Metabolic Profiling of CSF from People Suffering from Sporadic and LRRK2 Parkinson’s Disease: A Pilot Study


 ,
, 

Abstract
1. Introduction
2. Materials and Methods
2.1. CSF Samples
2.2. 1H NMR Analysis
2.3. Targeted Mass Spectrometry Analysis
2.4. Bile-Acid Analysis
2.5. Statistical Analysis
2.5.1. Univariate Data Analysis
2.5.2. Multivariate Data Analysis
2.5.3. Machine Learning-Based Regression Analysis
2.5.4. Metabolite Pathway Enrichment Analysis
3. Results
3.1. Statistical and Metabolite Pathway Enrichment Analysis
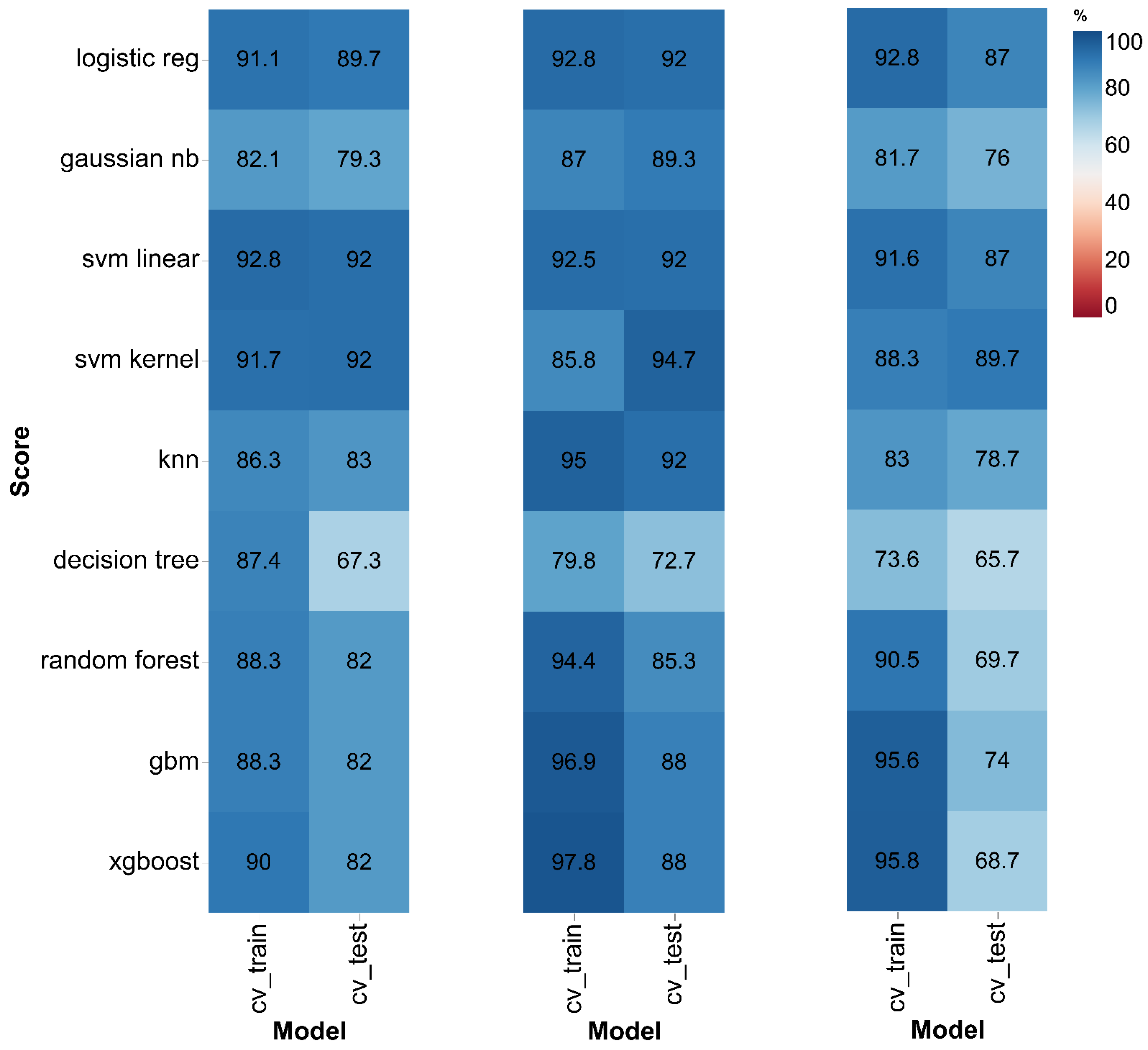
3.2. Machine Learning-Based Classification Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Availability
References
- Graham, S.F.; Rey, N.L.; Ugur, Z.; Yilmaz, A.; Sherman, E.; Maddens, M.; Bahado-Singh, R.O.; Becker, K.; Schulz, E.; Meyerdirk, L.K.; et al. Metabolomic profiling of bile acids in an experimental model of prodromal parkinson’s disease. Metabolites 2018, 8, 71. [Google Scholar] [CrossRef] [PubMed]
- de Lau, L.M.L.; Breteler, M.M.B. Epidemiology of parkinson’s disease. Lancet Neurol. 2006, 5, 525–535. [Google Scholar] [CrossRef]
- Klein, C.; Westenberger, A. Genetics of parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a008888. [Google Scholar] [CrossRef]
- Li, J.-Q.; Tan, L.; Yu, J.-T. The role of the lrrk2 gene in parkinsonism. Mol. Neurodegener. 2014, 9, 47. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.-H.; Cheng, M.-L.; Tang, H.-Y.; Huang, C.-Y.; Wu, Y.-R.; Chen, C.-M. Alternations of Metabolic Profile and Kynurenine Metabolism in the Plasma of Parkinson’s Disease. Mol. Neurobiol. 2018, 55, 6319–6328. [Google Scholar] [CrossRef]
- Jenner, P. Molecular mechanisms of l-dopa-induced dyskinesia. Nat. Rev. Neurosci. 2008, 9, 665–677. [Google Scholar] [CrossRef]
- Trezzi, J.P.; Galozzi, S.; Jaeger, C.; Barkovits, K.; Brockmann, K.; Maetzler, W.; Berg, D.; Marcus, K.; Betsou, F.; Hiller, K.; et al. Distinct metabolomic signature in cerebrospinal fluid in early parkinson’s disease. Mov. Disord Off. J. Mov. Disord. Soc. 2017, 32, 1401–1408. [Google Scholar] [CrossRef]
- Goldman, J.G.; Postuma, R. Premotor and nonmotor features of parkinson’s disease. Curr. Opin. Neurol. 2014, 27, 434–441. [Google Scholar] [CrossRef]
- Patti, G.J.; Yanes, O.; Siuzdak, G. Innovation: Metabolomics: The apogee of the omics trilogy. Nat. Rev. Mol. Cell Biol. 2012, 13, 263–269. [Google Scholar] [CrossRef]
- Hatano, T.; Saiki, S.; Okuzumi, A.; Mohney, R.P.; Hattori, N. Identification of novel biomarkers for parkinson’s disease by metabolomic technologies. J. Neurol. Neurosurg. Psychiatry 2016, 87, 295–301. [Google Scholar] [CrossRef]
- Graham, S.F.; Kumar, P.; Bahado-Singh, R.O.; Robinson, A.; Mann, D.; Green, B.D. Novel metabolite biomarkers of huntington’s disease as detected by high-resolution mass spectrometry. J. Proteome Res. 2016, 15, 1592–1601. [Google Scholar] [CrossRef] [PubMed]
