Sex Differences in Diabetes- and TGF-β1-Induced Renal Damage

 ,
, 

 ,
,  ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Assessment of Renal Damage
2.3. Assessment of Sex Hormone Concentration in Blood Plasma
2.4. Ex Vivo Model and Study Protocol
2.5. Cell Line and In Vitro Protocol
2.6. Immunofluorescence Staining
2.7. Characterization of TKPTS Cells
2.8. Reverse Transcription and Real-Time PCR
2.9. Statistical Analysis
3. Results and Discussion
3.1. Diabetes Mellitus Causes Changes in Laboratory Parameters and Development of Diabetic Nephropathy
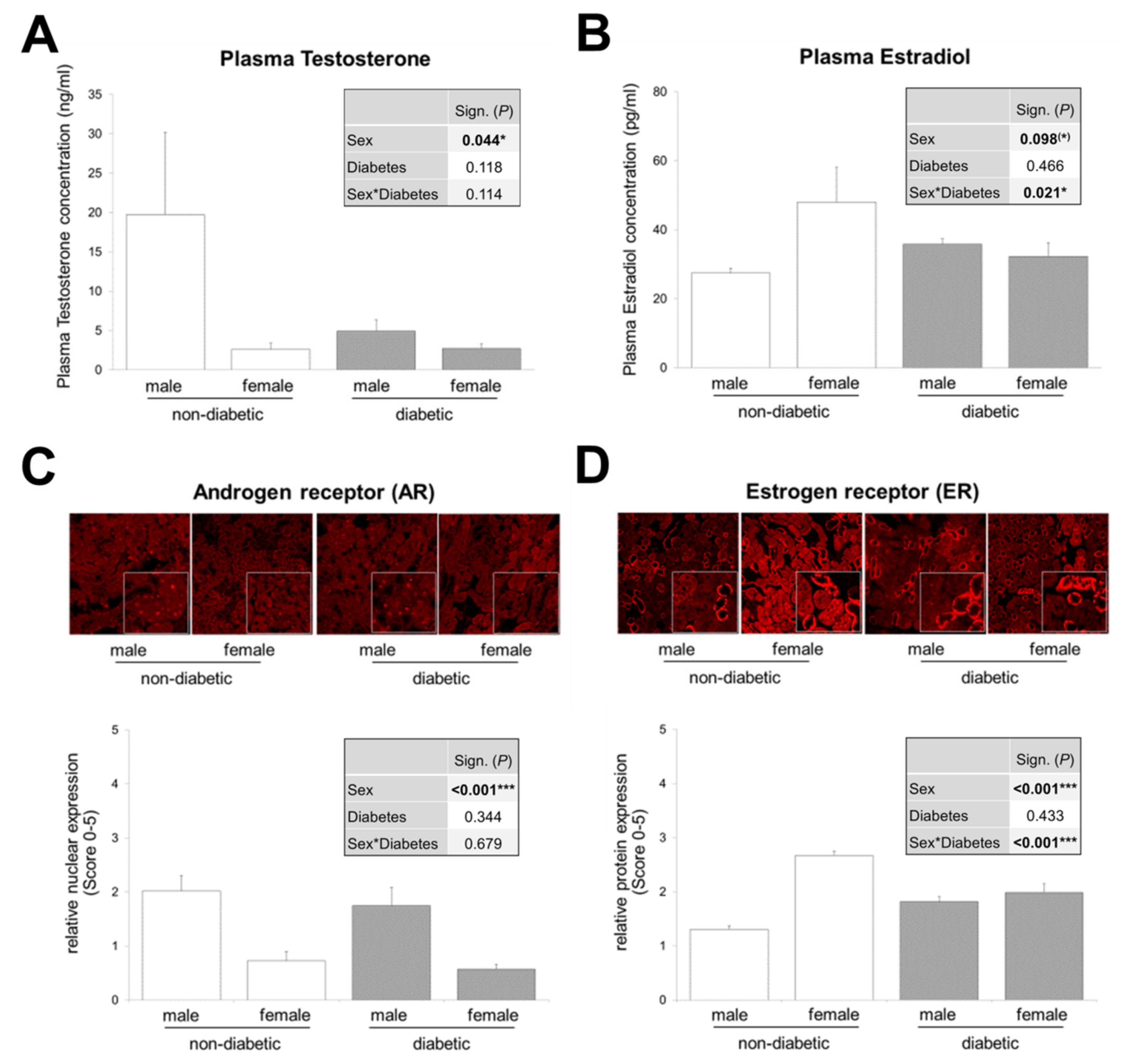
3.2. Interaction between Sex and T2DM in Plasma Estradiol Levels and Renal Estrogen Receptor Expression
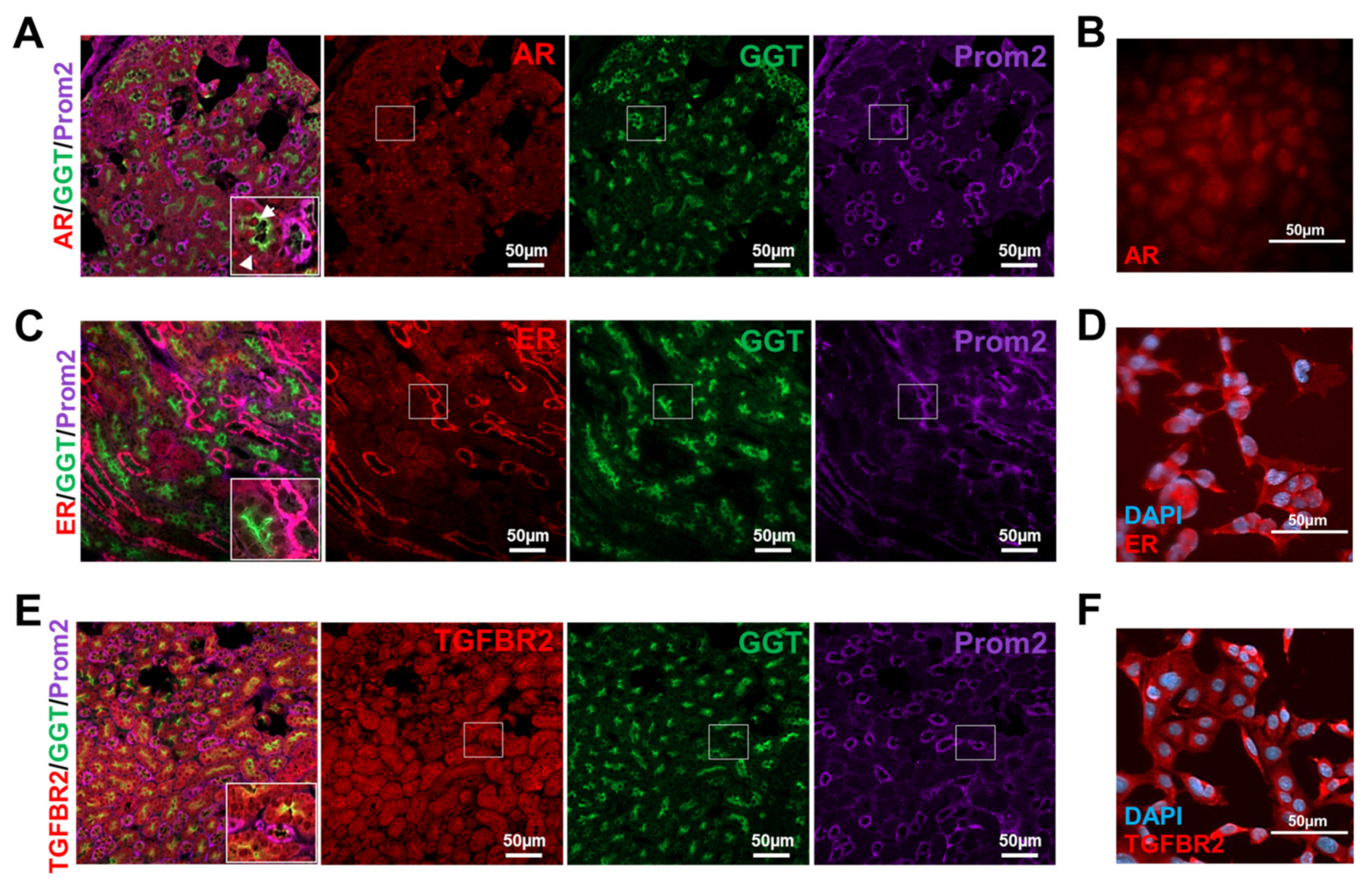
3.3. Murine Tubular Cells Express the Receptors Required for Sex Hormone and TGF-β1 Signaling
3.4. Sex and Diabetic Conditions Cause Changes in Expression Pattern of Molecular Markers for TIF in DN
3.5. Sex Hormones Directly Influence the Expression of TGF-β1 and BMP7 in Cortical Tissue from Mouse Kidney
3.6. In the Presence of Sex Hormones, the TGF-β1-Induced CTGF Expression Is Differentially Regulated in Both Sexes and Additionally Inversed Depending on TGF-β1 Concentration
3.7. The Opposite Effects of Sex Hormones in Dependency of TGF-β1-Concentration Is Confirmed in TKPTS Cells
3.8. TGF-β1-Regulated TGFBR1 and TGFBR2 Expression Is Influenced by Sex Hormones
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Martinez-Castelao, A.; Navarro-Gonzalez, J.F.; Gorriz, J.L.; de Alvaro, F. The Concept and the epidemiology of diabetic nephropathy have changed in recent years. J. Clin. Med. 2015, 4, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Wolf, G. New insights into the pathophysiology of diabetic nephropathy: From haemodynamics to molecular pathology. Eur. J. Clin. Investig. 2004, 34, 785–796. [Google Scholar] [CrossRef] [PubMed]
- Hasslacher, C.; Ritz, E.; Wahl, P.; Michael, C. Similar risks of nephropathy in patients with type I or type II diabetes mellitus. Nephrol. Dial. Transpl. Off. Publ. Eur. Dial. Transpl. Assoc. Eur. Ren. Assoc. 1989, 4, 859–863. [Google Scholar] [CrossRef] [PubMed]
- Alicic, R.Z.; Rooney, M.T.; Tuttle, K.R. Diabetic kidney disease: Challenges, progress, and possibilities. Clin. J. Am. Soc. Nephrol. CJASN 2017, 12, 2032–2045. [Google Scholar] [CrossRef] [PubMed]
