The Emerging Role of the Lysosome in Parkinson’s Disease


Abstract
1. Introduction
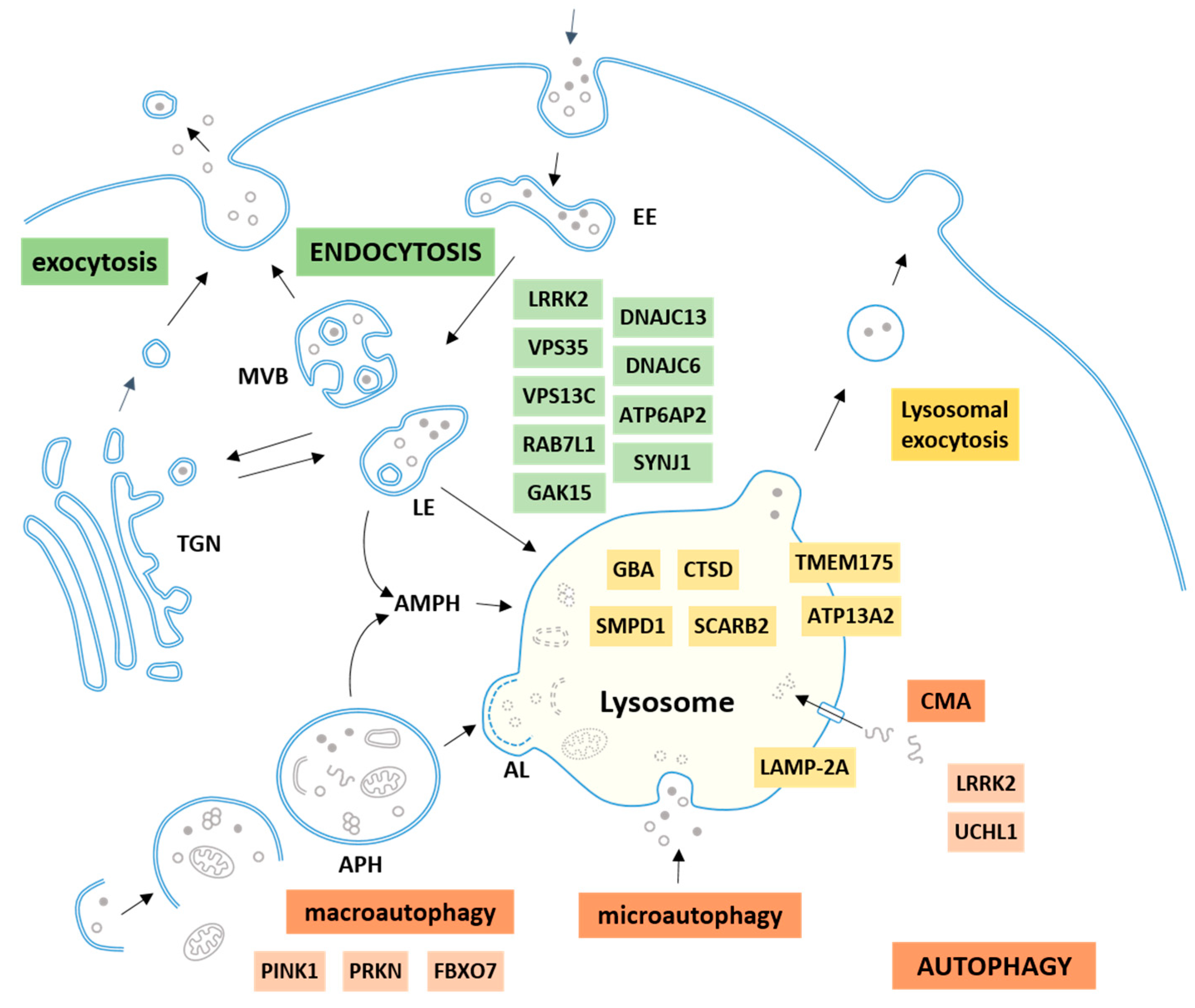
2. The Autophagy–Lysosomal–Endosomal System
2.1. Autophagy
2.2. Endocytosis
3. Parkinson’s Disease and Alpha-Synuclein
4. Lysosomal-Dependent Degradation of Synuclein
5. Lysosomal Dysfunction in Parkinson’s Disease
6. Lysosomal Storage Disorders and Parkinson’s Disease
7. Autophagy Dysfunction in Parkinson’s Disease
8. Endocytosis Dysfunction in Parkinson’s Disease
9. Role of Glial Cells in PD
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Futerman, A.H.; Van Meer, G. The cell biology of lysosomal storage disorders. Nat. Rev. Mol. Cell Biol. 2004, 5, 554–565. [Google Scholar] [CrossRef]
- Fraldi, A.; Klein, A.D.; Medina, D.L.; Settembre, C. Brain Disorders Due to Lysosomal Dysfunction. Annu. Rev. Neurosci. 2016, 39, 277–295. [Google Scholar] [CrossRef] [PubMed]
- Bonam, S.R.; Wang, F.; Muller, S. Lysosomes as a therapeutic target. Nat. Rev. Drug Discov. 2019, 18, 923–948. [Google Scholar] [CrossRef]
- Xu, H.; Ren, D. Lysosomal Physiology. Annu. Rev. Physiol. 2015, 77, 57–80. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Klionsky, D.J. Autophagy and neurodegeneration. ACS Chem. Biol. 2006, 1, 211–213. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Autophagy in the Pathogenesis of Disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef]
- Kim, I.; Rodriguez-Enriquez, S.; Lemasters, J.J. Selective degradation of mitochondria by mitophagy. Arch. Biochem. Biophys. 2007, 462, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Dunn, W.A.; Cregg, J.M.; Kiel, J.A.K.W.; van der Klei, I.J.; Oku, M.; Sakai, Y.; Sibirny, A.A.; Stasyk, O.V.; Veenhuis, M. Pexophagy: The selective autophagy of peroxisomes. Autophagy 2005, 1, 75–83. [Google Scholar] [CrossRef]
- Bernales, S.; Schuck, S.; Walter, P. ER-phagy: Selective autophagy of the endoplasmic reticulum. Autophagy 2007, 3, 285–287. [Google Scholar] [CrossRef]
- Kraft, C.; Deplazes, A.; Sohrmann, M.; Peter, M. Mature ribosomes are selectively degraded upon starvation by an autophagy pathway requiring the Ubp3p/Bre5p ubiquitin protease. Nat. Cell Biol. 2008, 10, 602–610. [Google Scholar] [CrossRef]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef]
- Levine, B. Eating oneself and uninvited guests: Autophagy-related pathways in cellular defense. Cell 2005, 120, 159–162. [Google Scholar] [PubMed]
- Lamark, T.; Johansen, T. Aggrephagy: Selective Disposal of Protein Aggregates by Macroautophagy. Int. J. Cell Biol. 2012, 2012, 736905. [Google Scholar] [CrossRef]
- Jung, C.H.; Ro, S.-H.; Cao, J.; Otto, N.M.; Kim, D.-H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef]
- Dice, J.F.; Terlecky, S.R.; Chiang, H.L.; Olson, T.S.; Isenman, L.D.; Short-Russell, S.R.; Freundlieb, S.; Terlecky, L.J. A selective pathway for degradation of cytosolic proteins by lysosomes. Semin. Cell Biol. 1990, 1, 449–455. [Google Scholar]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef]
- Ahlberg, J.; Glaumann, H. Uptake-Microautophagy-and degradation of exogenous proteins by isolated rat liver lysosomes. Effects of pH, ATP, and inhibitors of proteolysis. Exp. Mol. Pathol. 1985, 42, 78–88. [Google Scholar] [CrossRef]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.I.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef]
- Kumari, S.; Mg, S.; Mayor, S. Endocytosis unplugged: Multiple ways to enter the cell. Cell Res. 2010, 20, 256–275. [Google Scholar] [CrossRef] [PubMed]
- Scott, C.C.; Vacca, F.; Gruenberg, J. Endosome maturation, transport and functions. Semin. Cell Dev. Biol. 2014, 31, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Elkin, S.R.; Lakoduk, A.M.; Schmid, S.L. Endocytic pathways and endosomal trafficking: A primer. Wiener Med. Wochenschr. 2016, 166, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Dittman, J.; Ryan, T.A. Molecular Circuitry of Endocytosis at Nerve Terminals. Annu. Rev. Cell Dev. Biol. 2009, 25, 133–160. [Google Scholar] [CrossRef]
- Cosker, K.E.; Segal, R.A. Neuronal signaling through endocytosis. Cold Spring Harb. Perspect. Biol. 2014, 6, a020669. [Google Scholar] [CrossRef]
- Yap, C.C.; Winckler, B. Harnessing the Power of the Endosome to Regulate Neural Development. Neuron 2012, 74, 440–451. [Google Scholar] [CrossRef]
- Tysnes, O.B.; Storstein, A. Epidemiology of Parkinson’s disease. J. Neural Transm. 2017, 124, 901–905. [Google Scholar] [CrossRef]
- Klein, A.D.; Mazzulli, J.R. Is Parkinson’s disease a lysosomal disorder? Brain 2018, 141, 2255–2262. [Google Scholar] [CrossRef]
- Arotcarena, M.-L.; Teil, M.; Dehay, B. Autophagy in Synucleinopathy: The Overwhelmed and Defective Machinery. Cells 2019, 8, 565. [Google Scholar] [CrossRef]
- Sassone, J.; Reale, C.; Dati, G.; Regoni, M.; Pellecchia, M.T.; Garavaglia, B. The Role of VPS35 in the Pathobiology of Parkinson’s Disease. Cell. Mol. Neurobiol. 2020. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K. Neuropathological Staging of Brain Pathology in Sporadic Parkinson’s disease: Separating the Wheat from the Chaff. J. Parkinsons. Dis. 2017, 7, S73–S87. [Google Scholar] [CrossRef] [PubMed]
- Bandres-Ciga, S.; Diez-Fairen, M.; Kim, J.J.; Singleton, A.B. Genetics of Parkinson’s disease: An introspection of its journey towards precision medicine. Neurobiol. Dis. 2020, 137, 104782. [Google Scholar] [CrossRef]
- Lesage, S.; Brice, A. Parkinson’s disease: From monogenic forms to genetic susceptibility factors. Hum. Mol. Genet. 2009, 18, R48–R59. [Google Scholar] [CrossRef] [PubMed]
- Van Den Eeden, S.K.; Tanner, C.M.; Bernstein, A.L.; Fross, R.D.; Bloch, D.A.; Nelson, L.M. Incidence of Parkinson’s Disease: Variation by Age, Gender, and Race/Ethnicity. Am. J. Epidemiol. 2003, 157, 1015–1022. [Google Scholar] [CrossRef]
- Simón-Sánchez, J.; Schulte, C.; Bras, J.M.; Sharma, M.; Gibbs, J.R.; Berg, D.; Paisan-Ruiz, C.; Lichtner, P.; Scholz, S.W.; Hernandez, D.G.; et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat. Genet. 2009, 41, 1308–1312. [Google Scholar] [CrossRef]
- Chang, D.; Nalls, M.A.; Hallgrímsdóttir, I.B.; Hunkapiller, J.; van der Brug, M.; Cai, F.; Kerchner, G.A.; Ayalon, G.; Bingol, B.; Sheng, M.; et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat. Genet. 2017, 49, 1511–1516. [Google Scholar] [CrossRef]
- Nalls, M.A.; Blauwendraat, C.; Vallerga, C.L.; Heilbron, K.; Bandres-Ciga, S.; Chang, D.; Tan, M.; Kia, D.A.; Noyce, A.J.; Xue, A.; et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet Neurol. 2019, 18, 1091–1102. [Google Scholar] [CrossRef]
- Nalls, M.A.; Pankratz, N.; Lill, C.M.; Do, C.B.; Hernandez, D.G.; Saad, M.; Destefano, A.L.; Kara, E.; Bras, J.; Sharma, M.; et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat. Genet. 2014, 46, 989–993. [Google Scholar] [CrossRef] [PubMed]