- Glenn, T.C.; Hirt, D.; Mendez, G.; McArthur, D.L.; Sturtevant, R.; Wolahan, S.; Fazlollahi, F.; Ordon, M.; Bilgin-Freiert, A.; Ellingson, B.; et al. Metabolomic analysis of cerebral spinal fluid from patients with severe brain injury. Acta Neurochir. Suppl. 2013, 118, 115–119. [Google Scholar]
- Nagata, Y.; Hirayama, A.; Ikeda, S.; Shirahata, A.; Shoji, F.; Maruyama, M.; Kayano, M.; Bundo, M.; Hattori, K.; Yoshida, S.; et al. Comparative analysis of cerebrospinal fluid metabolites in alzheimer’s disease and idiopathic normal pressure hydrocephalus in a japanese cohort. Biomark. Res. 2018, 6, 5. [Google Scholar] [CrossRef] [PubMed]
- Amara, A.W.; Chahine, L.M.; Caspell-Garcia, C.; Long, J.D.; Coffey, C.; Högl, B.; Videnovic, A.; Iranzo, A.; Mayer, G.; Foldvary-Schaefer, N.; et al. Longitudinal assessment of excessive daytime sleepiness in early parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 2017, 88, 653–662. [Google Scholar] [CrossRef]
- Ravanbakhsh, S.; Liu, P.; Bjorndahl, T.C.; Mandal, R.; Grant, J.R.; Wilson, M.; Eisner, R.; Sinelnikov, I.; Hu, X.; Luchinat, C.; et al. Accurate, fully-automated nmr spectral profiling for metabolomics. PLoS ONE 2015, 10, e0124219. [Google Scholar] [CrossRef] [PubMed]
- Marksteiner, J.; Blasko, I.; Kemmler, G.; Koal, T.; Humpel, C. Bile acid quantification of 20 plasma metabolites identifies lithocholic acid as a putative biomarker in alzheimer’s disease. Metab. Off. J. Metab. Soc. 2018, 14, 1. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Mandal, R.; Sinelnikov, I.V.; Broadhurst, D.; Wishart, D.S. Metaboanalyst 2.0--a comprehensive server for metabolomic data analysis. Nucleic Acids Res. 2012, 40, W127–W133. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Psychogios, N.; Young, N.; Wishart, D.S. Metaboanalyst: A web server for metabolomic data analysis and interpretation. Nucleic Acids Res. 2009, 37, W652–W660. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Sinelnikov, I.V.; Han, B.; Wishart, D.S. Metaboanalyst 3.0--making metabolomics more meaningful. Nucleic Acids Res. 2015, 43, W251–W257. [Google Scholar] [CrossRef]
- Richhariya, B.; Tanveer, M.; Rashid, A.H. Diagnosis of alzheimer’s disease using universum support vector machine based recursive feature elimination (usvm-rfe). Biomed. Signal Process. Control 2020, 59, 101903. [Google Scholar] [CrossRef]
- Lin, X.; Yang, F.; Zhou, L.; Yin, P.; Kong, H.; Xing, W.; Lu, X.; Jia, L.; Wang, Q.; Xu, G. A support vector machine-recursive feature elimination feature selection method based on artificial contrast variables and mutual information. J. Chromatogr. B 2012, 910, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Guyon, I.; Weston, J.; Barnhill, S.; Vapnik, V. Gene selection for cancer classification using support vector machines. Mach. Learn. 2002, 46, 389–422. [Google Scholar] [CrossRef]
