acid sphingomyelinase, cathepsin D, and ROS in mycobacterial infection (
Figure 8) and indicates a pathogen-triggered signaling cascade that leads to bacterial clearance in vitro and in vivo.
A recent study indicated that Asm-mediated maturation of phagosomes is important for controlling mycobacterial infection [
19]. Sortilin (
Sort1), also named neurotensin receptor 3, is a transmembrane receptor that transports lysosomal proteins from the trans-Golgi network into lysosomes. Sortilin is upregulated during infection of macrophages with mycobacteria and is required for the delivery of both prosaposin and Asm from the Golgi complex to phagosomes. Studies on
Sort1-deficient macrophages revealed a reduced association of Asm with phagosomes in
Sort1-deficient cells compared to wild-type (wt) macrophages [
20]. In vivo,
Sort1-deficient mice exhibited substantially increased cellular infiltration of neutrophils and higher bacterial burden after infection with
Mycobacterium tuberculosis in the lungs, suggesting that
Sort1-dependent delivery of Asm into phagosomes is crucial for restricting bacterial growth [
19].
Further studies demonstrated that Asm is required for the proper fusion of late phagosomes with lysosomes, which is crucial for efficient transfer of lysosomal antibacterial hydrolases into phagosomes [
11]. Additional studies on zebrafish implied that Asm together with mitochondrial cyclophilin D induces necroptosis of macrophages upon infection with
Mycobacterium tuberculosis, which might contribute to enhanced accessibility to the innate and specific immune system and thereby the elimination of intracellular mycobacteria [
21].
Recognition of microbial pathogens by specific cell surface receptors and their internalization by professional macrophages is the first line of defense in bacterial infection [
31]. Previous work has linked Asm with phagocytosis, reporting either involvement in bacterial internalization or maturation of phagosomes and lysosomes [
32,
34]. However, using a different range of assays, we show here that both binding and internalization of
Mycobacterium bovis Bacillus Calmette-Guérin (BCG) into macrophages are independent of Asm. Neither the initial steps of phagosome maturation, such as Rab7 or Lamp1 expression, nor the localization of BCG in bone-marrow-derived macrophages differed between wt and Asm-deficient macrophages. Our flow cytometry studies employing Trypan blue quenching of adherent GFP-BCG confirm that the higher bacterial load we observed in Asm-deficient cells was not caused by defective BCG internalization by macrophages. This implies that Asm is not involved in adherence and internalization, at least in our model.
Following the uptake of microbes by phagocytes, bacteria containing phagosomes mature into phagolysosomes by gaining bactericidal factors such as active cathepsins to degrade microbes. A common defense mechanism used by mycobacteria is disruption of phagosome maturation and blockade of phagosomal acidification. It has been shown that
Mycobacterium tuberculosis infection induces a general downregulation of cathepsins B, D, and S within macrophages, favoring increased intracellular survival of the pathogen [
19,
33]. Other studies suggested that Asm-derived ceramide specifically binds to cathepsin D (CTSD), resulting in enhanced enzymatic activity and proteolytic activation of proteins to be secreted [
16].