- Neumann, J.; Bras, J.; Deas, E.; O’sullivan, S.S.; Parkkinen, L.; Lachmann, R.H.; Li, A.; Holton, J.; Guerreiro, R.; Paudel, R.; et al. Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease. Brain 2009, 132, 1783–1794. [Google Scholar] [CrossRef]
- Sidransky, E.; Nalls, M.A.; Aasly, J.O.; Aharon-Peretz, J.; Annesi, G.; Barbosa, E.R.; Bar-Shira, A.; Berg, D.; Bras, J.; Brice, A.; et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N. Engl. J. Med. 2009, 361, 1651–1661. [Google Scholar] [CrossRef]
- Singleton, A.; Hardy, J. A generalizable hypothesis for the genetic architecture of disease: Pleomorphic risk loci. Hum. Mol. Genet. 2011, 20, R158–R162. [Google Scholar] [CrossRef]
- Goedert, M.; Jakes, R.; Spillantini, M.G. The Synucleinopathies: Twenty Years on. J. Parkinsons. Dis. 2017, 7, S51–S69. [Google Scholar] [CrossRef] [PubMed]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [PubMed]
- Krüger, R.; Kuhn, W.; Müller, T.; Woitalla, D.; Graeber, M.; Kösel, S.; Przuntek, H.; Epplen, J.T.; Schöls, L.; Riess, O. Ala30Pro mutation in the gene encoding α-synuclein in Parkinson’s disease. Nat. Genet. 1998, 18, 106–108. [Google Scholar] [CrossRef]
- Zarranz, J.J.; Alegre, J.; Go, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Hoenicka, J.; Rodriguez, O.; Ser, T.; Mun, D.G. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 2004, 55, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Giasson, B.I.; Murray, I.V.J.; Trojanowski, J.Q.; Lee, V.M.Y. A Hydrophobic Stretch of 12 Amino Acid Residues in the Middle of α-Synuclein Is Essential for Filament Assembly. J. Biol. Chem. 2001, 276, 2380–2386. [Google Scholar] [CrossRef]
- Pan, P.Y.; Zhu, Y.; Shen, Y.; Yue, Z. Crosstalk between presynaptic trafficking and autophagy in Parkinson’s disease. Neurobiol. Dis. 2019, 122, 64–71. [Google Scholar] [CrossRef]
- Lashuel, H.A.; Overk, C.R.; Oueslati, A.; Masliah, E. The many faces of α-synuclein: From structure and toxicity to therapeutic target. Nat. Rev. Neurosci 2013, 14, 38–48. [Google Scholar] [CrossRef]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science 2003, 302, 841. [Google Scholar] [CrossRef]
- Chartier-Harlin, M.C.; Kachergus, J.; Roumier, C.; Mouroux, V.; Douay, X.; Lincoln, S.; Levecque, C.; Larvor, L.; Andrieux, J.; Hulihan, M.; et al. A-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 2004, 364, 1167–1169. [Google Scholar] [CrossRef]
- Mutez, E.; Leprêtre, F.; Le Rhun, E.; Larvor, L.; Duflot, A.; Mouroux, V.; Kerckaert, J.P.; Figeac, M.; Dujardin, K.; Destée, A.; et al. SNCA locus duplication carriers: From genetics to Parkinson disease phenotypes. Hum. Mutat. 2011, 32, 2079–2090. [Google Scholar] [CrossRef] [PubMed]
- Maraganore, D.M.; De Andrade, M.; Elbaz, A.; Farrer, M.J.; Ioannidis, J.P.; Krüger, R.; Rocca, W.A.; Schneider, N.K.; Lesnick, T.G.; Lincoln, S.J.; et al. Collaborative analysis of α-synuclein gene promoter variability and Parkinson disease. J. Am. Med. Assoc. 2006, 296, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.K.; Chen, C.M.; Chang, C.Y.; Chang, K.H.; Chen, I.C.; Li, M.L.; Lee-Chen, G.J.; Wu, Y.R. alpha-Synuclein promoter RsaI T-to-C polymorphism and the risk of Parkinson’s disease. J. Neural Transm. 2006, 113, 1425–1433. [Google Scholar] [CrossRef]
- Fuchs, J.; Nilsson, C.; Kachergus, J.; Munz, M.; Larsson, E.M.; Schüle, B.; Langston, J.W.; Middleton, F.A.; Ross, O.A.; Hulihan, M.; et al. Phenotypic variation in a large Swedish pedigree due to SNCA duplication and triplication. Neurology 2007, 68, 916–922. [Google Scholar] [CrossRef] [PubMed]
- Lesage, S.; Anheim, M.; Letournel, F.; Bousset, L.; Honoré, A.; Rozas, N.; Pieri, L.; Madiona, K.; Dürr, A.; Melki, R.; et al. G51D α-synuclein mutation causes a novel Parkinsonian-pyramidal syndrome. Ann. Neurol. 2013, 73, 459–471. [Google Scholar] [CrossRef]
- Proukakis, C.; Dudzik, C.G.; Brier, T.; Mackay, D.S.; Cooper, J.M.; Millhauser, G.L.; Houlden, H. A novel a-synuclein missense mutation in Parkinson disease. Neurology 2013, 80, 1062–1064. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Stafanis, L.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired degradation of mutant α-synuclein by chaperone-mediated autophagy. Science 2004, 305, 1292–1295. [Google Scholar] [CrossRef]
- Vogiatzi, T.; Xilouri, M.; Vekrellis, K.; Stefanis, L. Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J. Biol Chem 2008, 283, 23542–23556. [Google Scholar] [CrossRef]
- Webb, J.L.; Ravikumar, B.; Atkins, J.; Skepper, J.N.; Rubinsztein, D.C. α-synuclein Is Degraded by Both Autophagy and the Proteasome. J. Biol. Chem. 2003, 278, 25009–25013. [Google Scholar] [CrossRef]
- Gan-Or, Z.; Dion, P.A.; Rouleau, G.A. Genetic perspective on the role of the Autophagy-Lysosome Pathway in Parkinson disease. Autophagy 2015, 11, 1443–1457. [Google Scholar] [CrossRef]
- Mak, S.K.; McCormack, A.L.; Manning-Bog, A.B.; Cuervo, A.M.; Di Monte, D.A. Lysosomal degradation of α-synuclein in vivo. J. Biol. Chem. 2010, 285, 13621–13629. [Google Scholar] [CrossRef] [PubMed]
- Xilouri, M.; Vogiatzi, T.; Vekrellis, K.; Park, D.; Stefanis, L. Abberant alpha-synuclein confers toxicity to neurons in part through inhibition of chaperone-mediated autophagy. PLoS ONE 2009, 4, e5515. [Google Scholar] [CrossRef] [PubMed]
- Martinez-vicente, M.; Talloczy, Z.; Kaushik, S.; Massey, A.C.; Mazzulli, J.; Mosharov, E.V.; Hodara, R.; Fredenburg, R.; Wu, D.; Follenzi, A.; et al. Dopamine-modified α-synuclein blocks chaperone-mediated autophagy. Nat. Neurosci. 2008, 118, 777–788. [Google Scholar] [CrossRef]
- Xilouri, M.; Vogiatzi, T.; Vekrellis, K.; Stefanis, L. alpha-synuclein degradation by autophagic pathways: A potential key to Parkinson’s disease pathogenesis. Autophagy 2008, 4, 917–919. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Rodriguez-Oroz, M.C.; Cooper, J.M.; Caballero, C.; Ferrer, I.; Obeso, J.A.; Schapira, A.H. V Chaperone-mediated autophagy markers in Parkinson disease brains. Arch. Neurol. 2010, 67, 1464–1472. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.E.; Gysbers, A.M.; Abbott, S.K.; Spiro, A.S.; Furuta, A.; Cooper, A.; Garner, B.; Kabuta, T.; Halliday, G.M. Lysosomal-associated membrane protein 2 Isoforms Are Differentially Affected in Early Parkinson’s Disease. Mov. Disord. 2015, 30, 1639–1647. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. Proteostasis and aging. Nat. Med. 2015, 21, 1406–1415. [Google Scholar] [CrossRef]
- Malik, B.R.; Maddison, D.C.; Smith, G.A.; Peters, O.M. Autophagic and endo-lysosomal dysfunction in neurodegenerative disease. Mol. Brain 2019, 12, 100. [Google Scholar] [CrossRef]
- Peng, W.; Minakaki, G.; Nguyen, M.; Krainc, D. Preserving Lysosomal Function in the Aging Brain: Insights from Neurodegeneration. Neurotherapeutics 2019, 16, 611–634. [Google Scholar] [CrossRef]
- Ramirez, A.; Heimbach, A.; Gründemann, J.; Stiller, B.; Hampshire, D.; Cid, L.P.; Goebel, I.; Mubaidin, A.F.; Wriekat, A.L.; Roeper, J.; et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat. Genet. 2006, 38, 1184–1191. [Google Scholar] [CrossRef]