- Ina, K.; Kitamura, H.; Tatsukawa, S.; Takayama, T.; Fujikura, Y.; Shimada, T. Transformation of interstitial fibroblasts and tubulointerstitial fibrosis in diabetic nephropathy. Med. Electron. Microsc. 2002, 35, 87–95. [Google Scholar] [CrossRef]
- Kolset, S.O.; Reinholt, F.P.; Jenssen, T. Diabetic nephropathy and extracellular matrix. J. Histochem. Cytochem. 2012, 60, 976–986. [Google Scholar] [CrossRef]
- Iwano, M.; Neilson, E.G. Mechanisms of tubulointerstitial fibrosis. Curr. Opin. Nephrol. Hypertens. 2004, 13, 279–284. [Google Scholar] [CrossRef]
- Li, R.; Chung, A.C.; Dong, Y.; Yang, W.; Zhong, X.; Lan, H.Y. The microRNA miR-433 promotes renal fibrosis by amplifying the TGF-beta/Smad3-Azin1 pathway. Kidney Int. 2013, 84, 1129–1144. [Google Scholar] [CrossRef]
- Vallon, V. The proximal tubule in the pathophysiology of the diabetic kidney. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, 14. [Google Scholar] [CrossRef]
- Yang, L.; Besschetnova, T.Y.; Brooks, C.R.; Shah, J.V.; Bonventre, J.V. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat. Med. 2010, 16, 535–543. [Google Scholar] [CrossRef]
- Lovisa, S.; Zeisberg, M.; Kalluri, R. Partial Epithelial-to-mesenchymal transition and other new mechanisms of kidney fibrosis. Trends Endocrinol. Metab. 2016, 27, 681–695. [Google Scholar] [CrossRef]
- Bonventre, J.V. Can we target tubular damage to prevent renal function decline in diabetes? Semin. Nephrol. 2012, 32, 452–462. [Google Scholar] [CrossRef]
- Loeffler, I.; Wolf, G. Transforming growth factor-beta and the progression of renal disease. Nephrol. Dial. Transpl. Off. Publ. Eur. Dial. Transpl. Assoc. Eur. Ren. Assoc. 2014, 29, 37–45. [Google Scholar]
- Mason, R.M.; Wahab, N.A. Extracellular matrix metabolism in diabetic nephropathy. J. Am. Soc. Nephrol. JASN 2003, 14, 1358–1373. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.; Chen, X.; Poronnik, P.; Pollock, C.A. The renal cortical fibroblast in renal tubulointerstitial fibrosis. Int. J. Biochem. Cell. Biol. 2006, 38, 1–5. [Google Scholar] [CrossRef]
- Wolf, G.; Schroeder, R.; Ziyadeh, F.N.; Stahl, R.A. Albumin up-regulates the type II transforming growth factor-beta receptor in cultured proximal tubular cells. Kidney Int. 2004, 66, 1849–1858. [Google Scholar] [CrossRef]
- Qi, W.; Twigg, S.; Chen, X.; Polhill, T.S.; Poronnik, P.; Gilbert, R.E.; Pollock, C.A. Integrated actions of transforming growth factor-beta1 and connective tissue growth factor in renal fibrosis. Am. J. Physiol. Ren. Physiol. 2005, 288, 800–809. [Google Scholar] [CrossRef] [PubMed]