- Chong, J.; Wishart, D.S.; Xia, J. Using metaboanalyst 4.0 for comprehensive and integrative metabolomics data analysis. Curr. Protoc. Bioinform. 2019, 68, e86. [Google Scholar] [CrossRef] [PubMed]
- Stoessel, D.; Schulte, C.; Teixeira dos Santos, M.C.; Scheller, D.; Rebollo-Mesa, I.; Deuschle, C.; Walther, D.; Schauer, N.; Berg, D.; Nogueira da Costa, A.; et al. Promising metabolite profiles in the plasma and csf of early clinical parkinson’s disease. Front. Aging Neurosci. 2018, 10, 51. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Lewis, M.J.; Morrissey, J.A.; Flegel, M.D.; Jeroncic, K.; Xiong, Y.; Cheng, D.; Eisner, R.; Gautam, B.; Tzur, D.; et al. The human cerebrospinal fluid metabolome. J. Chromatography. Banalytical Technol. Biomed. Life Sci. 2008, 871, 164–173. [Google Scholar] [CrossRef]
- Goldstein, D.S.; Holmes, C.; Sharabi, Y. Cerebrospinal fluid biomarkers of central catecholamine deficiency in parkinson’s disease and other synucleinopathies. Brain A J. Neurol. 2012, 135, 1900–1913. [Google Scholar] [CrossRef]
- Hong, Z.; Shi, M.; Chung, K.A.; Quinn, J.F.; Peskind, E.R.; Galasko, D.; Jankovic, J.; Zabetian, C.P.; Leverenz, J.B.; Baird, G.; et al. Dj-1 and alpha-synuclein in human cerebrospinal fluid as biomarkers of parkinson’s disease. Brain A J. Neurol. 2010, 133, 713–726. [Google Scholar] [CrossRef]
- Mondello, S.; Constantinescu, R.; Zetterberg, H.; Andreasson, U.; Holmberg, B.; Jeromin, A. Csf α-synuclein and uch-l1 levels in parkinson’s disease and atypical parkinsonian disorders. Parkinsonism Relat. Disord. 2014, 20, 382–387. [Google Scholar] [CrossRef]
- Saiki, S.; Hatano, T.; Fujimaki, M.; Ishikawa, K.I.; Mori, A.; Oji, Y.; Okuzumi, A. Decreased long-chain acylcarnitines from insufficient β-oxidation as potential early diagnostic markers for parkinson’s disease. Sci. Rep. 2017, 7, 7328. [Google Scholar] [CrossRef]
- Hajri, T.; Abumrad, N.A. Fatty acid transport across membranes: Relevance to nutrition and metabolic pathology. Annu. Rev. Nutr. 2002, 22, 383–415. [Google Scholar] [CrossRef] [PubMed]
- Jones, L.L.; McDonald, D.A.; Borum, P.R. Acylcarnitines: Role in brain. Prog. Lipid Res. 2010, 49, 61–75. [Google Scholar] [CrossRef] [PubMed]
- Mergenthaler, P.; Lindauer, U.; Dienel, G.A.; Meisel, A. Sugar for the brain: The role of glucose in physiological and pathological brain function. Trends Neurosci. 2013, 36, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Burté, F.; Houghton, D.; Lowes, H.; Pyle, A.; Nesbitt, S.; Yarnall, A.; Yu-Wai-Man, P.; Burn, D.J.; Santibanez-Koref, M.; Hudson, G. Metabolic profiling of parkinson’s disease and mild cognitive impairment. Mov. Disord. Off. J. Mov. Disord. Soc. 2017, 32, 927–932. [Google Scholar]
- Ishmayana, S.; Kennedy, U.J.; Learmonth, R.P. Preliminary evidence of inositol supplementation effect on cell growth, viability and plasma membrane fluidity of the yeast saccharomyces cerevisiae. Procedia Chem. 2015, 17, 162–169. [Google Scholar] [CrossRef][Green Version]