- Gan-Or, Z.; Bar-Shira, A.; Dahary, D.; Mirelman, A.; Kedmi, M.; Gurevich, T.; Giladi, N.; Orr-Urtreger, A. Association of sequence alterations in the putative promoter of RAB7L1 with a reduced Parkinson disease risk. Arch. Neurol. 2012, 69, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Do, C.B.; Tung, J.Y.; Dorfman, E.; Kiefer, A.K.; Drabant, E.M.; Francke, U.; Mountain, J.L.; Goldman, S.M.; Tanner, C.M.; Langston, J.W.; et al. Web-based genome-wide association study identifies two novel loci and a substantial genetic component for parkinson’s disease. PLoS Genet. 2011, 7, e1002141. [Google Scholar] [CrossRef] [PubMed]
- Robak, L.A.; Jansen, I.E.; van Rooij, J.; Uitterlinden, A.G.; Kraaij, R.; Jankovic, J.; Heutink, P.; Shulman, J.M.; Nalls, M.A.; Plagnol, V.; et al. Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease. Brain 2017, 140, 3191–3203. [Google Scholar] [CrossRef] [PubMed]
- Hopfner, F.; Mueller, S.H.; Szymczak, S.; Junge, O.; Tittmann, L.; May, S.; Lohmann, K.; Grallert, H.; Lieb, W.; Strauch, K.; et al. Rare Variants in Specific Lysosomal Genes Are Associated with Parkinson’s Disease. Mov. Disord. 2020, 35, 1245–1248. [Google Scholar] [CrossRef]
- Yun, S.P.; Kim, D.; Kim, S.; Kim, S.; Karuppagounder, S.S.; Kwon, S.H.; Lee, S.; Kam, T.I.; Lee, S.; Ham, S.; et al. α-Synuclein accumulation and GBA deficiency due to L444P GBA mutation contributes to MPTP-induced parkinsonism. Mol. Neurodegener. 2018, 13, 1–19. [Google Scholar] [CrossRef]
- Adler, C.H.; Beach, T.G.; Shill, H.A.; Caviness, J.N.; Driver-Dunckley, E.; Sabbagh, M.N.; Patel, A.; Sue, L.I.; Serrano, G.; Jacobson, S.A.; et al. GBA mutations in Parkinson disease: Earlier death but similar neuropathological features. Eur. J. Neurol. 2017, 24, 1363–1368. [Google Scholar] [CrossRef]
- Madero-Pérez, J.; Fdez, E.; Fernández, B.; Lara Ordóñez, A.J.; Blanca Ramírez, M.; Gómez-Suaga, P.; Waschbüsch, D.; Lobbestael, E.; Baekelandt, V.; Nairn, A.C.; et al. Parkinson disease-associated mutations in LRRK2 cause centrosomal defects via Rab8a phosphorylation. Mol. Neurodegener. 2018, 13, 3. [Google Scholar] [CrossRef]
- Bae, E.J.; Lee, S.J. The LRRK2-RAB axis in regulation of vesicle trafficking and α-synuclein propagation. Biochim. Biophys. Acta 2020, 1866, 165632. [Google Scholar] [CrossRef]
- Kuwahara, T.; Iwatsubo, T. The Emerging Functions of LRRK2 and Rab GTPases in the Endolysosomal System. Front. Neurosci. 2020, 14, 227. [Google Scholar] [CrossRef]
- Abeliovich, A.; Gitler, A.D. Defects in trafficking bridge Parkinson’s disease pathology and genetics. Nature 2016, 539, 207–216. [Google Scholar] [CrossRef]
- Schreij, A.M.; Fon, E.A.; McPherson, P.S. Endocytic membrane trafficking and neurodegenerative disease. Cell. Mol. Life Sci. 2016, 73, 1529–1545. [Google Scholar] [CrossRef]
- Zhang, Y.; Sloan, S.A.; Clarke, L.E.; Caneda, C.; Plaza, C.A.; Blumenthal, P.D.; Vogel, H.; Steinberg, G.K.; Edwards, M.S.B.; Li, G.; et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 2016, 89, 37–53. [Google Scholar] [CrossRef] [PubMed]
- Booth, H.D.E.; Hirst, W.D.; Wade-Martins, R. The Role of Astrocyte Dysfunction in Parkinson’s Disease Pathogenesis. Trends Neurosci. 2017, 40, 358–370. [Google Scholar] [CrossRef]
- Chuang, W.L.; Pacheco, J.; Cooper, S.; McGovern, M.M.; Cox, G.F.; Keutzer, J.; Zhang, X.K. Lyso-sphingomyelin is elevated in dried blood spots of Niemann-Pick B patients. Mol. Genet. Metab. 2014, 111, 209–211. [Google Scholar] [CrossRef]
- Foo, J.N.; Liany, H.; Bei, J.X.; Yu, X.Q.; Liu, J.; Au, W.L.; Prakash, K.M.; Tan, L.C.; Tan, E.K. A rare lysosomal enzyme gene SMPD1 variant (p.R591C) associates with Parkinson’s disease. Neurobiol. Aging 2013, 34, 2890.e13-5. [Google Scholar] [CrossRef]
- Alcalay, R.N.; Mallett, V.; Vanderperre, B.; Tavassoly, O.; Dauvilliers, Y.; Wu, R.Y.J.; Ruskey, J.A.; Leblond, C.S.; Ambalavanan, A.; Laurent, S.B.; et al. SMPD1 mutations, activity, and α-synuclein accumulation in Parkinson’s disease. Mov. Disord. 2019, 34, 526–535. [Google Scholar] [CrossRef] [PubMed]
- Steinfeld, R.; Reinhardt, K.; Schreiber, K.; Hillebrand, M.; Kraetzner, R.; Brück, W.; Saftig, P.; Gärtner, J. Cathepsin D deficiency is associated with a human neurodegenerative disorder. Am. J. Hum. Genet. 2006, 78, 988–998. [Google Scholar] [CrossRef]
- Kam, T.I.; Hinkle, J.T.; Dawson, T.M.; Dawson, V.L. Microglia and astrocyte dysfunction in parkinson’s disease. Neurobiol. Dis. 2020, 144, 105028. [Google Scholar] [CrossRef]
- Lee, H.J.; Suk, J.E.; Patrick, C.; Bae, E.J.; Cho, J.H.; Rho, S.; Hwang, D.; Masliah, E.; Lee, S.J. Direct transfer of α-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J. Biol. Chem. 2010, 285, 9262–9272. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Suk, J.E.; Bae, E.J.; Lee, S.J. Clearance and deposition of extracellular α-synuclein aggregates in microglia. Biochem. Biophys. Res. Commun. 2008, 372, 423–428. [Google Scholar] [CrossRef]
- Aflaki, E.; Stubblefield, B.K.; McGlinchey, R.P.; McMahon, B.; Ory, D.S.; Sidransky, E. A characterization of Gaucher iPS-derived astrocytes: Potential implications for Parkinson’s disease. Neurobiol. Dis. 2020, 134, 104647. [Google Scholar] [CrossRef]
- Del Tredici, K.; Ludolph, A.C.; Feldengut, S.; Jacob, C.; Reichmann, H.; Bohl, J.R.; Braak, H. Fabry disease with concomitant Lewy body disease. J. Neuropathol. Exp. Neurol. 2020, 79, 378–392. [Google Scholar] [CrossRef] [PubMed]
- di Domenico, A.; Carola, G.; Calatayud, C.; Pons-Espinal, M.; Muñoz, J.P.; Richaud-Patin, Y.; Fernandez-Carasa, I.; Gut, M.; Faella, A.; Parameswaran, J.; et al. Patient-Specific iPSC-Derived Astrocytes Contribute to Non-Cell-Autonomous Neurodegeneration in Parkinson’s Disease. Stem Cell Rep. 2019, 12, 213–229. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.; DeAndrade, M.P.; Novis, H.S.; Lin, S.; Chang, J.; Lengacher, N.; Tomlinson, J.J.; Tansey, M.G.; LaVoie, M.J. Lysosome and Inflammatory Defects in GBA1-Mutant Astrocytes Are Normalized by LRRK2 Inhibition. Mov. Disord. 2020, 35, 760–773. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.; Kim, J.; Jeong, H.K.; Kim, B.; Jou, I.; Park, M.; Chen, L.; Kang, U.J.; Zhuang, X.; Joe, E. hye PINK1 deficiency attenuates astrocyte proliferation through mitochondrial dysfunction, reduced AKT and increased p38 MAPK activation, and downregulation of EGFR. Glia 2013, 61, 800–812. [Google Scholar] [CrossRef]
- Solano, R.M.; Casarejos, M.J.; Menéndez-Cuervo, J.; Rodriguez-Navarro, J.A.; De Yébenes, J.G.; Mena, M.A. Glial dysfunction in parkin null mice: Effects of aging. J. Neurosci. 2008, 28, 598–611. [Google Scholar] [CrossRef]
- He, C.; Bassik, M.C.; Moresi, V.; Sun, K.; Wei, Y.; Zou, Z.; An, Z.; Loh, J.; Fisher, J.; Sun, Q.; et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 2012, 481, 511–515. [Google Scholar] [CrossRef]