- Hills, C.E.; Squires, P.E. TGF-beta1-induced epithelial-to-mesenchymal transition and therapeutic intervention in diabetic nephropathy. Am. J. Nephrol. 2010, 31, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, Q. Bone morphogenetic protein-7 and Gremlin: New emerging therapeutic targets for diabetic nephropathy. Biochem. Biophys. Res. Commun. 2009, 383, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.N.; Lapage, J.; Hirschberg, R. Loss of tubular bone morphogenetic protein-7 in diabetic nephropathy. J. Am. Soc. Nephrol. JASN 2001, 12, 2392–2399. [Google Scholar]
- Nguyen, T.Q.; Roestenberg, P.; van Nieuwenhoven, F.A.; Bovenschen, N.; Li, Z.; Xu, L.; Oliver, N.; Aten, J.; Joles, J.A.; Vial, C.; et al. CTGF inhibits BMP-7 signaling in diabetic nephropathy. J. Am. Soc. Nephrol. JASN 2008, 19, 2098–2107. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, M.; Hanai, J.; Sugimoto, H.; Mammoto, T.; Charytan, D.; Strutz, F.; Kalluri, R. BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat. Med. 2003, 9, 964–968. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Chen, Q.; Simon, T.C.; Strebeck, F.; Chaudhary, L.; Morrissey, J.; Liapis, H.; Klahr, S.; Hruska, K.A. Bone morphogenic protein-7 (BMP-7), a novel therapy for diabetic nephropathy. Kidney Int. 2003, 63, 2037–2049. [Google Scholar] [CrossRef] [PubMed]
- Neugarten, J.; Acharya, A.; Silbiger, S.R. Effect of gender on the progression of nondiabetic renal disease: A meta-analysis. J. Am. Soc. Nephrol. JASN 2000, 11, 319–329. [Google Scholar] [PubMed]
- Maric, C. Sex, diabetes and the kidney. Am. J. Physiol. Ren. Physiol. 2009, 296, 680–688. [Google Scholar] [CrossRef] [PubMed]
- Maric, C.; Sullivan, S. Estrogens and the diabetic kidney. Gend. Med. 2008, 5, 103–113. [Google Scholar] [CrossRef]
- Harvey, J.N. The influence of sex and puberty on the progression of diabetic nephropathy and retinopathy. Diabetologia 2011, 54, 1943–1945. [Google Scholar] [CrossRef][Green Version]
- Kautzky-Willer, A.; Harreiter, J.; Pacini, G. Sex and gender differences in risk, pathophysiology and complications of type 2 diabetes mellitus. Endocr. Rev. 2016, 37, 278–316. [Google Scholar] [CrossRef]
- Silbiger, S.; Neugarten, J. Gender and human chronic renal disease. Gend. Med. 2008, 5, 310. [Google Scholar] [CrossRef]
- Kautzky-Willer, A.; Handisurya, A. Metabolic diseases and associated complications: Sex and gender matter! Eur. J. Clin. Investig. 2009, 39, 631–648. [Google Scholar] [CrossRef]
