Pathways for Sensing and Responding to Hydrogen Peroxide at the Endoplasmic Reticulum


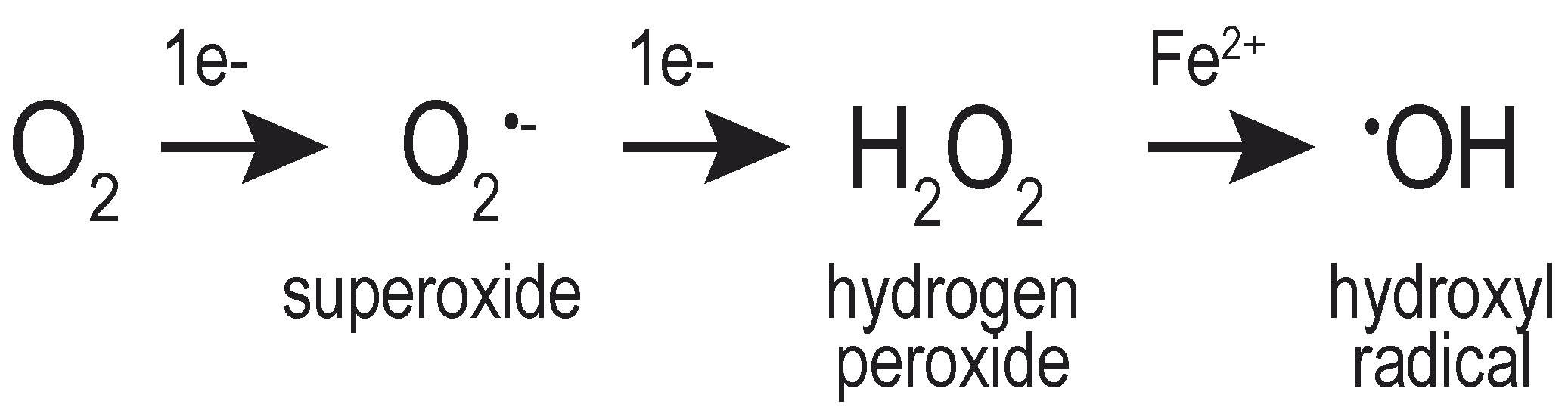{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Properties of H2O2
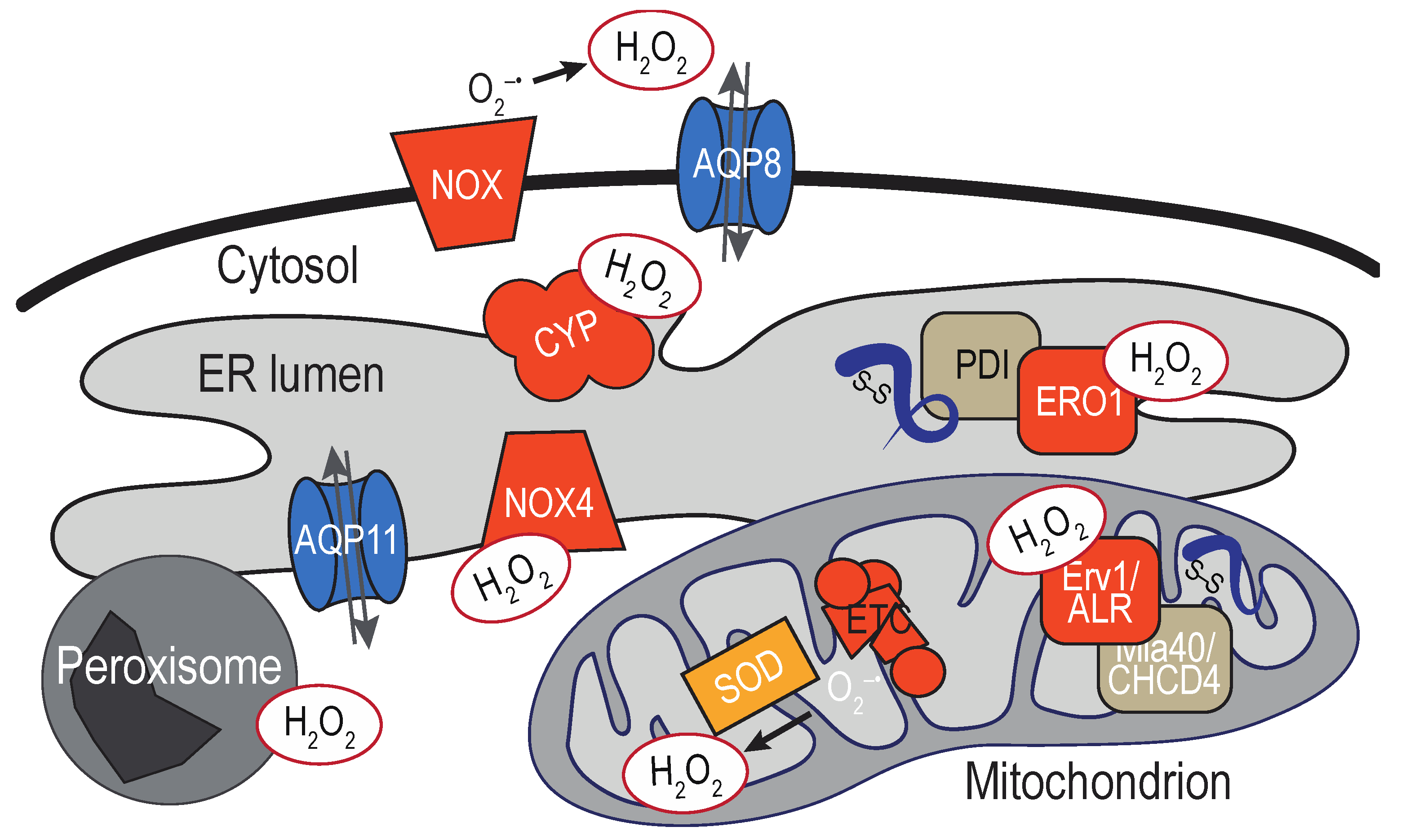
3. Sources of ER-localized H2O2
3.1. ER-Localized Enzymes
3.2. Mitochondria-Associated Membranes
3.3. Additional Proximal Sources of ER H2O2
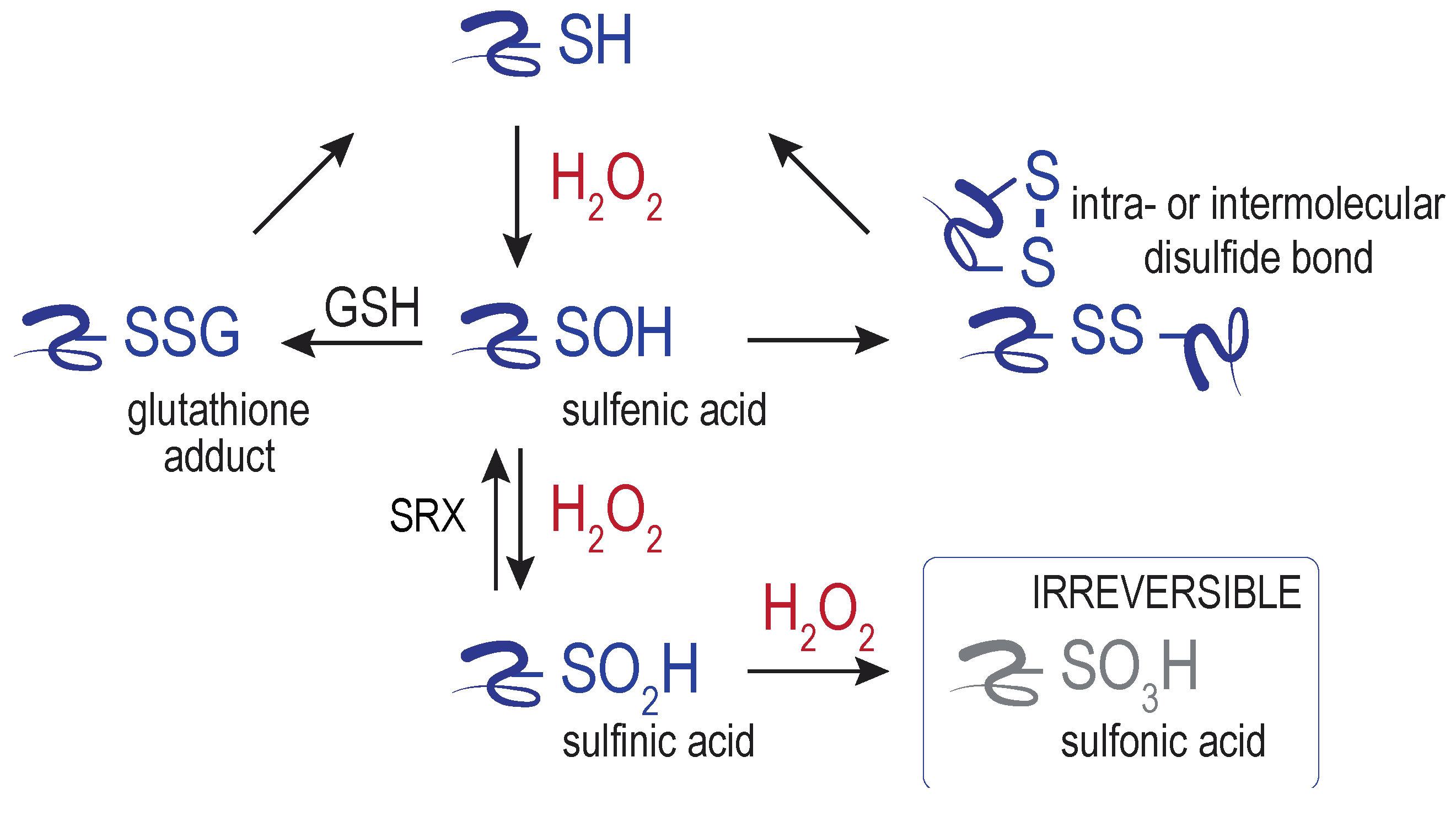
4. ER Targets of H2O2
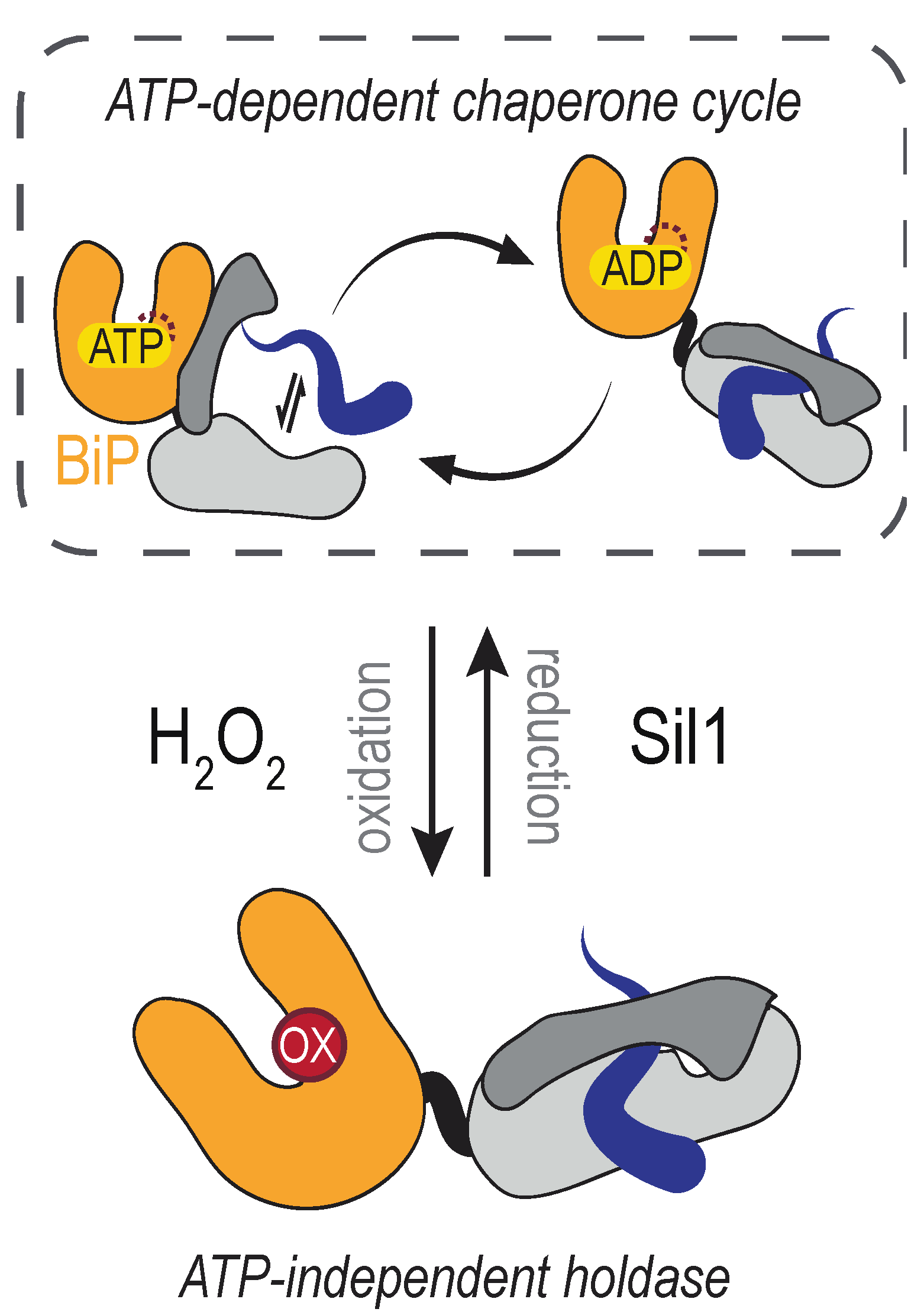
4.1. The Molecular Chaperone BiP
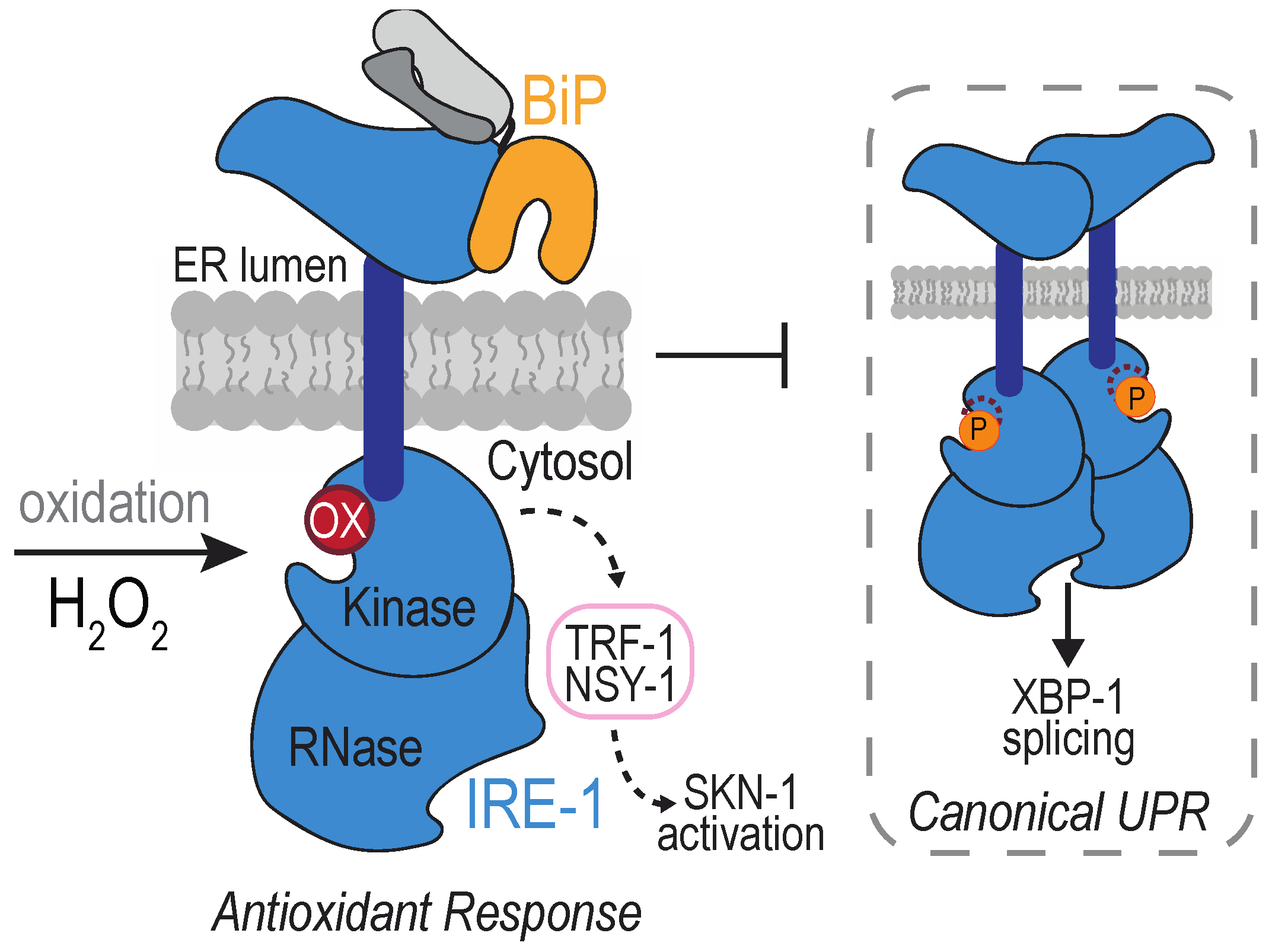
4.2. The Transmembrane Protein Kinase IRE1
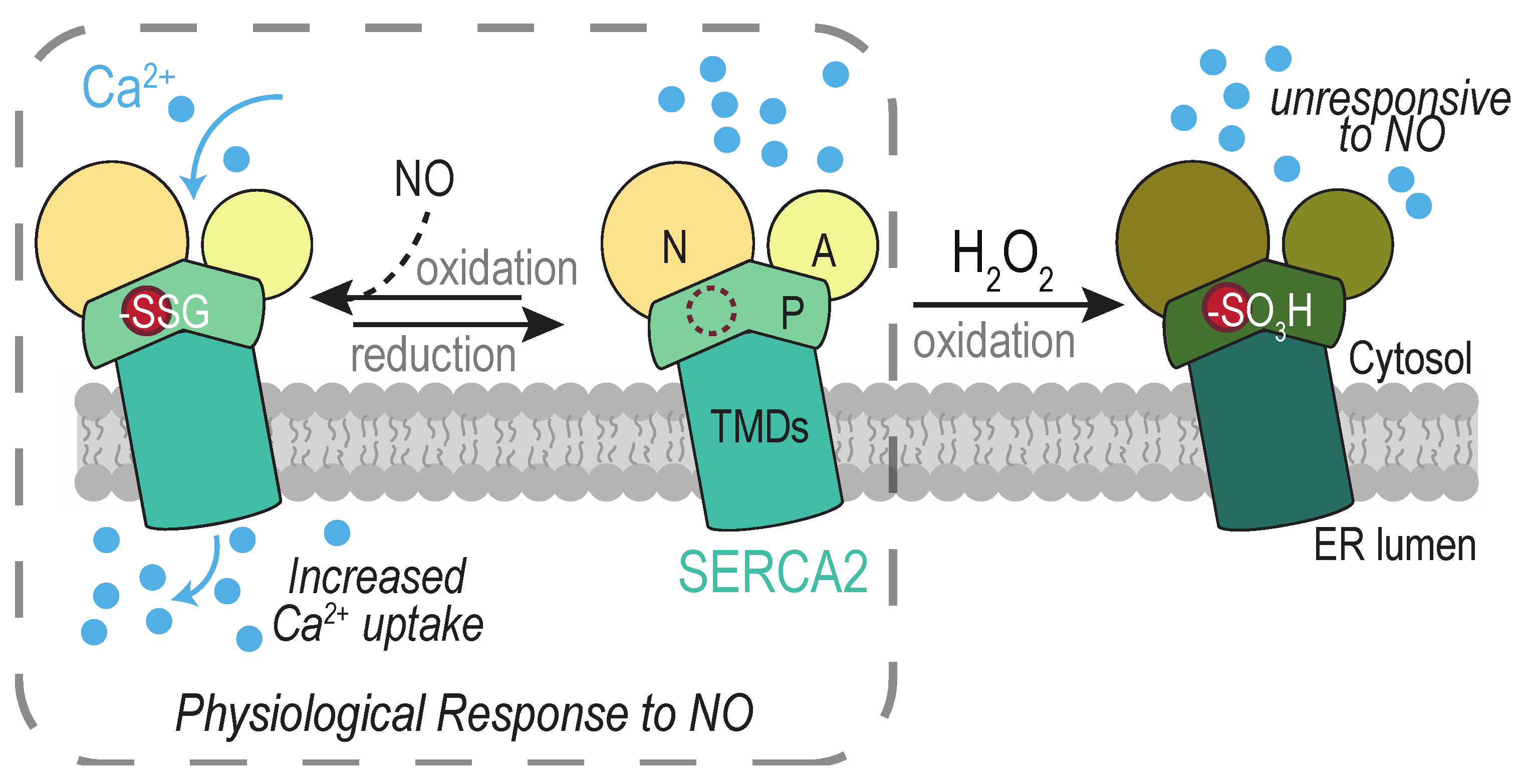
4.3. The SERCA2 Calcium Pump
4.3.1. SERCA2 Cys674
4.3.2. SERCA2 Cys875 and Cys887
5. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef]
- Reczek, C.R.; Chandel, N.S. ROS-dependent signal transduction. Curr. Opin. Cell Biol. 2015, 33, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C. Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol. 2008, 4, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C. The biological chemistry of hydrogen peroxide. Methods Enzymol. 2013, 528, 3–25. [Google Scholar] [PubMed]
- Imlay, J.A. Pathways of oxidative damage. Annu. Rev. Microbiol. 2003, 57, 395–418. [Google Scholar] [CrossRef] [PubMed]
- Jomova, K.; Valko, M. Advances in metal-induced oxidative stress and human disease. Toxicology 2011, 283, 65–87. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Branicky, R.; Noë, A.; Hekimi, S. Superoxide dismutases: Dual roles in controlling ROS damage and regulating ROS signaling. J. Cell Biol. 2018, 217, 1915–1928. [Google Scholar] [CrossRef]
- Brown, D. The Discovery of Water Channels (Aquaporins). Ann. Nutr. Metab. 2017, 70, 37–42. [Google Scholar] [CrossRef]
- Carbrey, J.M.; Agre, P. Discovery of the aquaporins and development of the field. In Aquaporins; Springer: Berlin, Germany, 2009; pp. 3–28. [Google Scholar]
- Bienert, G.P.; Chaumont, F. Aquaporin-facilitated transmembrane diffusion of hydrogen peroxide. Biochim. Biophys. Acta 2014, 1840, 1596–1604. [Google Scholar] [CrossRef] [PubMed]
- Bestetti, S.; Galli, M.; Sorrentino, I.; Pinton, P.; Rimessi, A.; Sitia, R.; Iria, M.F. Human aquaporin-11 guarantees efficient transport of H2O2 across the endoplasmic reticulum membrane. Redox Biol. 2020, 28, 101326. [Google Scholar] [CrossRef] [PubMed]
- Morishita, Y.; Matsuzaki, T.; Hara-chikuma, M.; Andoo, A.; Shimono, M.; Matsuki, A.; Kobayashi, K.; Ikeda, M.; Yamamoto, T.; Verkman, A.; et al. Disruption of aquaporin-11 produces polycystic kidneys following vacuolization of the proximal tubule. Mol. Cell. Biol. 2005, 25, 7770–7779. [Google Scholar] [CrossRef]
- Bertolotti, M.; Bestetti, S.; García-Manteiga, J.M.; Medrano-Fernandez, I.; Dal Mas, A.; Malosio, M.L.; Sitia, R. Tyrosine kinase signal modulation: A matter of H2O2 membrane permeability. Antioxid. Redox Signal 2013, 19, 1447–1451. [Google Scholar] [CrossRef] [PubMed]
- Ozu, M.; Galizia, L.; Acuña, C.; Amodeo, G. Aquaporins: More Than Functional Monomers in a Tetrameric Arrangement. Cells 2018, 7, 209. [Google Scholar] [CrossRef]
- Kreida, S.; Törnroth-Horsefield, S. Structural insights into aquaporin selectivity and regulation. Curr. Opin. Struct. Biol. 2015, 33, 126–134. [Google Scholar] [CrossRef]
- Bestetti, S.; Medraño-Fernandez, I.; Galli, M.; Bienert, G.P.; Musco, G.; Orsi, A.; Rubartelli, A.; Sitia, R. A persulfidation-based mechanism controls aquaporin-8 conductance. Sci. Adv. 2018, 4, eaar5770. [Google Scholar] [CrossRef]
- Biteau, B.; Labarre, J.; Toledano, M.B. ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae sulphiredoxin. Nature 2003, 425, 980–984. [Google Scholar] [CrossRef]
- Woo, H.A.; Jeong, W.; Chang, T.S.; Park, K.J.; Park, S.J.; Yang, J.S.; Rhee, S.G. Reduction of cysteine sulfinic acid by sulfiredoxin is specific to 2-cys peroxiredoxins. J. Biol. Chem. 2005, 280, 3125–3128. [Google Scholar] [CrossRef]
- Akter, S.; Fu, L.; Jung, Y.; Conte, M.L.; Lawson, J.R.; Lowther, W.T.; Sun, R.; Liu, K.; Yang, J.; Carroll, K.S. Chemical proteomics reveals new targets of cysteine sulfinic acid reductase. Nat. Chem. Biol. 2018, 14, 995–1004. [Google Scholar] [CrossRef]
- Finkel, T. From sulfenylation to sulfhydration: What a thiolate needs to tolerate. Sci. Signal. 2012, 5, pe10. [Google Scholar] [CrossRef] [PubMed]