- Clark, L.N.; Ross, B.M.; Wang, Y.; Mejia-Santana, H.; Harris, J.; Louis, E.D.; Cote, L.J.; Andrews, H.; Fahn, S.; Waters, C.; et al. Mutations in the glucocerebrosidase gene are associated with early-onset Parkinson disease. Neurology 2007, 69, 1270–1277. [Google Scholar] [CrossRef]
- Cilia, R.; Tunesi, S.; Marotta, G.; Cereda, E.; Siri, C.; Tesei, S.; Zecchinelli, A.L.; Canesi, M.; Mariani, C.B.; Meucci, N.; et al. Survival and dementia in GBA-associated Parkinson’s disease: The mutation matters. Ann. Neurol. 2016, 80, 662–673. [Google Scholar] [CrossRef]
- Brockmann, K.; Srulijes, K.; Pflederer, S.; Hauser, A.K.; Schulte, C.; Maetzler, W.; Gasser, T.; Berg, D. GBA-associated Parkinson’s disease: Reduced survival and more rapid progression in a prospective longitudinal study. Mov. Disord. 2015, 30, 407–411. [Google Scholar] [CrossRef]
- Mullin, S.; Beavan, M.; Bestwick, J.; McNeill, A.; Proukakis, C.; Cox, T.; Hughes, D.; Mehta, A.; Zetterberg, H.; Schapira, A.H.V. Evolution and clustering of prodromal parkinsonian features in GBA1 carriers. Mov. Disord. 2019, 34, 1365–1373. [Google Scholar] [CrossRef]
- Kinghorn, K.J. Pathological looping in the synucleinopathies: Investigating the link between Parkinson’s disease and Gaucher disease. DMM Dis. Model. Mech. 2011, 4, 713–715. [Google Scholar] [CrossRef]
- Stojkovska, I.; Krainc, D.; Mazzulli, J.R. Molecular mechanisms of α-synuclein and GBA1 in Parkinson’s disease. Cell Tissue Res. 2018, 373, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Mazzulli, J.R.; Xu, Y.H.; Sun, Y.; Knight, A.L.; McLean, P.J.; Caldwell, G.A.; Sidransky, E.; Grabowski, G.A.; Krainc, D. Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 2011, 146, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Cleeter, M.W.J.; Chau, K.-Y.; Gluck, C.; Mehta, A.; Hughes, D.A.; Duchen, M.; Wood, N.W.; Hardy, J.; Mark Cooper, J.; Schapira, A.H. Glucocerebrosidase inhibition causes mitochondrial dysfunction and free radical damage. Neurochem. Int. 2013, 62, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Rocha, E.M.; Smith, G.A.; Park, E.; Cao, H.; Graham, A.R.; Brown, E.; McLean, J.R.; Hayes, M.A.; Beagan, J.; Izen, S.C.; et al. Sustained Systemic Glucocerebrosidase Inhibition Induces Brain α-Synuclein Aggregation, Microglia and Complement C1q Activation in Mice. Antioxid. Redox Signal. 2015, 23, 550–564. [Google Scholar] [CrossRef]
- Rockenstein, E.; Clarke, J.; Viel, C.; Panarello, N.; Treleaven, C.M.; Kim, C.; Spencer, B.; Adame, A.; Park, H.; Dodge, J.C.; et al. Glucocerebrosidase modulates cognitive and motor activities in murine models of Parkinson’s disease. Hum. Mol. Genet. 2016, 25, 2645–2660. [Google Scholar] [CrossRef]
- Xu, Y.H.; Sun, Y.; Ran, H.; Quinn, B.; Witte, D.; Grabowski, G.A. Accumulation and distribution of a-synuclein and ubiquitin in the CNS of Gaucher disease mouse models. Mol. Genet. Metab. 2011, 102, 436–447. [Google Scholar] [CrossRef]
- Schöndorf, D.C.; Aureli, M.; McAllister, F.E.; Hindley, C.J.; Mayer, F.; Schmid, B.; Sardi, S.P.; Valsecchi, M.; Hoffmann, S.; Schwarz, L.K.; et al. IPSC-derived neurons from GBA1-associated Parkinson’s disease patients show autophagic defects and impaired calcium homeostasis. Nat. Commun. 2014, 5, 1–17. [Google Scholar] [CrossRef]
- Velayati, A.; Yu, W.H.; Sidransky, E. The role of glucocerebrosidase mutations in parkinson disease and lewy body disorders. Curr. Neurol. Neurosci. Rep. 2010, 10, 190–198. [Google Scholar] [CrossRef]
- Bendikov-Bar, I.; Ron, I.; Filocamo, M.; Horowitz, M. Characterization of the ERAD process of the L444P mutant glucocerebrosidase variant. Blood Cells Mol. Dis. 2011, 46, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Cullen, V.; Sardi, S.P.; Ng, J.; Xu, Y.H.; Sun, Y.; Tomlinson, J.J.; Kolodziej, P.; Kahn, I.; Saftig, P.; Woulfe, J.; et al. Acid β-glucosidase mutants linked to gaucher disease, parkinson disease, and lewy body dementia alter α-synuclein processing. Ann. Neurol. 2011, 69, 940–953. [Google Scholar] [CrossRef] [PubMed]
- McNeill, A.; Magalhaes, J.; Shen, C.; Chau, K.-Y.Y.; Hughes, D.; Mehta, A.; Foltynie, T.; Cooper, J.M.; Abramov, A.Y.; Gegg, M.; et al. Ambroxol improves lysosomal biochemistry in glucocerebrosidase mutation-linked Parkinson disease cells. Brain 2014, 137, 1481–1495. [Google Scholar] [CrossRef]
- de la Mata, M.; Cotán, D.; Oropesa-Ávila, M.; Garrido-Maraver, J.; Cordero, M.D.; Villanueva Paz, M.; Delgado Pavón, A.; Alcocer-Gómez, E.; de Lavera, I.; Ybot-González, P.; et al. Pharmacological Chaperones and Coenzyme Q10 Treatment Improves Mutant β-Glucocerebrosidase Activity and Mitochondrial Function in Neuronopathic Forms of Gaucher Disease. Sci. Rep. 2015, 5, 10903. [Google Scholar] [CrossRef]
- Collins, L.M.; Drouin-Ouellet, J.; Kuan, W.L.; Cox, T.; Barker, R.A. Dermal fibroblasts from patients with Parkinson’s disease have normal GCase activity and autophagy compared to patients with PD and GBA mutations. F1000Research 2018, 6, 1751. [Google Scholar] [CrossRef] [PubMed]
- Gegg, M.E.; Burke, D.; Heales, S.J.R.; Cooper, J.M.; Hardy, J.; Wood, N.W.; Schapira, A.H.V. Glucocerebrosidase deficiency in substantia nigra of parkinson disease brains. Ann. Neurol. 2012, 72, 455–463. [Google Scholar] [CrossRef]
- Moors, T.E.; Paciotti, S.; Ingrassia, A.; Quadri, M.; Breedveld, G.; Tasegian, A.; Chiasserini, D.; Eusebi, P.; Duran-Pacheco, G.; Kremer, T.; et al. Characterization of Brain Lysosomal Activities in GBA-Related and Sporadic Parkinson’s Disease and Dementia with Lewy Bodies. Mol. Neurobiol. 2019, 56, 1344–1355. [Google Scholar] [CrossRef]
- Chiasserini, D.; Paciotti, S.; Eusebi, P.; Persichetti, E.; Tasegian, A.; Kurzawa-Akanbi, M.; Chinnery, P.F.; Morris, C.M.; Calabresi, P.; Parnetti, L.; et al. Selective loss of glucocerebrosidase activity in sporadic Parkinson’s disease and dementia with Lewy bodies. Mol. Neurodegener. 2015, 10, 15. [Google Scholar] [CrossRef]
- Murphy, K.E.; Gysbers, A.M.; Abbott, S.K.; Tayebi, N.; Kim, W.S.; Sidransky, E.; Cooper, A.; Garner, B.; Halliday, G.M. Reduced glucocerebrosidase is associated with increased α-synuclein in sporadic Parkinson’s disease. Brain 2014, 137, 834–848. [Google Scholar] [CrossRef]
- Rocha, E.M.; Smith, G.A.; Park, E.; Cao, H.; Brown, E.; Hallett, P.; Isacson, O. Progressive decline of glucocerebrosidase in aging and Parkinson’s disease. Ann. Clin. Transl. Neurol. 2015, 2, 433–438. [Google Scholar] [CrossRef]
- van Veen, S.; Martin, S.; Van den Haute, C.; Benoy, V.; Lyons, J.; Vanhoutte, R.; Kahler, J.P.; Decuypere, J.P.; Gelders, G.; Lambie, E.; et al. ATP13A2 deficiency disrupts lysosomal polyamine export. Nature 2020, 578, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Bras, J.; Verloes, A.; Schneider, S.A.; Mole, S.E.; Guerreiro, R.J. Mutation of the parkinsonism gene ATP13A2 causes neuronal ceroid-lipofuscinosis. Hum. Mol. Genet. 2012, 21, 2646–2650. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.H.; Tan, E.K.; Chen, M.L.; Tan, L.C.; Lim, H.Q.; Chen, G.S.; Wu, R.M. Novel ATP13A2 variant associated with Parkinson disease in Taiwan and Singapore. Neurology 2008, 71, 1727–1732. [Google Scholar] [CrossRef] [PubMed]