- Lieu, T.; Jayaweera, G.; Bunnett, N.W. Gpba: A gpcr for bile acids and an emerging therapeutic target for disorders of digestion and sensation. Br J. Pharm. 2014, 171, 1156–1166. [Google Scholar] [CrossRef]
- Liu, Y.; Rong, Z.; Xiang, D.; Zhang, C.; Liu, D. Detection technologies and metabolic profiling of bile acids: A comprehensive review. Lipids Health Dis 2018, 17, 121. [Google Scholar] [CrossRef] [PubMed]
- Quinn, M.; McMillin, M.; Galindo, C.; Frampton, G.; Pae, H.Y.; DeMorrow, S. Bile acids permeabilize the blood brain barrier after bile duct ligation in rats via rac1-dependent mechanisms. Dig. Liver Dis. 2014, 46, 527–534. [Google Scholar] [CrossRef]
- Bron, B.; Waldram, R.; Silk, D.B.; Williams, R. Serum, cerebrospinal fluid, and brain levels of bile acids in patients with fulminant hepatic failure. Gut 1977, 18, 692–696. [Google Scholar] [CrossRef]
- Graham, S.F.; Rey, N.L.; Yilmaz, A. Biochemical profiling of the brain and blood metabolome in a mouse model of prodromal parkinson’s disease reveals distinct metabolic profiles. J. Proteome Res. 2018, 17, 2460–2469. [Google Scholar] [CrossRef]
- Bustos, B.I.; Krainc, D.; Lubbe, S.J. Whole-exome analysis in parkinson’s disease reveals a high burden of ultra rare variants in early onset cases. bioRxiv 2006. [Google Scholar] [CrossRef]
- Ruotsalainen, M.; Ahtee, L. Intrastriatal taurine increases striatal extracellular dopamine in a tetrodotoxin-sensitive manner in rats. Neurosci. Lett. 1996, 212, 175–178. [Google Scholar] [CrossRef]
- Che, Y.; Hou, L.; Sun, F.; Zhang, C.; Liu, X.; Piao, F.; Zhang, D.; Li, H.; Wang, Q. Taurine protects dopaminergic neurons in a mouse parkinson’s disease model through inhibition of microglial m1 polarization. Cell Death Dis. 2018, 9, 435. [Google Scholar] [CrossRef]
- Buervenich, S.; Sydow, O.; Carmine, A.; Zhang, Z.; Anvret, M.; Olson, L. Alcohol dehydrogenase alleles in parkinson’s disease. Mov. Disord. 2000, 15, 813–818. [Google Scholar] [CrossRef]
- Tan, E.K.; Nagamitsu, S.; Matsuura, T.; Khajavi, M.; Jankovic, J.; Ondo, W.; Ashizawa, T. Alcohol dehydrogenase polymorphism and parkinson’s disease. Neurosci. Lett. 2001, 305, 70–72. [Google Scholar] [CrossRef]
- Scheperjans, F.; Aho, V.; Pereira, P.A.; Koskinen, K.; Paulin, L.; Pekkonen, E.; Haapaniemi, E.; Kaakkola, S.; Eerola-Rautio, J.; Pohja, M.; et al. Gut microbiota are related to parkinson’s disease and clinical phenotype. Mov. Disord. Off. J. Mov. Disord. Soc. 2015, 30, 350–358. [Google Scholar] [CrossRef]
- De Craene, J.O.; Bertazzi, D.L.; Bär, S.; Friant, S. Phosphoinositides, major actors in membrane trafficking and lipid signaling pathways. Int J Mol Sci 2017, 18, 634. [Google Scholar] [CrossRef] [PubMed]