- Zivanovic, J.; Kouroussis, E.; Kohl, J.B.; Adhikari, B.; Bursac, B.; Schott-Roux, S.; Petrovic, D.; Miljkovic, j.; Thomas-Lopez, D.; Jung, Y.; et al. Selective Persulfide Detection Reveals Evolutionarily Conserved Antiaging Effects of S-Sulfhydration. Cell Metab. 2019, 30, 1152–1170. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.P. Radical-free biology of oxidative stress. Am. J. Physiol. Cell Physiol. 2008, 295, C849–C868. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, C.E.; Carroll, K.S. Orchestrating redox signaling networks through regulatory cysteine switches. ACS Chem. Biol. 2010, 5, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Roos, G.; Messens, J. Protein sulfenic acid formation: From cellular damage to redox regulation. Free Radic. Biol. Med. 2011, 51, 314–326. [Google Scholar] [CrossRef] [PubMed]
- Klomsiri, C.; Karplus, P.A.; Poole, L.B. Cysteine-based redox switches in enzymes. Antioxid. Redox Signal. 2011, 14, 1065–1077. [Google Scholar] [CrossRef]
- Stone, J.R. An assessment of proposed mechanisms for sensing hydrogen peroxide in mammalian systems. Arch. Biochem. Biophys. 2004, 422, 119–124. [Google Scholar] [CrossRef]
- Zeida, A.; Trujillo, M.; Ferrer-Sueta, G.; Denicola, A.; Estrin, D.A.; Radi, R. Catalysis of Peroxide Reduction by Fast Reacting Protein Thiols. Chem. Rev. 2019, 119, 10829–10855. [Google Scholar] [CrossRef]
- Kalyanaraman, B.; Darley-Usmar, V.; Davies, K.J.; Dennery, P.A.; Forman, H.J.; Grisham, M.B.; Mann, G.E.; Moore, K.; Roberts, L.J., II; Ischiropoulos, H. Measuring reactive oxygen and nitrogen species with fluorescent probes: Challenges and limitations. Free Radic. Biol. Med. 2012, 52, 1–6. [Google Scholar] [CrossRef]
- Winterbourn, C.C. The challenges of using fluorescent probes to detect and quantify specific reactive oxygen species in living cells. Biochim. Biophys. Acta 2014, 1840, 730–738. [Google Scholar] [CrossRef]
- Zhao, B.; Summers, F.A.; Mason, R.P. Photooxidation of Amplex Red to resorufin: Implications of exposing the Amplex Red assay to light. Free Radic. Biol. Med. 2012, 53, 1080–1087. [Google Scholar] [CrossRef]
- Meyer, A.J.; Dick, T.P. Fluorescent protein-based redox probes. Antioxid. Redox Signal. 2010, 13, 621–650. [Google Scholar] [CrossRef] [PubMed]
- Roma, L.P.; Duprez, J.; Takahashi, H.K.; Gilon, P.; Wiederkehr, A.; Jonas, J.C. Dynamic measurements of mitochondrial hydrogen peroxide concentration and glutathione redox state in rat pancreatic β-cells using ratiometric fluorescent proteins: Confounding effects of pH with HyPer but not roGFP1. Biochem. J. 2012, 441, 971–978. [Google Scholar] [CrossRef]
- Seaver, L.C.; Imlay, J.A. Hydrogen peroxide fluxes and compartmentalization inside growing Escherichia coli. J. Bacteriol. 2001, 183, 7182–7189. [Google Scholar] [CrossRef] [PubMed]
- Miki, T.; Orii, Y. The reaction of horseradish peroxidase with hydroperoxides derived from Triton X-100. Anal. Biochem. 1985, 146, 28–34. [Google Scholar] [CrossRef]
- Antunes, F.; Cadenas, E. Cellular titration of apoptosis with steady state concentrations of H(2)O(2): Submicromolar levels of H(2)O(2) induce apoptosis through Fenton chemistry independent of the cellular thiol state. Free Radic. Biol. Med. 2001, 30, 1008–1018. [Google Scholar] [CrossRef]
- Boveris, A.; Arnaiz, S.L.; Bustamante, J.; Alvarez, S.; Valdez, L.; Boveris, A.D.; Navarro, A. Pharmacological regulation of mitochondrial nitric oxide synthase. In Methods in Enzymology 359; Elsevier: Cham, Switzerland, 2002; pp. 328–339. [Google Scholar]
- Chance, B.; Sies, H.; Boveris, A. Hydroperoxide metabolism in mammalian organs. Physiol. Rev. 1979, 59, 527–605. [Google Scholar] [CrossRef] [PubMed]
- Lyublinskaya, O.; Antunes, F. Measuring intracellular concentration of hydrogen peroxide with the use of genetically encoded H2O2 biosensor HyPer. Redox Biol. 2019, 24, 101200. [Google Scholar] [CrossRef] [PubMed]
- Matsusaki, M.; Kanemura, S.; Kinoshita, M.; Lee, Y.H.; Inaba, K.; Okumura, M. The Protein Disulfide Isomerase Family: From proteostasis to pathogenesis. Biochim. Biophys. Acta. General Subj. 2020, 1864, 129338. [Google Scholar] [CrossRef]