- Djarmati, A.; Hagenah, J.; Reetz, K.; Winkler, S.; Behrens, M.I.; Pawlack, H.; Lohmann, K.; Ramirez, A.; Tadić, V.; Brüggemann, N.; et al. ATP13A2 variants in early-onset Parkinson’s disease patients and controls. Mov. Disord. 2009, 24, 2104–2111. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.M.; Lin, C.H.; Juan, H.F.; Hu, F.J.; Hsiao, Y.C.; Chang, H.Y.; Chao, C.Y.; Chen, I.C.; Lee, L.C.; Wang, T.W.; et al. ATP13A2 variability in Taiwanese Parkinson’s disease. Am. J. Med. Genet. Part. B 2011, 156, 720–729. [Google Scholar] [CrossRef]
- Di Fonzo, A.; Chien, H.F.; Socal, M.; Giraudo, S.; Tassorelli, C.; Iliceto, G.; Fabbrini, G.; Marconi, R.; Fincati, E.; Abbruzzese, G.; et al. ATP13A2 missense mutations in juvenile parkinsonism and young onset Parkinson disease. Neurology 2007, 68, 1557–1562. [Google Scholar] [CrossRef]
- Vilariño-Güell, C.; Soto, A.I.; Lincoln, S.J.; Yahmed, S.B.; Kefi, M.; Heckman, M.G.; Hulihan, M.M.; Chai, H.; Diehl, N.N.; Amouri, R.; et al. ATP13A2 variability in Parkinson disease. Hum. Mutat. 2009, 30, 406–410. [Google Scholar] [CrossRef]
- Fei, Q.Z.; Cao, L.; Xiao, Q.; Zhang, T.; Zheng, L.; Wang, X.J.; Wang, G.; Zhou, H.Y.; Wang, Y.; Chen, S. Di Lack of association between ATP13A2 Ala746Thr variant and Parkinson’s disease in Han population of mainland China. Neurosci. Lett. 2010, 475, 61–63. [Google Scholar] [CrossRef]
- Mao, X.Y.; Burgunder, J.M.; Zhang, Z.J.; Chang, X.L.; Peng, R.; Burgunder, J.M.; Yang, Y.; Wang, Y.C.; Li, T.; Zhang, Z.J. ATP13A2 G2236A variant is rare in patients with early-onset Parkinson’s disease and familial Parkinson’s disease from mainland China. Park. Relat. Disord. 2010, 16, 235–236. [Google Scholar] [CrossRef]
- Ramonet, D.; Podhajska, A.; Stafa, K.; Sonnay, S.; Trancikova, A.; Tsika, E.; Pletnikova, O.; Troncoso, J.C.; Glauser, L.; Moore, D.J. PARK9-associated ATP13A2 localizes to intracellular acidic vesicles and regulates cation homeostasis and neuronal integrity. Hum. Mol. Genet. 2012, 21, 1725–1743. [Google Scholar] [CrossRef]
- Dehay, B.; Ramirez, A.; Martinez-Vicente, M.; Perier, C.; Canron, M.H.; Doudnikoff, E.; Vital, A.; Vila, M.; Klein, C.; Bezard, E. Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc. Natl. Acad. Sci. USA 2012, 109, 9611–9616. [Google Scholar] [CrossRef] [PubMed]
- Dehay, B.; Martinez-Vicente, M.; Ramirez, A.; Perier, C.; Klein, C.; Vila, M.; Bezard, E. Lysosomal dysfunction in Parkinson disease: ATP13A2 gets into the groove. Autophagy 2012, 8, 1389–1391. [Google Scholar] [CrossRef] [PubMed]
- Usenovic, M.; Tresse, E.; Mazzulli, J.R.; Taylor, J.P.; Krainc, D. Deficiency of ATP13A2 leads to lysosomal dysfunction, α-synuclein accumulation, and neurotoxicity. J. Neurosci. 2012, 32, 4240–4246. [Google Scholar] [CrossRef] [PubMed]
- Kett, L.R.; Stiller, B.; Bernath, M.M.; Tasset, I.; Blesa, J.; Jackson-Lewis, V.; Chan, R.B.; Zhou, B.; Di Paolo, G.; Przedborski, S.; et al. α-Synuclein-independent histopathological and motor deficits in mice lacking the endolysosomal Parkinsonism protein Atp13a2. J. Neurosci. 2015, 35, 5724–5742. [Google Scholar] [CrossRef] [PubMed]
- Schultheis, P.J.; Fleming, S.M.; Clippinger, A.K.; Lewis, J.; Tsunemi, T.; Giasson, B.; Dickson, D.W.; Mazzulli, J.R.; Bardgett, M.E.; Haik, K.L.; et al. Atp13a2-deficient mice exhibit neuronal ceroid lipofuscinosis, limited a-synuclein accumulation and age-dependent sensorimotor deficits. Hum. Mol. Genet. 2013, 22, 2067–2082. [Google Scholar] [CrossRef]
- Dirr, E.R.; Ekhator, O.R.; Blackwood, R.; Holden, J.G.; Masliah, E.; Schultheis, P.J.; Fleming, S.M. Exacerbation of sensorimotor dysfunction in mice deficient in Atp13a2 and overexpressing human wildtype alpha-synuclein. Behav. Brain Res. 2018, 343, 41–49. [Google Scholar] [CrossRef]
- Yang, X.; Xu, Y. Review Article Mutations in the ATP13A2 Gene and Parkinsonism: A Preliminary Review. BioMed Res. Int. 2014, 2014, 371256. [Google Scholar] [CrossRef]
- Jinn, S.; Drolet, R.E.; Cramer, P.E.; Wong, A.H.-K.; Toolan, D.M.; Gretzula, C.A.; Voleti, B.; Vassileva, G.; Disa, J.; Tadin-Strapps, M.; et al. TMEM175 deficiency impairs lysosomal and mitochondrial function and increases α-synuclein aggregation. Proc. Natl. Acad. Sci. USA 2017, 114, 2389–2394. [Google Scholar] [CrossRef]
- Schuchman, E.H.; Desnick, R.J. Types A and B Niemann-Pick disease. Mol. Genet. Metab. 2017, 120, 27–33. [Google Scholar] [CrossRef]
- Klenk, E. Über die Natur der Phosphatide der Milz bei der Niemann-Pickschen Krankheit. 10. Mitteilung über Phosphatide. Hoppe. Seylers. Z. Physiol. Chem. 1934, 229, 151–156. [Google Scholar] [CrossRef]
- Gan-Or, Z.; Ozelius, L.J.; Bar-Shira, A.; Saunders-Pullman, R.; Mirelman, A.; Kornreich, R.; Gana-Weisz, M.; Raymond, D.; Rozenkrantz, L.; Deik, A.; et al. The p.L302P mutation in the lysosomal enzyme gene SMPD1 is a risk factor for Parkinson disease. Neurology 2013, 80, 1606–1610. [Google Scholar] [CrossRef]
- Gan-Or, Z.; Orr-Urtreger, A.; Alcalay, R.N.; Bressman, S.; Giladi, N.; Rouleau, G.A. The emerging role of SMPD1 mutations in Parkinson’s disease: Implications for future studies. Park. Relat. Disord. 2015, 21, 1294–1295. [Google Scholar] [CrossRef] [PubMed]
- Dagan, E.; Schlesinger, I.; Ayoub, M.; Mory, A.; Nassar, M.; Kurolap, A.; Peretz-Aharon, J.; Gershoni-Baruch, R. The contribution of Niemann-Pick SMPD1 mutations to Parkinson disease in Ashkenazi Jews. Parkinsonism Relat. Disord. 2015, 21, 1067–1071. [Google Scholar] [CrossRef]
- Nelson, M.P.; Boutin, M.; Tse, T.E.; Lu, H.; Haley, E.D.; Ouyang, X.; Zhang, J.; Auray-Blais, C.; Shacka, J.J. The lysosomal enzyme alpha-Galactosidase A is deficient in Parkinson’s disease brain in association with the pathologic accumulation of alpha-synuclein. Neurobiol. Dis. 2018, 110, 68–81. [Google Scholar] [CrossRef]
- Chu, Y.; Dodiya, H.; Aebischer, P.; Olanow, C.W.; Kordower, J.H. Alterations in lysosomal and proteasomal markers in Parkinson’s disease: Relationship to alpha-synuclein inclusions. Neurobiol. Dis. 2009, 35, 385–398. [Google Scholar] [CrossRef]
- Sevlever, D.; Jiang, P.; Yen, S.H.C. Cathepsin D is the main lysosomal enzyme involved in the degradation of α-synuclein and generation of its carboxy-terminally truncated species. Biochemistry 2008, 47, 9678–9687. [Google Scholar] [CrossRef]
- Qiao, L.; Hamamichi, S.; Caldwell, K.A.; Caldwell, G.A.; Yacoubian, T.A.; Wilson, S.; Xie, Z.L.; Speake, L.D.; Parks, R.; Crabtree, D.; et al. Lysosomal enzyme cathepsin D protects against alpha-synuclein aggregation and toxicity. Mol. Brain 2008, 1, 17. [Google Scholar] [CrossRef]