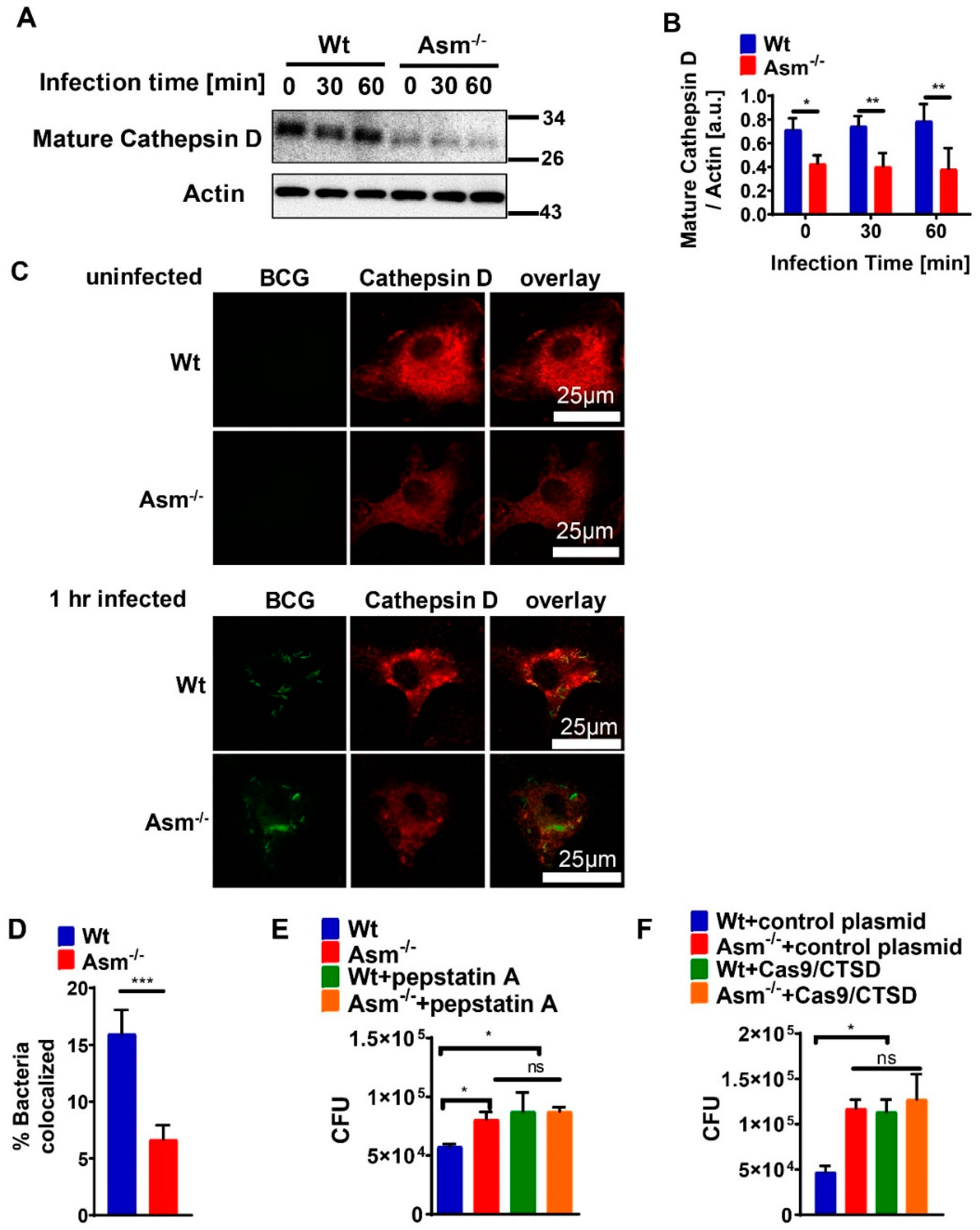
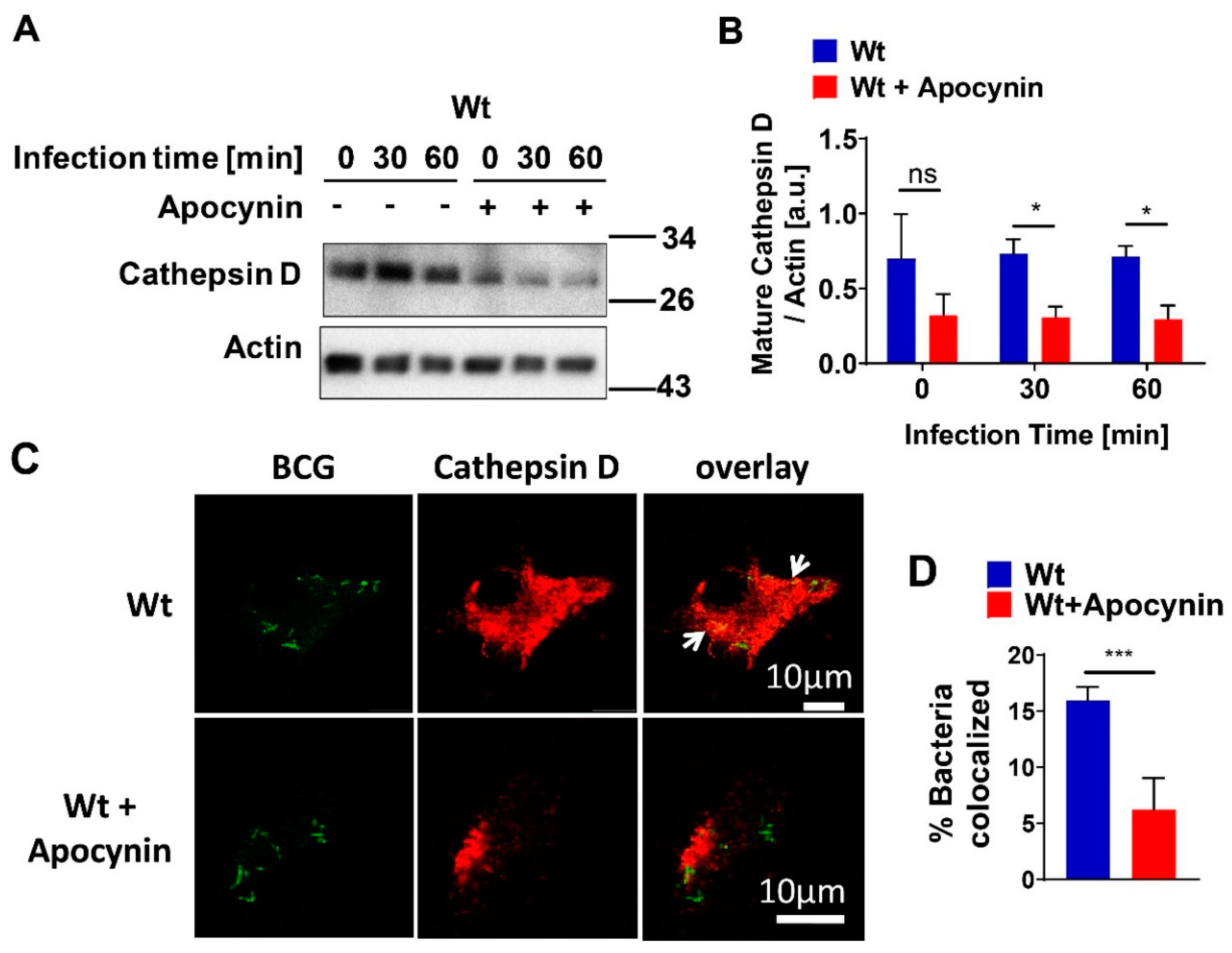
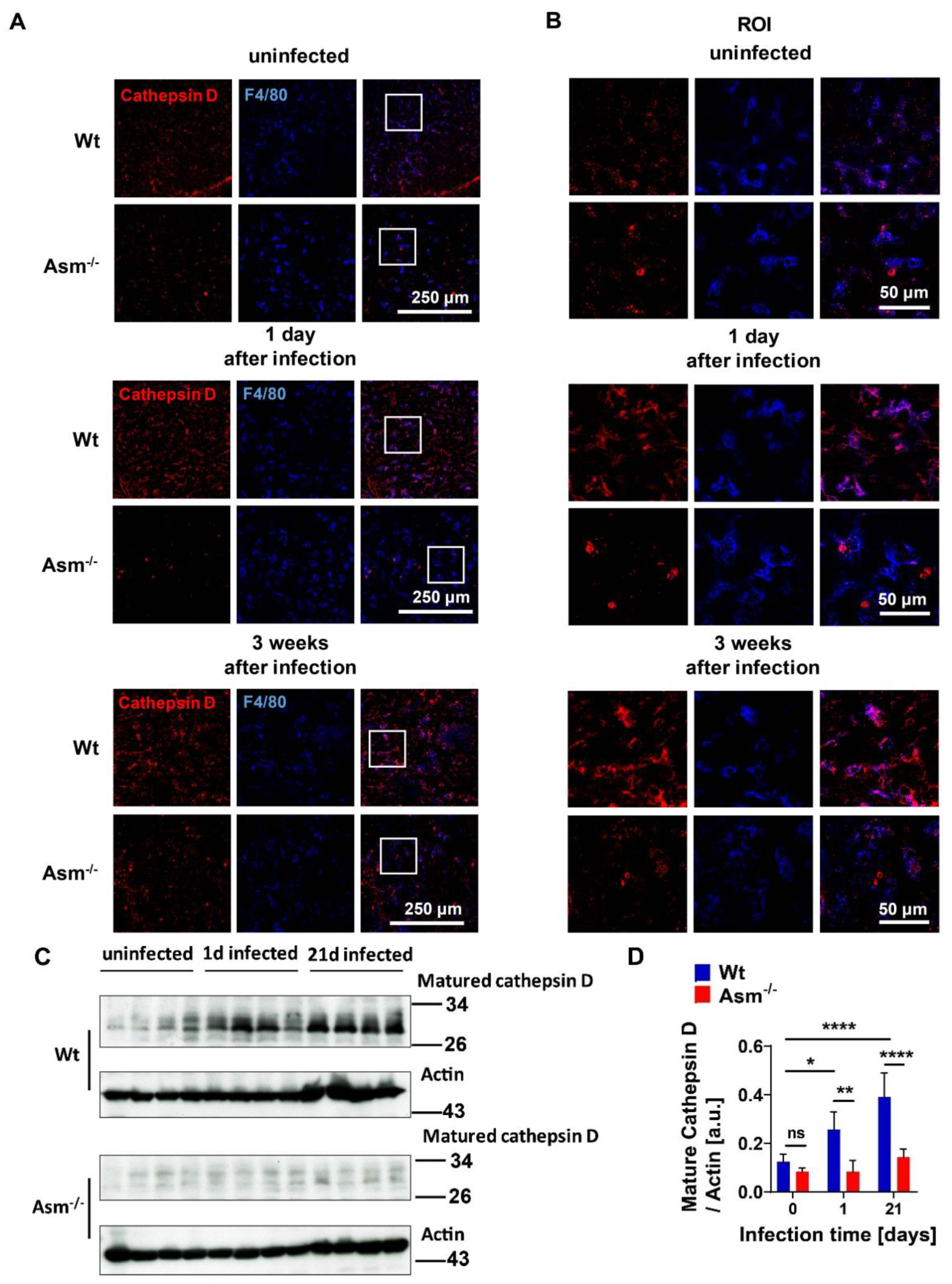
Our presented results show that the expression of mature cathepsin D (CTSD) in wild-type (wt) macrophages was constitutively high and absent in cells with acid sphingomyelinase (Asm) deficiency, while other cathepsins, such as B, L, K, and S, did not differ between wt and Asm-deficient macrophages. The results also reveal that CTSD was relevant for the killing of BCG in macrophages as the downregulation/inhibition of mature cathepsin D increased bacterial load in wt macrophages to the level of Asm-deficient macrophages. In addition, immunofluorescence stainings for CTSD and bacteria in wt macrophages showed a higher co-localization between BCG and CTSD than in Asm-deficient cells. These results indicate that while CTSD expression/maturation was not modulated in vitro with BCG infection, it was regulated by acid sphingomyelinase (Asm) and was required to target BCG to CTSD and properly kill bacteria. Instead, in vivo the expression/maturation of CTSD in the liver of wt mice was significantly upregulated after BCG infection, whereas it was absent in the liver of Asm-deficient mice. These data support the in vitro results with macrophages and suggest that Asm controls CTSD, a process that leads to the killing of bacteria. The function of CTSD as a mediator of mycobacterial antigen presentation in macrophages may explain our finding that Asm-deficient mice were unable to control BCG infection. Due to a lack or delay of antigen presentation in Asm-deficient macrophages, the adaptive immune response in vivo may be retarded.