- Zito, E. ERO1: A protein disulfide oxidase and H2O2 producer. Free Radic. Biol. Med. 2015, 83, 299–304. [Google Scholar] [CrossRef]
- Gross, E.; Sevier, C.S.; Heldman, N.; Vitu, E.; Bentzur, M.; Kaiser, C.A.; Thorpe, C.; Fass, D. Generating disulfides enzymatically: Reaction products and electron acceptors of the endoplasmic reticulum thiol oxidase Ero1p. Proc. Natl. Acad. Sci. USA 2006, 103, 299–304. [Google Scholar] [CrossRef]
- Tu, B.P.; Weissman, J.S. The FAD- and O(2)-dependent reaction cycle of Ero1-mediated oxidative protein folding in the endoplasmic reticulum. Mol. Cell 2002, 10, 983–994. [Google Scholar] [CrossRef]
- Nguyen, V.D.; Saaranen, M.J.; Karala, A.R.; Lappi, A.; Wang, L.; Raykhel, I.B.; Alanen, H.I.; Salo, K.E.H.; Wang, C.; Ruddock, L.W. Two endoplasmic reticulum PDI peroxidases increase the efficiency of the use of peroxide during disulfide bond formation. J. Mol. Biol. 2011, 406, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Zito, E.; Melo, E.P.; Yang, Y.; Wahlander, Å.; Neubert, T.A.; Ron, D. Oxidative protein folding by an endoplasmic reticulum-localized peroxiredoxin. Mol. Cell. 2010, 40, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Tavender, T.J.; Springate, J.J.; Bulleid, N.J. Recycling of peroxiredoxin IV provides a novel pathway for disulphide formation in the endoplasmic reticulum. EMBO J. 2010, 29, 4185–4197. [Google Scholar] [CrossRef]
- Kakihana, T.; Nagata, K.; Sitia, R. Peroxides and peroxidases in the endoplasmic reticulum: Integrating redox homeostasis and oxidative folding. Antioxid. Redox Signal. 2012, 16, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Bertolotti, M.; Yim, S.H.; Garcia-Manteiga, J.M.; Masciarelli, S.; Kim, Y.; Kang, M.; Iuchi, Y.; Fujii, J.; Roberta, V.; Rubartelli, A. B- to plasma-cell terminal differentiation entails oxidative stress and profound reshaping of the antioxidant responses. Antioxid. Redox Signal. 2010, 13, 1133–1144. [Google Scholar] [CrossRef]
- Shimizu, Y.; Hendershot, L.M. Oxidative folding: Cellular strategies for dealing with the resultant equimolar production of reactive oxygen species. Antioxid. Redox Signal. 2009, 11, 2317–2331. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Tian, Y.; Zhang, R.; Jing, J.; Zhang, X. Endoplasmic Reticulum-Directed Ratiometric Fluorescent Probe for Quantitive Detection of Basal H2O2. Anal. Chem. 2017, 89, 12945–12950. [Google Scholar] [CrossRef] [PubMed]
- Malinouski, M.; Zhou, Y.; Belousov, V.V.; Hatfield, D.L.; Gladyshev, V.N. Hydrogen peroxide probes directed to different cellular compartments. PLoS ONE 2011, 6, e14564. [Google Scholar] [CrossRef]
- Mehmeti, I.; Lortz, S.; Lenzen, S. The H2O2-sensitive HyPer protein targeted to the endoplasmic reticulum as a mirror of the oxidizing thiol-disulfide milieu. Free Radic. Biol. Med. 2012, 53, 1451–1458. [Google Scholar] [CrossRef]
- Melo, E.P.; Lopes, C.; Gollwitzer, P.; Lortz, S.; Lenzen, S.; Mehmeti, I.; Kaminski, C.F.; Ron, D.; Avezov, E. TriPer, an optical probe tuned to the endoplasmic reticulum tracks changes in luminal H2O2. BMC Biol. 2017, 15, 24. [Google Scholar] [CrossRef] [PubMed]
- Buvelot, H.; Jaquet, V.; Krause, K.H. Mammalian NADPH Oxidases. Methods Mol. Biol. 2019, 1982, 17–36. [Google Scholar]
- Guo, S.; Chen, X. The human Nox4: Gene, structure, physiological function and pathological significance. J. Drug Target 2015, 23, 888–896. [Google Scholar] [CrossRef] [PubMed]
- Laurindo, F.R.; Araujo, T.L.; Abrahão, T.B. Nox NADPH oxidases and the endoplasmic reticulum. Antioxid. Redox Signal. 2014, 20, 2755–2775. [Google Scholar] [CrossRef] [PubMed]
- Rinnerthaler, M.; Büttner, S.; Laun, P.; Heeren, G.; Felder, T.K.; Klinger, H.; Weinberger, M.; Stolze, K.; Grousl, T.; Hasek, J.; et al. Yno1p/Aim14p, a NADPH-oxidase ortholog, controls extramitochondrial reactive oxygen species generation, apoptosis, and actin cable formation in yeast. Proc. Natl. Acad. Sci. USA 2012, 109, 8658–8663. [Google Scholar] [CrossRef]
- Anilkumar, N.; San Jose, G.; Sawyer, I.; Santos, C.X.C.; Sand, C.; Brewer, A.C.; Warren, D.; Shah, A.M. A 28-kDa splice variant of NADPH oxidase-4 is nuclear-localized and involved in redox signaling in vascular cells. Arterioscler. Thromb. Vasc. Biol. 2013, 33, e104–e112. [Google Scholar] [CrossRef]