- Cullen, V.; Lindfors, M.; Ng, J.; Paetau, A.; Swinton, E.; Kolodziej, P.; Boston, H.; Saftig, P.; Woulfe, J.; Feany, M.B.; et al. Cathepsin D expression level affects alpha-synuclein processing, aggregation, and toxicity in vivo. Mol. Brain 2009, 2, 5. [Google Scholar] [CrossRef]
- Crabtree, D.; Dodson, M.; Ouyang, X.; Boyer-Guittaut, M.; Liang, Q.; Ballestas, M.E.; Fineberg, N.; Zhang, J. Over-expression of an inactive mutant cathepsin D increases endogenous alpha-synuclein and cathepsin B activity in SH-SY5Y cells. J. Neurochem. 2014, 128, 950–961. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.Y.; Gegg, M.; Chau, D.; Schapira, A. Glucocerebrosidase activity, cathepsin D and monomeric α-synuclein interactions in a stem cell derived neuronal model of a PD associated GBA1 mutation. Neurobiol. Dis. 2020, 134, 104620. [Google Scholar] [CrossRef]
- Buechner, S.; De Cristofaro, M.T.R.; Ramat, S.; Borsini, W. Parkinsonism and Anderson Fabry’s disease: A case report. Mov. Disord. 2006, 21, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Wise, A.H.; Yang, A.; Naik, H.; Stauffer, C.; Zeid, N.; Liong, C.; Balwani, M.; Desnick, R.J.; Alcalay, R.N. Parkinson’s disease prevalence in Fabry disease: A survey study. Mol. Genet. Metab. Rep. 2018, 14, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Huebecker, M.; Moloney, E.B.; Van Der Spoel, A.C.; Priestman, D.A.; Isacson, O.; Hallett, P.J.; Platt, F.M. Reduced sphingolipid hydrolase activities, substrate accumulation and ganglioside decline in Parkinson’s disease. Mol. Neurodegener. 2019, 14, 40. [Google Scholar] [CrossRef]
- Nelson, M.P.; Tse, T.E.; O’Quinn, D.B.; Percival, S.M.; Jaimes, E.A.; Warnock, D.G.; Shacka, J.J. Autophagy-lysosome pathway associated neuropathology and axonal degeneration in the brains of alpha-galactosidase A-deficient mice. Acta Neuropathol. Commun. 2014, 2, 20. [Google Scholar] [CrossRef] [PubMed]
- Dehay, B.; Bové, J.; Rodríguez-Muela, N.; Perier, C.; Recasens, A.; Boya, P.; Vila, M. Pathogenic lysosomal depletion in Parkinson’s disease. J. Neurosci. 2010, 30, 12535–12544. [Google Scholar] [CrossRef]
- Inzelberg, R.; Korczyn, A.D. Parkinsonism in adult-onset GM2 gangliosidosis. Mov. Disord. 1994, 9, 375–377. [Google Scholar] [CrossRef]
- Nijssen, P.C.G.; Brusse, E.; Leyten, A.C.M.; Martin, J.J.; Teepen, J.L.J.M.; Roos, R.A.C. Autosomal dominant adult neuronal ceroid lipofuscinosis: Parkinsonism due to both striatal and nigral dysfunction. Mov. Disord. 2002, 17, 482–487. [Google Scholar] [CrossRef]
- Wong, K.; Sidransky, E.; Verma, A.; Mixon, T.; Sandberg, G.D.; Wakefield, L.K.; Morrison, A.; Lwin, A.; Colegial, C.; Allman, J.M.; et al. Neuropathology provides clues to the pathophysiology of Gaucher disease. Mol. Genet. Metab. 2004, 82, 192–207. [Google Scholar] [CrossRef]
- Saito, Y.; Kawashima, A.; Ruberu, N.N.; Fujiwara, H.; Koyama, S.; Sawabe, M.; Arai, T.; Nagura, H.; Yamanouchi, H.; Hasegawa, M.; et al. Accumulation of phosphorylated alpha-synuclein in aging human brain. J. Neuropathol. Exp. Neurol. 2003, 62, 644–654. [Google Scholar] [CrossRef]
- Saito, Y.; Suzuki, K.; Hulette, C.M.; Murayama, S. Aberrant phosphorylation of alpha-synuclein in human Niemann-Pick type C1 disease. J. Neuropathol. Exp. Neurol. 2004, 63, 323–328. [Google Scholar] [CrossRef]
- Suzuki, K.; Iseki, E.; Togo, T.; Yamaguchi, A.; Katsuse, O.; Katsuyama, K.; Kanzaki, S.; Shiozaki, K.; Kawanishi, C.; Yamashita, S.; et al. Neuronal and glial accumulation of α- and β-synucleins in human lipidoses. Acta Neuropathol. 2007, 114, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.R.; Santos, M.B.; Marshall, M.S.; Cantuti-Castelvetri, L.; Lopez-Rosas, A.; Li, G.; Van Breemen, R.; Claycomb, K.I.; Gallea, J.I.; Celej, M.S.; et al. Neuronal inclusions of α-synuclein contribute to the pathogenesis of Krabbe disease. J. Pathol. 2014, 232, 509–521. [Google Scholar] [CrossRef] [PubMed]
- Bae, E.J.; Yang, N.Y.; Lee, C.; Kim, S.; Lee, H.J.; Lee, S.J. Haploinsufficiency of cathepsin D leads to lysosomal dysfunction and promotes cell-to-cell transmission of α-synuclein aggregates. Cell Death Dis. 2015, 6, e1901. [Google Scholar] [CrossRef] [PubMed]
- Nalysnyk, L.; Rotella, P.; Simeone, J.C.; Hamed, A.; Weinreb, N. Gaucher disease epidemiology and natural history: A comprehensive review of the literature. Hematology 2017, 22, 65–73. [Google Scholar] [CrossRef]
- Gegg, M.E.; Sweet, L.; Wang, B.H.; Shihabuddin, L.S.; Sardi, S.P.; Schapira, A.H.V. No evidence for substrate accumulation in Parkinson brains with GBA mutations. Mov. Disord. 2015, 30, 1085–1089. [Google Scholar] [CrossRef] [PubMed]
- Pchelina, S.; Baydakova, G.; Nikolaev, M.; Senkevich, K.; Emelyanov, A.; Kopytova, A.; Miliukhina, I.; Yakimovskii, A.; Timofeeva, A.; Berkovich, O.; et al. Blood lysosphingolipids accumulation in patients with parkinson’s disease with glucocerebrosidase 1 mutations. Mov. Disord. 2018, 33, 1325–1330. [Google Scholar] [CrossRef]
- Blumenreich, S.; Barav, O.B.; Jenkins, B.J.; Futerman, A.H. Lysosomal storage disorders shed light on lysosomal dysfunction in Parkinson’s disease. Int. J. Mol. Sci. 2020, 21, 4966. [Google Scholar] [CrossRef]
- Cookson, M.R. LRRK2 Pathways Leading to Neurodegeneration. Curr. Neurol. Neurosci. Rep. 2015, 15, 42. [Google Scholar] [CrossRef]
- Paisán-Ruíz, C.; Jain, S.; Evans, E.W.; Gilks, W.P.; Simón, J.; Van Der Brug, M.; De Munain, A.L.; Aparicio, S.; Gil, A.M.; Khan, N.; et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 2004, 44, 595–600. [Google Scholar] [CrossRef]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef]
- Consortium, I.P.D.G. Imputation of sequence variants for identification of genetic risks for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet 2011, 377, 641–649. [Google Scholar]
- Manzoni, C. The LRRK2-macroautophagy axis and its relevance to Parkinson’s disease. Biochem. Soc. Trans. 2017, 45, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Orenstein, S.J.; Kuo, S.-H.; Tasset, I.; Arias, E.; Koga, H.; Fernandez-Carasa, I.; Cortes, E.; Honig, L.S.; Dauer, W.; Consiglio, A.; et al. Interplay of LRRK2 with chaperone-mediated autophagy. Nat. Neurosci. 2013, 16, 394–406. [Google Scholar] [CrossRef] [PubMed]
- Kabuta, T.; Setsuie, R.; Mitsui, T.; Kinugawa, A.; Sakurai, M.; Aoki, S.; Uchida, K.; Wada, K. Aberrant molecular properties shared by familial Parkinson’s disease-associated mutant UCH-L1 and carbonyl-modified UCH-L1. Hum. Mol. Genet. 2008, 17, 1482–1496. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Vicente, M. Autophagy in neurodegenerative diseases: From pathogenic dysfunction to therapeutic modulation. Semin. Cell Dev. Biol. 2015, 40, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.P.; Youle, R.J. Targeting mitochondrial dysfunction: Role for PINK1 and Parkin in mitochondrial quality control. Antioxid. Redox Signal. 2011, 14, 1929–1938. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef]