- Michell, R.H.; Kirk, C.J.; Jones, L.M.; Downes, C.P.; Creba, J.A. The stimulation of inositol lipid metabolism that accompanies calcium mobilization in stimulated cells: Defined characteristics and unanswered questions. Philos. Trans. R. Soc. London. Ser. Bbiological Sci. 1981, 296, 123–138. [Google Scholar]
- Luo, H.R.; Huang, Y.E.; Chen, J.C.; Saiardi, A.; Iijima, M.; Ye, K.; Huang, Y.; Nagata, E.; Devreotes, P.; Snyder, S.H. Inositol pyrophosphates mediate chemotaxis in dictyostelium via pleckstrin homology domain-ptdins(3,4,5)p3 interactions. Cell 2003, 114, 559–572. [Google Scholar] [CrossRef]
- Berridge, M.J. Regulation of ion channels by inositol trisphosphate and diacylglycerol. J. Exp. Biol. 1986, 124, 323–335. [Google Scholar]
- Prasad, N.K.; Decker, S.J. Sh2-containing 5’-inositol phosphatase, ship2, regulates cytoskeleton organization and ligand-dependent down-regulation of the epidermal growth factor receptor. J. Biol. Chem. 2005, 280, 13129–13136. [Google Scholar] [CrossRef]
- Salinas-Mondragon, R.E.; Kajla, J.D.; Perera, I.Y.; Brown, C.S.; Sederoff, H.W. Role of inositol 1,4,5-triphosphate signalling in gravitropic and phototropic gene expression. Plantcell Environ. 2010, 33, 2041–2055. [Google Scholar] [CrossRef] [PubMed]
- Bartke, N.; Hannun, Y.A. Bioactive sphingolipids: Metabolism and function. J. Lipid Res. 2009, 50, S91–S96. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| sPD Control | sPD | LRRK2 Control | LRRK2 PD | p-Value | |
|---|---|---|---|---|---|
| n | 20 | 20 | 20 | 20 | |
| Age, mean (SD) | 57.65 (9.56) | 58.85 (8.95) | 60.05 (9.39) | 59.43 (9.11) | 0.45 a |
| Gender | |||||
| Male | 10 | 11 | 10 | 10 | 0.26 b |
| Female | 10 | 9 | 10 | 10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yilmaz, A.; Ugur, Z.; Ustun, I.; Akyol, S.; Bahado-Singh, R.O.; Maddens, M.; Aasly, J.O.; Graham, S.F. Metabolic Profiling of CSF from People Suffering from Sporadic and LRRK2 Parkinson’s Disease: A Pilot Study. Cells 2020, 9, 2394. https://doi.org/10.3390/cells9112394
Yilmaz A, Ugur Z, Ustun I, Akyol S, Bahado-Singh RO, Maddens M, Aasly JO, Graham SF. Metabolic Profiling of CSF from People Suffering from Sporadic and LRRK2 Parkinson’s Disease: A Pilot Study. Cells. 2020; 9(11):2394. https://doi.org/10.3390/cells9112394
Chicago/Turabian StyleYilmaz, Ali, Zafer Ugur, Ilyas Ustun, Sumeyya Akyol, Ray O. Bahado-Singh, Michael Maddens, Jan O. Aasly, and Stewart F. Graham. 2020. "Metabolic Profiling of CSF from People Suffering from Sporadic and LRRK2 Parkinson’s Disease: A Pilot Study" Cells 9, no. 11: 2394. https://doi.org/10.3390/cells9112394
APA StyleYilmaz, A., Ugur, Z., Ustun, I., Akyol, S., Bahado-Singh, R. O., Maddens, M., Aasly, J. O., & Graham, S. F. (2020). Metabolic Profiling of CSF from People Suffering from Sporadic and LRRK2 Parkinson’s Disease: A Pilot Study. Cells, 9(11), 2394. https://doi.org/10.3390/cells9112394