- Zhang, L.; Nguyen, M.V.; Lardy, B.; Jesaitis, A.J.; Grichine, A.; Rousset, F.; Talbot, M.; Paclet, M.; Qian, G.; Morel, F. New insight into the Nox4 subcellular localization in HEK293 cells: First monoclonal antibodies against Nox4. Biochimie 2011, 93, 457–468. [Google Scholar] [CrossRef]
- Rousset, F.; Zhang, L.; Lardy, B.; Morel, F.; Nguyen, M.V.C. Transmembrane Nox4 topology revealed by topological determination by Ubiquitin Fusion Assay, a novel method to uncover membrane protein topology. Biochem. Biophys. Res. Commun. 2020, 521, 383–388. [Google Scholar] [CrossRef]
- Trevelin, S.C.; Lopes, L.R. Protein disulfide isomerase and Nox: New partners in redox signaling. Curr. Pharm. Des. 2015, 21, 5951–5963. [Google Scholar] [CrossRef]
- Albertolle, M.E.; Peter Guengerich, F. The relationships between cytochromes P450 and H2O2: Production, reaction, and inhibition. J. Inorg. Biochem. 2018, 186, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Veith, A.; Moorthy, B. Role of cytochrome P450s in the generation and metabolism of reactive oxygen species. Curr. Opin. Toxicol. 2018, 7, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Guengerich, F.P. Human cytochrome P450 enzymes. In Cytochrome P450: Structure, Mechanism, and Biochemistry; Springer: New York, NY, USA, 2015; pp. 523–785. [Google Scholar]
- Gillette, J.R.; Brodie, B.B.; La Du, B.N. The oxidation of drugs by liver microsomes: On the role of TPNH and oxygen. J. Pharmacol. Exp. Ther. 1957, 119, 532–540. [Google Scholar] [PubMed]
- Black, S.D. Membrane topology of the mammalian P450 cytochromes. FASEB J. 1992, 6, 680–685. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Finger, Y.; Riemer, J. Protein import by the mitochondrial disulfide relay in higher eukaryotes. Biol. Chem. 2020, 401, 749–763. [Google Scholar] [CrossRef]
- Backes, S.; Herrmann, J.M. Protein Translocation into the Intermembrane Space and Matrix of Mitochondria: Mechanisms and Driving Forces. Front. Mol. Biosci. 2017, 4, 83. [Google Scholar] [CrossRef]
- Allen, S.; Balabanidou, V.; Sideris, D.P.; Lisowsky, T.; Tokatlidis, K. Erv1 mediates the Mia40-dependent protein import pathway and provides a functional link to the respiratory chain by shuttling electrons to cytochrome c. J. Mol. Biol 2005, 353, 937–944. [Google Scholar] [CrossRef]
- Bihlmaier, K.; Mesecke, N.; Terziyska, N.; Bien, M.; Hell, K.; Herrmann, J.M. The disulfide relay system of mitochondria is connected to the respiratory chain. J. Cell Biol. 2007, 179, 389–395. [Google Scholar] [CrossRef]
- Dabir, D.V.; Leverich, E.P.; Kim, S.K.; Tsai, F.D.; Hirasawa, M.; Knaff, D.B.; Koehler, C.M. A role for cytochrome c and cytochrome c peroxidase in electron shuttling from Erv1. EMBO J. 2007, 26, 4801–4811. [Google Scholar] [CrossRef]
- Farrell, S.R.; Thorpe, C. Augmenter of liver regeneration: A flavin-dependent sulfhydryl oxidase with cytochrome c reductase activity. Biochemistry 2005, 44, 1532–1541. [Google Scholar] [CrossRef]
- Mailloux, R.J. Mitochondrial Antioxidants and the Maintenance of Cellular Hydrogen Peroxide Levels. Oxid. Med. Cell. Longev. 2018, 2018, 7857251. [Google Scholar] [CrossRef] [PubMed]
- Rowland, A.A.; Voeltz, G.K. Endoplasmic reticulum-mitochondria contacts: Function of the junction. Nat. Rev. Mol. Cell Biol. 2012, 13, 607–625. [Google Scholar] [CrossRef]
- Gilady, S.Y.; Bui, M.; Lynes, E.M.; Benson, M.D.; Watts, R.; Vance, J.E.; Simmen, T. Ero1alpha requires oxidizing and normoxic conditions to localize to the mitochondria-associated membrane (MAM). Cell Stress Chaperones 2010, 15, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Booth, D.M.; Enyedi, B.; Geiszt, M.; Várnai, P.; Hajnóczky, G. Redox Nanodomains Are Induced by and Control Calcium Signaling at the ER-Mitochondrial Interface. Mol. Cell 2016, 63, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Pak, V.V.; Ezeriņa, D.; Lyublinskaya, O.G.; Pedre, B.; Tyurin-Kuzmin, P.A.; Mishina, N.M.; Thauvin, M.; Young, D.; Wahni, K.; Gache, S.A.M.; et al. Ultrasensitive Genetically Encoded Indicator for Hydrogen Peroxide Identifies Roles for the Oxidant in Cell Migration and Mitochondrial Function. Cell Metab. 2020, 31, 642–653. [Google Scholar] [CrossRef]