- Kazlauskaite, A.; Martínez-torres, R.J.; Wilkie, S.; Kumar, A.; Peltier, J.; Johnson, C.; Zhang, J.; Hope, A.G.; Peggie, M.; Trost, M.; et al. Binding to serine 65-phosphorylated ubiquitin primes Parkin for optimal PINK 1 -dependent phosphorylation and activation. EMBO Rep. 2015, 16, 939–954. [Google Scholar] [CrossRef]
- Kane, L.A.; Lazarou, M.; Fogel, A.I.; Li, Y.; Yamano, K.; Sarraf, S.A.; Banerjee, S.; Youle, R.J. PINK1 phosphorylates ubiquitin to activate parkin E3 ubiquitin ligase activity. J. Cell Biol. 2014, 205, 143–153. [Google Scholar] [CrossRef]
- Koyano, F.; Okatsu, K.; Kosako, H.; Tamura, Y.; Go, E.; Kimura, M.; Kimura, Y.; Tsuchiya, H.; Yoshihara, H.; Hirokawa, T.; et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 2014, 510, 162–166. [Google Scholar] [CrossRef]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H. Mitochondria in the aetiology and pathogenesis of Parkinson’s disease. Lancet Neurol. 2008, 7, 97–109. [Google Scholar] [CrossRef]
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Chung, K.C. PINK1 positively regulates IL-1β-mediated signaling through Tollip and IRAK1 modulation. J. Neuroinflammation 2012, 9, 271. [Google Scholar] [CrossRef] [PubMed]
- Torres-Odio, S.; Key, J.; Hoepken, H.H.; Canet-Pons, J.; Valek, L.; Roller, B.; Walter, M.; Morales-Gordo, B.; Meierhofer, D.; Harter, P.N.; et al. Progression of pathology in PINK1-deficient mouse brain from splicing via ubiquitination, ER stress, and mitophagy changes to neuroinflammation. J. Neuroinflammation 2017, 14, 154. [Google Scholar] [CrossRef]
- Sun, L.; Shen, R.; Agnihotri, S.K.; Chen, Y.; Huang, Z.; Büeler, H. Lack of PINK1 alters glia innate immune responses and enhances inflammation-induced, nitric oxide-mediated neuron death. Sci. Rep. 2018, 8, 383. [Google Scholar] [CrossRef]
- Mouton-Liger, F.; Rosazza, T.; Sepulveda-Diaz, J.; Ieang, A.; Hassoun, S.M.; Claire, E.; Mangone, G.; Brice, A.; Michel, P.P.; Corvol, J.C.; et al. Parkin deficiency modulates NLRP3 inflammasome activation by attenuating an A20-dependent negative feedback loop. Glia 2018, 66, 1736–1751. [Google Scholar] [CrossRef]
- Fonzo, A.D.; Dekker, M.C.J.; Montagna, P.; Baruzzi, A.; Yonova, E.H.; Guedes, L.C.; Szczerbinska, A.; Zhao, T.; Dubbel-Hulsman, L.O.M.; Wouters, C.H.; et al. FBXO7 mutations cause autosomal recessive, early-onset parkinsonian- pyramidal syndrome. Neurology 2009, 72, 240–245. [Google Scholar] [CrossRef]
- Paisán-Ruiz, C.; Guevara, R.; Federoff, M.; Hanagasi, H.; Sina, F.; Elahi, E.; Schneider, S.A.; Schwingenschuh, P.; Bajaj, N.; Emre, M.; et al. Early-onset L-dopa-responsive Parkinsonism with pyramidal signs due to ATP13A2, PLA2G6, FBXO7 and Spatacsin mutations. Mov. Disord. 2010, 25, 1791–1800. [Google Scholar] [CrossRef]
- Vingill, S.; Brockelt, D.; Lancelin, C.; Tatenhorst, L.; Dontcheva, G.; Preisinger, C.; Schwedhelm-Domeyer, N.; Joseph, S.; Mitkovski, M.; Goebbels, S.; et al. Loss of FBXO 7 (PARK 15) results in reduced proteasome activity and models a parkinsonism-like phenotype in mice. EMBO J. 2016, 35, 2008–2025. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.D.; Lee, J.C.T.; Tan, E.K. Pathophysiological mechanisms linking F-box only protein 7 (FBXO7) and Parkinson’s disease (PD). Mutat. Res. 2018, 778, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.D.; Xie, S.P.; Sathiyamoorthy, S.; Saw, W.T.; Sing, T.Y.; Ng, S.H.; Chua, H.P.H.; Tang, A.M.Y.; Shaffra, F.; Li, Z.; et al. F-box protein 7 mutations promote protein aggregation in mitochondria and inhibit mitophagy. Hum. Mol. Genet. 2015, 24, 6314–6330. [Google Scholar] [CrossRef]
- Burchell, V.S.; Nelson, D.E.; Sanchez-martinez, A.; Delgado, M.; Ivatt, R.M.; Pogson, J.H.; Randle, S.J.; Wray, S.; Lewis, A.; Houlden, H.; et al. The Parkinson’s disease genes Fbxo7 and Parkin interact to mediate mitophagy. Nat. Neurosci 2014, 16, 1–28. [Google Scholar]
- Galvez, T.; Gilleron, J.; Zerial, M.; O’Sullivan, G. A SnapShot: Mammalian Rab proteins in endocytic trafficking. Cell 2012, 151, 234. [Google Scholar] [CrossRef]
- Seaman, M.N.J.; Marcusson, E.G.; Cereghino, J.L.; Emr, S.D. Endosome to Golgi retrieval of the vacuolar protein sorting receptor, Vps10p, requires the function of the VPS29, VPS30, and VPS35 gene products. J. Cell Biol. 1997, 137, 79–92. [Google Scholar] [CrossRef]
- Wieffer, M.; Maritzen, T.; Haucke, V. SnapShot: Endocytic trafficking. Cell 2009, 137. [Google Scholar] [CrossRef]
- Hierro, A.; Rojas, A.L.; Rojas, R.; Murthy, N.; Effantin, G.; Kajava, A.V.; Steven, A.C.; Bonifacino, J.S.; Hurley, J.H. Functional architecture of the retromer cargo-recognition complex. Nature 2007, 449, 1063–1067. [Google Scholar] [CrossRef]
- Cui, Y.; Carosi, J.M.; Yang, Z.; Ariotti, N.; Kerr, M.C.; Parton, R.G.; Sargeant, T.J.; Teasdale, R.D. Retromer has a selective function in cargo sorting via endosome transport carriers. J. Cell Biol. 2019, 218, 615–631. [Google Scholar] [CrossRef] [PubMed]
- Zavodszky, E.; Seaman, M.N.J.; Moreau, K.; Jimenez-Sanchez, M.; Breusegem, S.Y.; Harbour, M.E.; Rubinsztein, D.C. Mutation in VPS35 associated with Parkinson’s disease impairs WASH complex association and inhibits autophagy. Nat. Commun. 2014, 5, 3828. [Google Scholar] [CrossRef]
- Zimprich, A.; Benet-Pagès, A.; Struhal, W.; Graf, E.; Eck, S.H.; Offman, M.N.; Haubenberger, D.; Spielberger, S.; Schulte, E.C.; Lichtner, P.; et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset parkinson disease. Am. J. Hum. Genet. 2011, 89, 168–175. [Google Scholar] [CrossRef]
- Vilariño-Güell, C.; Wider, C.; Ross, O.A.; Dachsel, J.C.; Kachergus, J.M.; Lincoln, S.J.; Soto-Ortolaza, A.I.; Cobb, S.A.; Wilhoite, G.J.; Bacon, J.A.; et al. VPS35 mutations in parkinson disease. Am. J. Hum. Genet. 2011, 89, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.R.; Weissbach, A.; Heldmann, M.; Kasten, M.; Tunc, S.; Sue, C.M.; Svetel, M.; Kostić, V.S.; Segura-Aguilar, J.; Ramirez, A.; et al. Frequency of the D620N mutation in VPS35 in Parkinson disease. Arch. Neurol. 2012, 69, 1360–1364. [Google Scholar] [CrossRef] [PubMed]
- Edvardson, S.; Cinnamon, Y.; Ta-Shma, A.; Shaag, A.; Yim, Y.I.; Zenvirt, S.; Jalas, C.; Lesage, S.; Brice, A.; Taraboulos, A.; et al. A deleterious mutation in DNAJC6 encoding the neuronal-specific clathrin-uncoating Co-chaperone auxilin, is associated with juvenile parkinsonism. PLoS ONE 2012, 7, e36458. [Google Scholar] [CrossRef] [PubMed]
- Vilariño-Güell, C.; Rajput, A.; Milnerwood, A.J.; Shah, B.; Szu-Tu, C.; Trinh, J.; Yu, I.; Encarnacion, M.; Munsie, L.N.; Tapia, L.; et al. DNAJC13 mutations in Parkinson disease. Hum. Mol. Genet. 2014, 23, 1794–1801. [Google Scholar] [CrossRef]
- Wang, S.; Ma, Z.; Xu, X.; Wang, Z.; Sun, L.; Zhou, Y.; Lin, X.; Hong, W.; Wang, T. A role of Rab29 in the integrity of the trans-golgi network and retrograde trafficking of mannose-6-phosphate receptor. PLoS ONE 2014, 9, e96242. [Google Scholar] [CrossRef]