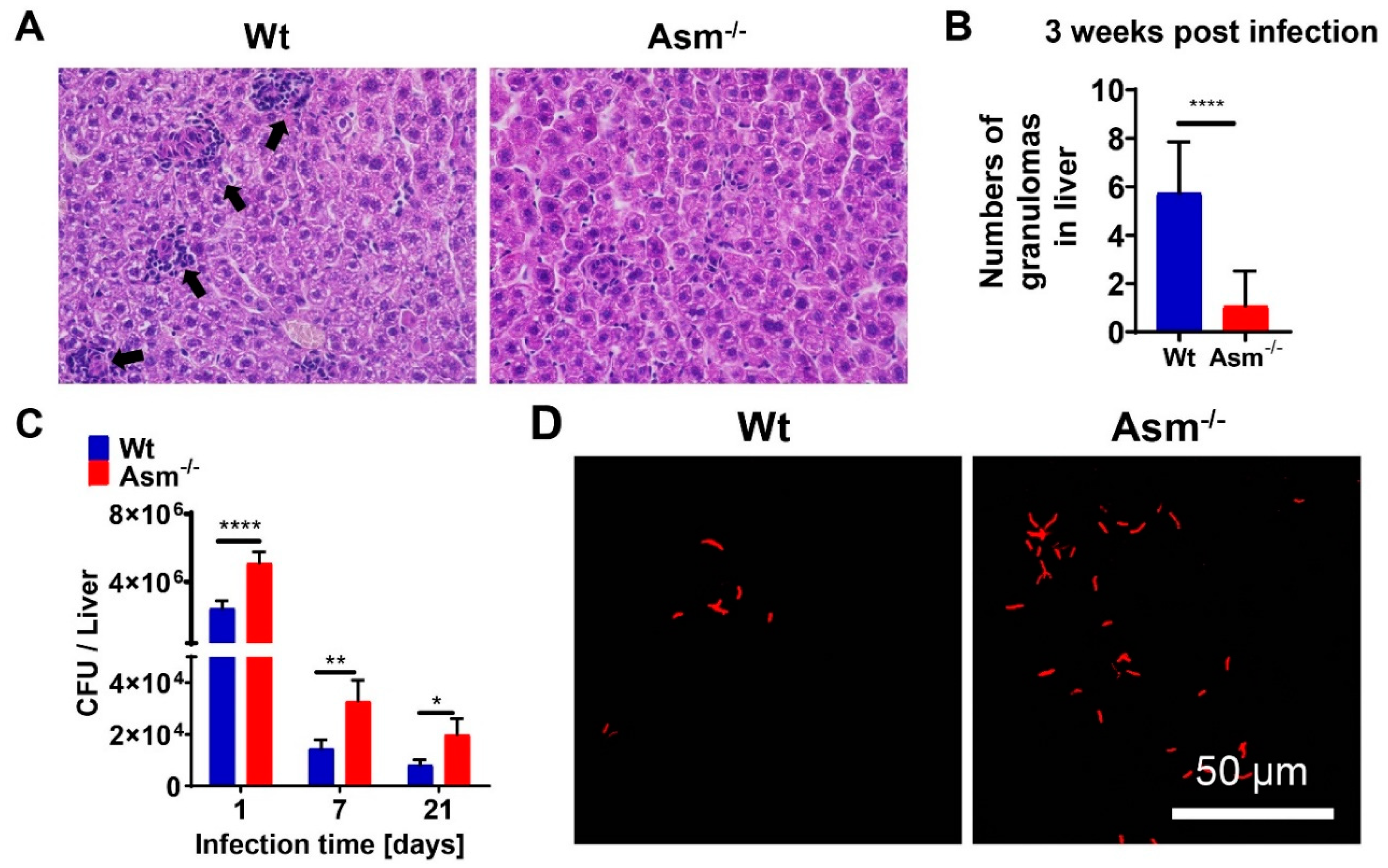
Granulomas, organized aggregates of immune cells, form in response to persistent stimuli/bacteria and are hallmarks of tuberculosis. Tuberculosis granulomas are most likely to be considered as host-protective structures formed to contain infection [
35]. This notion is in accordance with our observation that in Asm-deficient mice, which were unable to clear BCG infection, hardly any granulomas were detectable. Since we observed no effect of Asm on the in vitro clustering of bone-marrow-derived macrophages after 24 h of infection (not shown), we assume that the in vivo granulomas could be affected by some other cell types, as mycobacterial granulomas are organized aggregates from different cell groups, including mature macrophages, differentiated or epithelial macrophages, foamy macrophages, and multinucleated (or Langerhans) giant cells [
36].