- Zandalinas, S.I.; Mittler, R. ROS-induced ROS release in plant and animal cells. Free Radic. Biol. Med. 2018, 122, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Prinz, W.A.; Toulmay, A.; Balla, T. The functional universe of membrane contact sites. Nat. Rev. Mol. Cell Biol. 2020, 21, 7–24. [Google Scholar] [CrossRef]
- Nordzieke, D.E.; Medraño-Fernandez, I. The Plasma Membrane: A Platform for Intra- and Intercellular Redox Signaling. Antioxidants 2018, 7, 168. [Google Scholar] [CrossRef]
- Chen, C.; Li, J.; Qin, X.; Wang, W. Peroxisomal Membrane Contact Sites in Mammalian Cells. Front. Cell Dev. Biol. 2020, 8, 512. [Google Scholar] [CrossRef]
- Santos, C.X.; Nabeebaccus, A.A.; Shah, A.M.; Camargo, L.L.; Filho, S.V.; Lopes, L.R. Endoplasmic reticulum stress and Nox-mediated reactive oxygen species signaling in the peripheral vasculature: Potential role in hypertension. Antioxid. Redox Signal. 2014, 20, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Hwang, I.; Uddin, M.J.; Pak, E.S.; Kang, H.; Jin, E.; Jo, S.; Kang, D.; Lee, H.; Ha, H. The impaired redox balance in peroxisomes of catalase knockout mice accelerates nonalcoholic fatty liver disease through endoplasmic reticulum stress. Free Radic. Biol. Med. 2020, 148, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Yoboue, E.D.; Sitia, R.; Simmen, T. Redox crosstalk at endoplasmic reticulum (ER) membrane contact sites (MCS) uses toxic waste to deliver messages. Cell Death Dis. 2018, 9, 331. [Google Scholar] [CrossRef] [PubMed]
- Braakman, I.; Bulleid, N.J. Protein folding and modification in the mammalian endoplasmic reticulum. Annu. Rev. Biochem. 2011, 80, 71–99. [Google Scholar] [CrossRef] [PubMed]
- Zhang, I.X.; Raghavan, M.; Satin, L.S. The Endoplasmic Reticulum and Calcium Homeostasis in Pancreatic Beta Cells. Endocrinology 2020, 161, bqz028. [Google Scholar] [CrossRef]
- Clerico, E.M.; Meng, W.; Pozhidaeva, A.; Bhasne, K.; Petridis, C.; Gierasch, L.M. Hsp70 molecular chaperones: Multifunctional allosteric holding and unfolding machines. Biochem. J. 2019, 476, 1653–1677. [Google Scholar] [CrossRef]
- Pobre, K.F.R.; Poet, G.J.; Hendershot, L.M. The endoplasmic reticulum (ER) chaperone BiP is a master regulator of ER functions: Getting by with a little help from ERdj friends. J. Biol. Chem. 2019, 294, 2098–2108. [Google Scholar] [CrossRef]
- Lewy, T.G.; Grabowski, J.M.; Bloom, M.E. BiP: Master Regulator of the Unfolded Protein Response and Crucial Factor in Flavivirus Biology. Yale J. Biol. Med. 2017, 90, 291–300. [Google Scholar]
- Srivastava, R.; Deng, Y.; Shah, S.; Rao, A.G.; Howell, S.H. BINDING PROTEIN is a master regulator of the endoplasmic reticulum stress sensor/transducer bZIP28 in Arabidopsis. Plant. Cell 2013, 25, 1416–1429. [Google Scholar] [CrossRef]
- Luo, S.; Mao, C.; Lee, B.; Lee, A.S. GRP78/BiP is required for cell proliferation and protecting the inner cell mass from apoptosis during early mouse embryonic development. Mol. Cell Biol. 2006, 26, 5688–5697. [Google Scholar] [CrossRef]
- Rose, M.D.; Misra, L.M.; Vogel, J.P. KAR2, a karyogamy gene, is the yeast homolog of the mammalian BiP/GRP78 gene. Cell 1989, 57, 1211–1221. [Google Scholar] [CrossRef]
- Normington, K.; Kohno, K.; Kozutsumi, Y.; Gething, M.J.; Sambrook, J.S. cerevisiae encodes an essential protein homologous in sequence and function to mammalian BiP. Cell 1989, 57, 1223–1236. [Google Scholar] [CrossRef]
- Sevier, C.S.; Qu, H.; Heldman, N.; Gross, E.; Fass, D.; Kaiser, C.A. Modulation of cellular disulfide-bond formation and the ER redox environment by feedback regulation of Ero1. Cell 2007, 129, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Appenzeller-Herzog, C.; Riemer, J.; Christensen, B.; Sørensen, E.S.; Ellgaard, L. A novel disulphide switch mechanism in Ero1alpha balances ER oxidation in human cells. EMBO J. 2008, 27, 2977–2987. [Google Scholar] [CrossRef] [PubMed]
- Baker, K.M.; Chakravarthi, S.; Langton, K.P.; Sheppard, A.M.; Lu, H.; Bulleid, N.J. Low reduction potential of Ero1alpha regulatory disulphides ensures tight control of substrate oxidation. EMBO J. 2008, 27, 2988–2997. [Google Scholar] [CrossRef]
- Wang, J.; Pareja, K.A.; Kaiser, C.A.; Sevier, C.S. Redox signaling via the molecular chaperone BiP protects cells against endoplasmic reticulum-derived oxidative stress. Elife 2014, 3, e03496. [Google Scholar] [CrossRef]