- Zhang, L.; Krzentowski, G.; Albert, A.; Lefebvre, P.J. Factors predictive of nephropathy in DCCT Type 1 diabetic patients with good or poor metabolic control. Diabet. Med. 2003, 20, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Holl, R.W.; Grabert, M.; Thon, A.; Heinze, E. Urinary excretion of albumin in adolescents with type 1 diabetes: Persistent versus intermittent microalbuminuria and relationship to duration of diabetes, sex, and metabolic control. Diabetes Care 1999, 22, 1555–1560. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Wells, C.C.; Garman, J.H.; Asico, L.; Escano, C.S.; Maric, C. Imbalance in sex hormone levels exacerbates diabetic renal disease. Hypertension 2008, 51, 1218–1224. [Google Scholar] [CrossRef] [PubMed]
- Shah, T.A.; Rogers, M.B. Unanswered questions regarding sex and BMP/TGF-beta signaling. J. Dev. Biol. 2018, 6, 14. [Google Scholar] [CrossRef]
- Alpers, C.E.; Hudkins, K.L. Mouse models of diabetic nephropathy. Curr. Opin. Nephrol. Hypertens. 2011, 20, 278–284. [Google Scholar] [CrossRef]
- Saito, M.; Kaneda, A.; Shigeto, H.; Hanata, N.; Otokuni, K.; Matsuoka, H. Development of an optimized 5-stage protocol for the in vitro preparation of insulin-secreting cells from mouse ES cells. Cytotechnology 2016, 68, 987–998. [Google Scholar] [CrossRef]
- Kumar, A.; Ruan, M.; Clifton, K.; Syed, F.; Khosla, S.; Oursler, M.J. TGF-beta mediates suppression of adipogenesis by estradiol through connective tissue growth factor induction. Endocrinology 2012, 153, 254–263. [Google Scholar] [CrossRef]
- Wang, S.H.; Chen, G.H.; Fan, Y.; Van Antwerp, M.; Baker, J.R., Jr. Tumor necrosis factor-related apoptosis-inducing ligand inhibits experimental autoimmune thyroiditis by the expansion of CD4+CD25+ regulatory T cells. Endocrinology 2009, 150, 2000–2007. [Google Scholar] [CrossRef]
- Kim, B.; Kim, Y.; Sakuma, R.; Hui, C.C.; Ruther, U.; Jorgensen, J.S. Primordial germ cell proliferation is impaired in Fused Toes mutant embryos. Dev. Biol. 2011, 349, 417–426. [Google Scholar] [CrossRef]
- Sultan, S.; Pascucci, M.; Ahmad, S.; Malik, I.A.; Bianchi, A.; Ramadori, P.; Ahmad, G.; Ramadori, G. LIPOCALIN-2 is a major acute-phase protein in a rat and mouse model of sterile abscess. Shock 2012, 37, 191–196. [Google Scholar] [CrossRef]
- Li, C.; Siragy, H.M. High glucose induces podocyte injury via enhanced (pro)renin receptor-Wnt-beta-catenin-snail signaling pathway. PLoS ONE 2014, 9, e89233. [Google Scholar]