- MacLeod, D.A.; Rhinn, H.; Kuwahara, T.; Zolin, A.; Di Paolo, G.; MacCabe, B.D.; Marder, K.S.; Honig, L.S.; Clark, L.N.; Small, S.A.; et al. RAB7L1 Interacts with LRRK2 to Modify Intraneuronal Protein Sorting and Parkinson’s Disease Risk. Neuron 2013, 77, 425–439. [Google Scholar] [CrossRef]
- Korvatska, O.; Strand, N.S.; Berndt, J.D.; Strovas, T.; Chen, D.H.; Leverenz, J.B.; Kiianitsa, K.; Mata, I.F.; Karakoc, E.; Greenup, J.L.; et al. Altered splicing of ATP6AP2 causes X-linked parkinsonism with spasticity (XPDS). Hum. Mol. Genet. 2013, 22, 3259–3268. [Google Scholar] [CrossRef]
- Krebs, C.E.; Karkheiran, S.; Powell, J.C.; Cao, M.; Makarov, V.; Darvish, H.; Di Paolo, G.; Walker, R.H.; Shahidi, G.A.; Buxbaum, J.D.; et al. The sac1 domain of SYNJ1 identified mutated in a family with early-onset progressive parkinsonism with generalized seizures. Hum. Mutat. 2013, 34, 1200–1207. [Google Scholar] [CrossRef]
- Quadri, M.; Fang, M.; Picillo, M.; Olgiati, S.; Breedveld, G.J.; Graafland, J.; Wu, B.; Xu, F.; Erro, R.; Amboni, M.; et al. Mutation in the SYNJ1 gene associated with autosomal recessive, early-onset parkinsonism. Hum. Mutat. 2013, 34, 1208–1215. [Google Scholar] [CrossRef]
- Williams, E.T.; Chen, X.; Moore, D.J. VPS35, the retromer complex and Parkinson’s disease. J. Parkinsons. Dis. 2017, 7, 219–233. [Google Scholar] [CrossRef]
- Beilina, A.; Bonet-Ponce, L.; Kumaran, R.; Kordich, J.J.; Ishida, M.; Mamais, A.; Kaganovich, A.; Saez-Atienzar, S.; Gershlick, D.C.; Roosen, D.A.; et al. The Parkinson’s Disease Protein LRRK2 Interacts with the GARP Complex to Promote Retrograde Transport to the trans-Golgi Network. Cell Rep. 2020, 31, 107614. [Google Scholar] [CrossRef] [PubMed]
- Beilina, A.; Rudenko, I.N.; Kaganovich, A.; Civiero, L.; Chau, H.; Kalia, S.K.; Kalia, L.V.; Lobbestael, E.; Chia, R.; Ndukwe, K.; et al. Unbiased screen for interactors of leucine-rich repeat kinase 2 supports a common pathway for sporadic and familial Parkinson disease. Proc. Natl. Acad. Sci. USA 2014, 111, 2626–2631. [Google Scholar] [CrossRef]
- Liu, Z.; Bryant, N.; Kumaran, R.; Beilina, A.; Abeliovich, A.; Cookson, M.R.; West, A.B. LRRK2 phosphorylates membrane-bound Rabs and is activated by GTP-bound Rab7L1 to promote recruitment to the trans-Golgi network. Hum. Mol. Genet. 2018, 27, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Trudler, D.; Weinreb, O.; Mandel, S.A.; Youdim, M.B.H.; Frenkel, D. DJ-1 deficiency triggers microglia sensitivity to dopamine toward a pro-inflammatory phenotype that is attenuated by rasagiline. J. Neurochem. 2014, 129, 434–447. [Google Scholar] [CrossRef]
- Barkhuizen, M.; Anderson, D.G.; Grobler, A.F. Advances in GBA-associated Parkinson’s disease - Pathology, presentation and therapies. Neurochem. Int. 2016, 93, 6–25. [Google Scholar] [CrossRef]
- McFarthing, K.; Buff, S.; Rafaloff, G.; Dominey, T.; Wyse, R.K.; Stott, S.R.W. Parkinson’s Disease Drug Therapies in the Clinical Trial Pipeline: 2020. J. Parkinsons. Dis. 2020, 10, 757–774. [Google Scholar] [CrossRef]
- Jung, O.; Patnaik, S.; Marugan, J.; Sidransky, E.; Westbroek, W. Progress and potential of non-inhibitory small molecule chaperones for the treatment of Gaucher disease and its implications for Parkinson disease. Expert Rev. Proteom. 2016, 13, 471–479. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Gene | Protein | Function | LSD | Type | Evidence | Ref. |
|---|---|---|---|---|---|---|
| GBA | Lysosomal acid GCase | Hydrolysis of glucosylceramide into ceramide and glucose | Gaucher disease | Risk factor | +++ | [36,37,38,39,75,76,77] |
| ATP13A2 | ATPase cation transporting 13A2 | Cation transporter and polyamine exporter | Neuronal ceroid lipofuscinosis | Risk factor/AR (Kufor Rakeb) | +++ | [76,78,79,80,81,82,83] |
| TMEM175 | Endosomal/lysosomal potassium channel TMEM175 | Conductance of potassium in lysosomes and endosomes | Risk factor | ++ | [36,37,38,76,84] | |
| SMPD1 | Sphingomyelin phosphodiesterase | Hydrolysis of sphingomyelin into ceramide | Niemann–Pick disease type A/B | Risk factor | ++ | [75,85,86,87,88] |
| SCARB2 | Lysosome membrane protein 2 | Receptor for lysosomal mannose-6-phosphate-independent targeting of GCase | Action myoclonus-renal failure syndrome | Risk factor | ++ | [38,61,74,89,90,91] |
| CTSD | Cathepsin D | Main aspartyl protease of the lysosome | Neuronal ceroid lipofuscinosis | Risk factor | + | [75] |
| GLA | α-galactosidase A | Hydrolysis the terminal alpha-galactosyl moieties from glycolipids and glycoproteins | Fabry disease | Risk factor | + | [92] |
| CTSB | Cathepsin B | Lysosomal cysteine protease | Risk factor | + | [36,37] | |
| GALC | Galacto-cerebrosidase | Hydrolysis of galactose ester bonds from glycolipids | Krabbe disease | Risk factor | + | [36,37] |
| ATP6V0A1 | V-ATPase 116 kDa subunit a1 | Subunit of a vacuolar ATPase that mediates acidification | Risk factor | + | [36] | |
| GUSB | β-glucuronidase | Hydrolysis of glycosaminoglycans | Mucopolysaccharidosis type VII | Risk factor | + | [37] |
| NEU1 | Sialidase-1 | Hydrolysis of the terminal sialic acid residues from sialylated glyco-conjugates | Sialidosis | Risk factor | + | [37] |
| SLC17A5 | Sialin | Free sialic acid exporter from lysosomes | Salla disease | Risk factor | + | [75] |
| ASAH1 | Acid ceramidase | Hydrolysis of sphingolipid ceramides into sphingosine and free fatty acids | Farber Lipogranulomatosis | Risk factor | + | [75] |
| LAMP1 | Lysosome-associated membrane glycoprotein 1 | Trafficking of cholesterol and lipids | Risk factor | + | [76] | |
| ARSA | Arylsulfatase A | Hydrolysis of cerebroside sulfate to cerebroside and sulfate | Metachromatic leukodystrophy | Risk factor/AR | + | [75,93,94] |
| NPC1 | NPC intracellular cholesterol transporter 1 | Intracellular trafficking of cholesterol and lipids | Niemann-Pick type C | Risk factor | + | [95,96] |
| NAGLU | α-N-acetyl glucosaminidase | Hydrolysis of terminal N-acetyl-d-glucosamine residues in N-acetyl-α-d-glucosaminides | San Filippo syndrome B | Risk factor | + | [37,97] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Navarro-Romero, A.; Montpeyó, M.; Martinez-Vicente, M. The Emerging Role of the Lysosome in Parkinson’s Disease. Cells 2020, 9, 2399. https://doi.org/10.3390/cells9112399
Navarro-Romero A, Montpeyó M, Martinez-Vicente M. The Emerging Role of the Lysosome in Parkinson’s Disease. Cells. 2020; 9(11):2399. https://doi.org/10.3390/cells9112399
Chicago/Turabian StyleNavarro-Romero, Alba, Marta Montpeyó, and Marta Martinez-Vicente. 2020. "The Emerging Role of the Lysosome in Parkinson’s Disease" Cells 9, no. 11: 2399. https://doi.org/10.3390/cells9112399
APA StyleNavarro-Romero, A., Montpeyó, M., & Martinez-Vicente, M. (2020). The Emerging Role of the Lysosome in Parkinson’s Disease. Cells, 9(11), 2399. https://doi.org/10.3390/cells9112399