- Wei, P.C.; Hsieh, Y.H.; Su, M.I.; Jiang, X.; Hsu, P.; Lo, W.; Weng, J.; Jeng, Y.; Wang, J.; Chen, P.; et al. Loss of the oxidative stress sensor NPGPx compromises GRP78 chaperone activity and induces systemic disease. Mol. Cell 2012, 48, 747–759. [Google Scholar] [CrossRef]
- Wang, J.; Sevier, C.S. Formation and Reversibility of BiP Protein Cysteine Oxidation Facilitate Cell Survival during and post Oxidative Stress. J. Biol. Chem. 2016, 291, 7541–7557. [Google Scholar] [CrossRef]
- Lind, C.; Gerdes, R.; Hamnell, Y.; Schuppe-Koistinen, I.; von Löwenhielm, H.B.; Holmgren, A.; Cotgreave, I.A. Identification of S-glutathionylated cellular proteins during oxidative stress and constitutive metabolism by affinity purification and proteomic analysis. Arch. Biochem. Biophys. 2002, 406, 229–240. [Google Scholar] [CrossRef]
- Ye, Z.W.; Zhang, J.; Ancrum, T.; Manevich, Y.; Townsend, D.M.; Tew, K.D. Glutathione S-Transferase P-Mediated Protein S-Glutathionylation of Resident Endoplasmic Reticulum Proteins Influences Sensitivity to Drug-Induced Unfolded Protein Response. Antioxid. Redox Signal. 2017, 26, 247–261. [Google Scholar] [CrossRef]
- Schäuble, N.; Lang, S.; Jung, M.; Cappel, S.; Schorr, S.; Ulucan, Ö.; Linxweiler, J.; Dudek, J.; Blum, R.; Helms, V.; et al. BiP-mediated closing of the Sec61 channel limits Ca2+ leakage from the ER. EMBO J. 2012, 31, 3282–3296. [Google Scholar] [CrossRef]
- Hamman, B.D.; Hendershot, L.M.; Johnson, A.E. BiP maintains the permeability barrier of the ER membrane by sealing the lumenal end of the translocon pore before and early in translocation. Cell 1998, 92, 747–758. [Google Scholar] [CrossRef]
- Ponsero, A.J.; Igbaria, A.; Darch, M.A.; Miled, S.; Outten, C.E.; Winther, J.R.; Palais, G.; D’autreaux, B.; Delaunay-Moisan, A.; Toledano, M.B. Endoplasmic Reticulum Transport of Glutathione by Sec61 Is Regulated by Ero1 and Bip. Mol. Cell 2017, 67, 962–973. [Google Scholar] [CrossRef] [PubMed]
- Siegenthaler, K.D.; Pareja, K.A.; Wang, J.; Sevier, C.S. An unexpected role for the yeast nucleotide exchange factor Sil1 as a reductant acting on the molecular chaperone BiP. Elife 2017, 6, e24141. [Google Scholar] [CrossRef]
- Chung, K.T.; Shen, Y.; Hendershot, L.M. BAP, a mammalian BiP-associated protein, is a nucleotide exchange factor that regulates the ATPase activity of BiP. J. Biol. Chem. 2002, 277, 47557–47563. [Google Scholar] [CrossRef] [PubMed]
- Karagöz, G.E.; Acosta-Alvear, D.; Walter, P. The Unfolded Protein Response: Detecting and Responding to Fluctuations in the Protein-Folding Capacity of the Endoplasmic Reticulum. Cold Spring Harbor Perspect. Biol. 2019, 11, a033886. [Google Scholar] [CrossRef]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef] [PubMed]
- Travers, K.J.; Patil, C.K.; Wodicka, L.; Lockhart, D.J.; Weissman, J.S.; Walter, P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell 2000, 101, 249–258. [Google Scholar] [CrossRef]
- Hetz, C.; Papa, F.R. The Unfolded Protein Response and Cell Fate Control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef]
- Shore, G.C.; Papa, F.R.; Oakes, S.A. Signaling cell death from the endoplasmic reticulum stress response. Curr. Opin. Cell Biol. 2011, 23, 143–149. [Google Scholar] [CrossRef]
- Hourihan, J.M.; Moronetti Mazzeo, L.E.; Fernández-Cárdenas, L.P.; Blackwell, T.K. Cysteine Sulfenylation Directs IRE-1 to Activate the SKN-1/Nrf2 Antioxidant Response. Mol. Cell 2016, 63, 553–566. [Google Scholar] [CrossRef]
- Camargo, L.L.; Harvey, A.P.; Rios, F.J.; Tsiropoulou, S.; da Silva, R.d.O.; Cao, Z.; Graham, D.; McMaster, C.; Burchmore, R.J.; Hartley, R.C.; et al. Vascular Nox (NADPH Oxidase) Compartmentalization, Protein Hyperoxidation, and Endoplasmic Reticulum Stress Response in Hypertension. Hypertension 2018, 72, 235–246. [Google Scholar] [CrossRef]
- Blackwell, T.K.; Steinbaugh, M.J.; Hourihan, J.M.; Ewald, C.Y.; Isik, M. SKN-1/Nrf, stress responses, and aging in Caenorhabditis elegans. Free Radic. Biol. Med. 2015, 88, 290–301. [Google Scholar] [CrossRef] [PubMed]
- Guerra-Moreno, A.; Ang, J.; Welsch, H.; Jochem, M.; Hanna, J. Regulation of the unfolded protein response in yeast by oxidative stress. FEBS Lett. 2019, 593, 1080–1088. [Google Scholar] [CrossRef] [PubMed]