- Su, J.Z.; Fukuda, N.; Jin, X.Q.; Lai, Y.M.; Suzuki, R.; Tahira, Y.; Takagi, H.; Ikeda, Y.; Kanmatsuse, K.; Miyazaki, H. Effect of AT2 receptor on expression of AT1 and TGF-beta receptors in VSMCs from SHR. Hypertension 2002, 40, 853–858. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Eddy, A.A. Molecular basis of renal fibrosis. Pediatr. Nephrol. 2000, 15, 290–301. [Google Scholar] [CrossRef] [PubMed]
- Clotet, S.; Riera, M.; Pascual, J.; Soler, M.J. RAS and sex differences in diabetic nephropathy. Am. J. Physiol. Ren. Physiol. 2016, 310, 945–957. [Google Scholar] [CrossRef]
- Grossmann, M.; Panagiotopolous, S.; Sharpe, K.; MacIsaac, R.J.; Clarke, S.; Zajac, J.D.; Jerums, G.; Thomas, M.C. Low testosterone and anaemia in men with type 2 diabetes. Clin. Endocrinol. Oxf. 2009, 70, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, M.; Thomas, M.C.; Panagiotopoulos, S.; Sharpe, K.; Macisaac, R.J.; Clarke, S.; Zajac, J.D.; Jerums, G. Low testosterone levels are common and associated with insulin resistance in men with diabetes. J. Clin. Endocrinol. Metab. 2008, 93, 1834–1840. [Google Scholar] [CrossRef]
- Tsai, E.C.; Matsumoto, A.M.; Fujimoto, W.Y.; Boyko, E.J. Association of bioavailable, free, and total testosterone with insulin resistance: Influence of sex hormone-binding globulin and body fat. Diabetes Care 2004, 27, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Stellato, R.K.; Feldman, H.A.; Hamdy, O.; Horton, E.S.; McKinlay, J.B. Testosterone, sex hormone-binding globulin, and the development of type 2 diabetes in middle-aged men: Prospective results from the Massachusetts male aging study. Diabetes Care 2000, 23, 490–494. [Google Scholar] [CrossRef] [PubMed]
- Ding, E.L.; Song, Y.; Malik, V.S.; Liu, S. Sex differences of endogenous sex hormones and risk of type 2 diabetes: A systematic review and meta-analysis. JAMA 2006, 295, 1288–1299. [Google Scholar] [CrossRef] [PubMed]
- Andersson, B.; Marin, P.; Lissner, L.; Vermeulen, A.; Bjorntorp, P. Testosterone concentrations in women and men with NIDDM. Diabetes Care 1994, 17, 405–411. [Google Scholar] [CrossRef]