Real-time observations of zebrafish embryos that were still at a stage prior to the appearance of T-lymphocytes revealed that
Mycobacterium marinum induced granuloma formation could be initiated with macrophages alone [
37]. In addition, studies with an in vitro model of mycobacterial granulomas, which enable cellular and molecular analysis of the very first steps in the host granulomatous response, indicated that macrophages were sufficient for the early stages of granuloma formation [
38]. On the other hand, Ramakrishnan’s group found that primary granulomas promoted early dissemination of infection via egress of infected macrophages, thus promoting infection [
39].
The increased susceptibility of Asm-deficient mice to BCG infection was reversed by transplantation with wt bone-marrow-derived macrophages in liver, suggesting that the reduced bacterial killing of Asm-deficient mice was due to an impaired microbicidal effect of Asm-deficient macrophages. In comparison to wt mice, the bacterial burden in the livers of Asm-deficient mice was enhanced after acute and chronic infection with BCG, whereas the bacterial load in spleens differed dramatically only after a short time of infection. Accordingly, the bacterial burden of Asm-deficient mice was significantly reduced after transplantation of wt macrophages in the liver and normalized to the level in wt mice, whereas the transplantation did not reverse susceptibility to BCG in the spleens of Asm-deficient mice. This discrepancy might be explained by different immune responses in different tissues.
A study that compared detailed phenotypical and functional analyses of murine Kupffer cells and splenic/peritoneal macrophages under steady-state conditions showed that liver macrophages exerted potent endocytic activity and displayed relatively high basal levels of ROS compared with splenic and peritoneal macrophages. Additional, ligation of TLR4, TLR7/8, and TLR9 on Kupffer cells resulted in a weak induction of IL-10; low or undetectable levels of IL-12, p40, and TNFα; and upregulation of CD40 on the surface compared to other macrophages [
40]. These results suggest that Kupffer cells are specialized as phagocytes but only play a limited immune-regulatory role [
35]. Bone-marrow-derived macrophages have been reported to have similar biological functions to Kupffer cells [
41]. Compared to splenic macrophages, bone-marrow-derived macrophages show a stronger capacity for proliferation and phagocytosis [
42]. Another aspect is that liver dendritic cells are generally weak activators of immunity, although they are capable of producing inflammatory cytokines, and certain subtypes potently activate T cells [
42]. A previous study showed that liver dendritic cells were less mature, captured less antigen, and induced less T cell stimulation than spleen dendritic cells because of differences of subtype composition [
43]. A study based on an investigation of in situ expression of MHC class II on liver dendritic cells identified that murine liver dendritic cells did not display measurable levels of the T cell-costimulatory molecules CD40, CD80, and CD86, which implied a low immune-stimulatory capacity of liver dendritic cells [
44]. Kupffer cells have been shown to play a critical role in mycobacterial infections. A recent study suggested that mouse Kupffer cells, in comparison to alveolar macrophages, are able to better restrict the growth of
Mycobacterium tuberculosis, because they are more capable of producing cytokines and molecules that modulate autophagy and cytoskeleton than dendritic cells [
45]. Together, these studies suggest different mechanisms of regulating immune responses in liver and spleen than our model. This also implies that other tissue immune cells, such as dendritic cells or T-lymphocytes, may have important functions predominantly in the spleen, while the immune response to BCG in the liver is mostly driven by macrophages.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