- Guerra-Moreno, A.; Prado, M.A.; Ang, J.; Schnell, H.M.; Micoogullari, Y.; Paulo, J.A.; Finley, D.; Gygi, S.P.; Hanna, J. Thiol-based direct threat sensing by the stress-activated protein kinase Hog1. Sci. Signal 2019, 12. [Google Scholar] [CrossRef] [PubMed]
- Fuse, Y.; Kobayashi, M. Conservation of the Keap1-Nrf2 System: An Evolutionary Journey through Stressful Space and Time. Molecules 2017, 22, 436. [Google Scholar] [CrossRef]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, T.; Imaizumi, K.; Oono, K.; Yui, D.; Gomi, F.; Katayama, T.; Tohyama, M. Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J. Biol. Chem. 2001, 276, 13935–13940. [Google Scholar] [CrossRef]
- Michalak, M.; Robert Parker, J.M.; Opas, M. Ca2+ signaling and calcium binding chaperones of the endoplasmic reticulum. Cell Calcium 2002, 32, 269–278. [Google Scholar] [CrossRef]
- Periasamy, M.; Kalyanasundaram, A. SERCA pump isoforms: Their role in calcium transport and disease. Muscle Nerve 2007, 35, 430–442. [Google Scholar] [CrossRef]
- Brini, M.; Carafoli, E. Calcium pumps in health and disease. Physiol. Rev. 2009, 89, 1341–1378. [Google Scholar] [CrossRef] [PubMed]
- Adachi, T.; Weisbrod, R.M.; Pimentel, D.R.; Ying, J.; Sharov, V.S.; Schöneich, C.; Cohen, R. A S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat. Med. 2004, 10, 1200–1207. [Google Scholar] [CrossRef] [PubMed]
- Lancel, S.; Qin, F.; Lennon, S.L.; Zhang, J.; Tong, X.; Mazzini, M.J.; Kang, Y.J.; Siwik, D.A.; Cohen, R.A.; Colucci, W.S. Oxidative posttranslational modifications mediate decreased SERCA activity and myocyte dysfunction in Galphaq-overexpressing mice. Circ. Res. 2010, 107, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Evangelista, A.M.; Thompson, M.D.; Bolotina, V.M.; Tong, X.; Cohen, R.A. Nox4- and Nox2-dependent oxidant production is required for VEGF-induced SERCA cysteine-674 S-glutathiolation and endothelial cell migration. Free Radic. Biol. Med. 2012, 53, 2327–2334. [Google Scholar] [CrossRef] [PubMed]
- Evangelista, A.M.; Thompson, M.D.; Weisbrod, R.M.; Pimental, D.R.; Tong, X.; Bolotina, V.M.; Cohen, R.A. Redox regulation of SERCA2 is required for vascular endothelial growth factor-induced signaling and endothelial cell migration. Antioxid. Redox Signal. 2012, 17, 1099–1108. [Google Scholar] [CrossRef] [PubMed]
- Qin, F.; Siwik, D.A.; Lancel, S.; Zhang, J.; Kuster, G.M.; Luptak, I.; Wang, L.; Tong, X.; Kang, Y.J.; Cohen, R.A.; et al. Hydrogen peroxide-mediated SERCA cysteine 674 oxidation contributes to impaired cardiac myocyte relaxation in senescent mouse heart. J. Am. Heart Assoc. 2013, 2, e000184. [Google Scholar] [CrossRef]
- Ying, J.; Tong, X.; Pimentel, D.R.; Weisbrod, R.M.; Trucillo, M.P.; Adachi, T.; Cohen, R. A Cysteine-674 of the sarco/endoplasmic reticulum calcium ATPase is required for the inhibition of cell migration by nitric oxide. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 783–790. [Google Scholar] [CrossRef]
- Tong, X.; Hou, X.; Jourd’heuil, D.; Weisbrod, R.M.; Cohen, R.A. Upregulation of Nox4 by TGF{beta}1 oxidizes SERCA and inhibits NO in arterial smooth muscle of the prediabetic Zucker rat. Circ. Res. 2010, 107, 975–983. [Google Scholar] [CrossRef]
- Weisbrod, R.M.; Shiang, T.; Al Sayah, L.; Fry, J.L.; Bajpai, S.; Reinhart-King, C.A.; Lob, H.E.; Santhanam, L.; Mitchell, G.; Cohen, R.A.; et al. Arterial stiffening precedes systolic hypertension in diet-induced obesity. Hypertension 2013, 62, 1105–1110. [Google Scholar] [CrossRef]
- Toya, T.; Ito, K.; Kagami, K.; Osaki, A.; Sato, A.; Kimura, T.; Horii, S.; Yasuda, R.; Namba, T.; Ido, Y.; et al. Impact of oxidative posttranslational modifications of SERCA2 on heart failure exacerbation in young patients with non-ischemic cardiomyopathy: A pilot study. Int. J. Cardiol. Heart Vasc. 2020, 26, 100437. [Google Scholar] [CrossRef]
- Thompson, M.D.; Mei, Y.; Weisbrod, R.M.; Silver, M.; Shukla, P.C.; Bolotina, V.M.; Cohen, R.A.; Tong, X. Glutathione adducts on sarcoplasmic/endoplasmic reticulum Ca2+ ATPase Cys-674 regulate endothelial cell calcium stores and angiogenic function as well as promote ischemic blood flow recovery. J. Biol. Chem. 2014, 289, 19907–19916. [Google Scholar] [CrossRef]