- Sun, J.; Devish, K.; Langer, W.J.; Carmines, P.K.; Lane, P.H. Testosterone treatment promotes tubular damage in experimental diabetes in prepubertal rats. Am. J. Physiol. Ren. Physiol. 2007, 292, 1681–1690. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Ohtani, K.I.; Uehara, Y.; Abe, Y.; Takahashi, H.; Tsuchiya, T.; Sato, N.; Ibuki, Y.; Mori, M. Orchiectomy and response to testosterone in the development of obesity in young Otsuka-Long-Evans-Tokushima Fatty (OLETF) rats. Int. J. Obes. Relat. Metab. Disord. 1998, 22, 318–324. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Prabhu, A.; Xu, Q.; Manigrasso, M.B.; Biswas, M.; Flynn, E.; Iliescu, R.; Lephart, E.D.; Maric, C. Expression of aromatase, androgen and estrogen receptors in peripheral target tissues in diabetes. Steroids 2010, 75, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Manigrasso, M.B.; Sawyer, R.T.; Marbury, D.C.; Flynn, E.R.; Maric, C. Inhibition of estradiol synthesis attenuates renal injury in male streptozotocin-induced diabetic rats. Am. J. Physiol. Ren. Physiol. 2011, 301, 634–640. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Prabhu, A.; Xu, S.; Manigrasso, M.B.; Maric, C. Dose-dependent effects of dihydrotestosterone in the streptozotocin-induced diabetic rat kidney. Am. J. Physiol. Ren. Physiol. 2009, 297, 307–315. [Google Scholar] [CrossRef]
- Salonia, A.; Lanzi, R.; Scavini, M.; Pontillo, M.; Gatti, E.; Petrella, G.; Licata, G.; Nappi, R.E.; Bosi, E.; Briganti, A.; et al. Sexual function and endocrine profile in fertile women with type 1 diabetes. Diabetes Care 2006, 29, 312–316. [Google Scholar] [CrossRef]
- Wells, C.C.; Riazi, S.; Mankhey, R.W.; Bhatti, F.; Ecelbarger, C.; Maric, C. Diabetic nephropathy is associated with decreased circulating estradiol levels and imbalance in the expression of renal estrogen receptors. Gend. Med. 2005, 2, 227–237. [Google Scholar] [CrossRef]
- Matsushita, M.; Tamura, K.; Osada, S.; Kogo, H. Effect of troglitazone on the excess testosterone and LH secretion in thyroidectomized, insulin-resistant, type 2 diabetic Goto-Kakizaki rats. Endocrine 2005, 27, 301–305. [Google Scholar] [CrossRef]
- Lamon-Fava, S.; Barnett, J.B.; Woods, M.N.; McCormack, C.; McNamara, J.R.; Schaefer, E.J.; Longcope, C.; Rosner, B.; Gorbach, S.L. Differences in serum sex hormone and plasma lipid levels in Caucasian and African-American premenopausal women. J. Clin. Endocrinol. Metab. 2005, 90, 4516–4520. [Google Scholar] [CrossRef]
- Stamataki, K.E.; Spina, J.; Rangou, D.B.; Chlouverakis, C.S.; Piaditis, G.P. Ovarian function in women with non-insulin dependent diabetes mellitus. Clin. Endocrinol. Oxf. 1996, 45, 615–621. [Google Scholar] [CrossRef]
- Kim, N.N.; Stankovic, M.; Cushman, T.T.; Goldstein, I.; Munarriz, R.; Traish, A.M. Streptozotocin-induced diabetes in the rat is associated with changes in vaginal hemodynamics, morphology and biochemical markers. BMC Physiol. 2006, 6, 4. [Google Scholar] [CrossRef] [PubMed]
- Chin, M.; Isono, M.; Isshiki, K.; Araki, S.; Sugimoto, T.; Guo, B.; Sato, H.; Haneda, M.; Kashiwagi, A.; Koya, D. Estrogen and raloxifene, a selective estrogen receptor modulator, ameliorate renal damage in db/db mice. Am. J. Pathol. 2005, 166, 1629–1636. [Google Scholar] [CrossRef][Green Version]
- Mankhey, R.W.; Bhatti, F.; Maric, C. 17beta-Estradiol replacement improves renal function and pathology associated with diabetic nephropathy. Am. J. Physiol. Ren. Physiol. 2005, 288, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Phillips, A.O.; Steadman, R. Diabetic nephropathy: The central role of renal proximal tubular cells in tubulointerstitial injury. Histol. Histopathol. 2002, 17, 247–252. [Google Scholar]
- Lindquist, J.A.; Mertens, P.R. Myofibroblasts, regeneration or renal fibrosis-is there a decisive hint? Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc. Eur. Ren. Assoc. 2013, 28, 2678–2681. [Google Scholar] [CrossRef]
- Anderberg, R.J.; Meek, R.L.; Hudkins, K.L.; Cooney, S.K.; Alpers, C.E.; Leboeuf, R.C.; Tuttle, K.R. Serum amyloid A and inflammation in diabetic kidney disease and podocytes. Lab. Invest. 2015, 95, 697. [Google Scholar] [CrossRef] [PubMed]
- Simon-Tillaux, N.; Hertig, A. Snail and kidney fibrosis. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc. Eur. Ren. Assoc. 2017, 32, 224–233. [Google Scholar] [CrossRef]
- Tan, R.J.; Zhou, D.; Liu, Y. Signaling crosstalk between tubular epithelial cells and interstitial fibroblasts after kidney injury. Kidney Dis. Basel 2016, 2, 136–144. [Google Scholar] [CrossRef]
- Yamamoto, T.; Nakamura, T.; Noble, N.A.; Ruoslahti, E.; Border, W.A. Expression of transforming growth factor beta is elevated in human and experimental diabetic nephropathy. Proc. Natl. Acad. Sci. USA 1993, 90, 1814–1818. [Google Scholar] [CrossRef]
- Lane, P.H.; Snelling, D.M.; Babushkina-Patz, N.; Langer, W.J. Sex differences in the renal transforming growth factor-beta 1 system after puberty. Pediatr. Nephrol. 2001, 16, 61–68. [Google Scholar] [CrossRef]