- Ying, J.; Sharov, V.; Xu, S.; Jiang, B.; Gerrity, R.; Christian, S.; Cohen, R.A. Cysteine-674 oxidation and degradation of sarcoplasmic reticulum Ca(2+) ATPase in diabetic pig aorta. Free Radic Biol Med. 2008, 45, 756–762. [Google Scholar] [CrossRef]
- Marino, M.; Stoilova, T.; Giorgi, C.; Bachi, A.; Cattaneo, A.; Auricchio, A.; Pinton, P.; Zito, E. SEPN1, an endoplasmic reticulum-localized selenoprotein linked to skeletal muscle pathology, counteracts hyperoxidation by means of redox-regulating SERCA2 pump activity. Hum. Mol. Genet. 2015, 24, 1843–1855. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.; Hu, J.; Liu, T.; Ago, T.; Sadoshima, J.; Li, H. Quantitative analysis of redox-sensitive proteome with DIGE and ICAT. J. Proteome Res. 2008, 7, 3789–3802. [Google Scholar] [CrossRef] [PubMed]
- Ushioda, R.; Miyamoto, A.; Inoue, M.; Watanabe, S.; Okumura, M.; Maegawa, K.; Uegaki, K.; Fujii, S.; Fukuda, Y.; Umitsu, M.; et al. Redox-assisted regulation of Ca2+ homeostasis in the endoplasmic reticulum by disulfide reductase ERdj5. Proc. Natl. Acad. Sci. USA 2016, 113, E6055–E6063. [Google Scholar] [CrossRef]
- Filipe, A.; Chernorudskiy, A.; Arbogast, S.; Varone, E.; Villar-Quiles, R.; Pozzer, D.; Moulin, M.; Fumagalli, S.; Cabet, E.; Dudhal, S.; et al. Defective endoplasmic reticulum-mitochondria contacts and bioenergetics in SEPN1-related myopathy. Cell Death Differ. 2020. [Google Scholar] [CrossRef] [PubMed]
- Pozzer, D.; Varone, E.; Chernorudskiy, A.; Schiarea, S.; Missiroli, S.; Giorgi, C.; Pinton, P.; Canato, M.; Germinario, E.; Nogara, L.; et al. A maladaptive ER stress response triggers dysfunction in highly active muscles of mice with SELENON loss. Redox Biol. 2019, 20, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Camacho, P. Ca2+-dependent redox modulation of SERCA 2b by ERp57. J. Cell Biol. 2004, 164, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Chernorudskiy, A.; Varone, E.; Colombo, S.F.; Fumagalli, S.; Cagnotto, A.; Cattaneo, A.; Briens, M.; Baltzinger, M.; Kuhn, L.; Bachi, A.; et al. Selenoprotein N is an endoplasmic reticulum calcium sensor that links luminal calcium levels to a redox activity. Proc. Natl. Acad. Sci. USA 2020, 117, 21288–21298. [Google Scholar] [CrossRef]
- Pozzer, D.; Favellato, M.; Bolis, M.; Invernizzi, R.W.; Solagna, F.; Blaauw, B.; Zito, E. Endoplasmic Reticulum Oxidative Stress Triggers Tgf-Beta-Dependent Muscle Dysfunction by Accelerating Ascorbic Acid Turnover. Sci. Rep. 2017, 7, 40993. [Google Scholar] [CrossRef]
- Raturi, A.; Gutiérrez, T.; Ortiz-Sandoval, C.; Ruangkittisakul, A.; Herrera-Cruz, M.S.; Rockley, J.P.; Gesson, K.; Ourdev, D.; Lou, P.; Lucchinetti, E.; et al. TMX1 determines cancer cell metabolism as a thiol-based modulator of ER-mitochondria Ca2+ flux. J. Cell Biol. 2016, 214, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Sakuta, N.; Watanabe, S.; Zhang, Y.; Yoshikaie, K.; Tanaka, Y.; Ushioda, R.; Kato, Y.; Takagi, J.; Tsukazaki, T.; et al. Structural Basis of Sarco/Endoplasmic Reticulum Ca2+-ATPase 2b Regulation via Transmembrane Helix Interplay. Cell Rep. 2019, 27, 1221–1230. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Inoue, M.; Tsutsumi, A.; Watanabe, S.; Nishizawa, T.; Nagata, K.; Kikkawa, M.; Inaba, K. Cryo-EM structures of SERCA2b reveal the mechanism of regulation by the luminal extension tail. Sci. Adv. 2020, 6, eabb0147. [Google Scholar] [CrossRef] [PubMed]
- Bechtel, T.J.; Li, C.; Kisty, E.A.; Maurais, A.J.; Weerapana, E. Profiling Cysteine Reactivity and Oxidation in the Endoplasmic Reticulum. ACS Chem. Biol. 2020, 15, 543–553. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roscoe, J.M.; Sevier, C.S. Pathways for Sensing and Responding to Hydrogen Peroxide at the Endoplasmic Reticulum. Cells 2020, 9, 2314. https://doi.org/10.3390/cells9102314
Roscoe JM, Sevier CS. Pathways for Sensing and Responding to Hydrogen Peroxide at the Endoplasmic Reticulum. Cells. 2020; 9(10):2314. https://doi.org/10.3390/cells9102314
Chicago/Turabian StyleRoscoe, Jennifer M., and Carolyn S. Sevier. 2020. "Pathways for Sensing and Responding to Hydrogen Peroxide at the Endoplasmic Reticulum" Cells 9, no. 10: 2314. https://doi.org/10.3390/cells9102314
APA StyleRoscoe, J. M., & Sevier, C. S. (2020). Pathways for Sensing and Responding to Hydrogen Peroxide at the Endoplasmic Reticulum. Cells, 9(10), 2314. https://doi.org/10.3390/cells9102314