- Hawse, J.R.; Subramaniam, M.; Ingle, J.N.; Oursler, M.J.; Rajamannan, N.M.; Spelsberg, T.C. Estrogen-TGFbeta cross-talk in bone and other cell types: Role of TIEG, Runx2, and other transcription factors. J. Cell Biochem. 2008, 103, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Diamond-Stanic, M.K.; You, Y.H.; Sharma, K. Sugar, sex, and TGF-beta in diabetic nephropathy. Semin. Nephrol. 2012, 32, 261–268. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Qi, W.; Gao, S.; Wang, Z. Transcriptional regulation of the TGF-beta1 promoter by androgen receptor. Biochem. J. 2008, 416, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Gewin, L.S. Renal fibrosis: Primacy of the proximal tubule. Matrix Biol. 2018, 68, 248–262. [Google Scholar] [CrossRef]
- Xu, Y.; Wan, J.; Jiang, D.; Wu, X. BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition in human renal proximal tubular epithelial cells. J. Nephrol. 2009, 22, 403–410. [Google Scholar]
- Kobayashi, T.; Inoue, T.; Okada, H.; Kikuta, T.; Kanno, Y.; Nishida, T.; Takigawa, M.; Sugaya, T.; Suzuki, H. Connective tissue growth factor mediates the profibrotic effects of transforming growth factor-beta produced by tubular epithelial cells in response to high glucose. Clin. Exp. Nephrol. 2005, 9, 114–121. [Google Scholar] [CrossRef]
- Rojas, A.; Padidam, M.; Cress, D.; Grady, W.M. TGF-beta receptor levels regulate the specificity of signaling pathway activation and biological effects of TGF-beta. Biochim. Biophys. Acta 2009, 1793, 1165–1173. [Google Scholar] [CrossRef]
- Derynck, R.; Feng, X.H. TGF-beta receptor signaling. Biochim. Biophys. Acta 1997, 1333, 105–150. [Google Scholar]
- Fafeur, V.; O’Hara, B.; Bohlen, P. A glycosylation-deficient endothelial cell mutant with modified responses to transforming growth factor-beta and other growth inhibitory cytokines: Evidence for multiple growth inhibitory signal transduction pathways. Mol. Biol. Cell 1993, 4, 135–144. [Google Scholar] [CrossRef][Green Version]
- Shi, Y.; Massague, J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Sense Primers | Antisense Primers | Tann. |
|---|---|---|---|
| HPRT1 | 5′-TGGATACAGGCCAGACTTTGTT-3′ | 5′-CAGATTCAACTTGCGCTCATC-3′ | 59 °C [36] |
| CTGF | 5′-TGCTGTGCAGGTGATAAAGC-3′ | 5′-AAGGCCATTTGTTCACCAAC-3′ | 58 °C [37] |
| TGF-β1 | 5′-AAGGGCTACCATGCCAACTT-3′ | 5′-CGGGTTGTGTTGGTTGTAGA-3′ | 62 °C [38] |
| BMP7 | 5′-GATTTCAGCCTGGACAACGAG-3′ | 5′-GGGCAACCCTAAGATGGACAG-3′ | 58 °C [39] |
| SAA | 5′-TCATTTGTTCACGAGGCTTTC-3′ | 5′-ATGGTGTCCTCATGTCCTCTG-3′ | 59 °C [40] |
| Snail1 | 5′-GCGGAAGATCTTCAACTGCAAATATTGTAA-3′ | 5′-GCAGTGGGAGCAGGAGAATGGCTTCTCAC-3′ | 54 °C [41] |
| TGFBR1 | 5′-GCTCTAGATTTCTGCCACCTCTGTAC-3′ | 5′-GCGAATTCGACAGTGCGGTTATGGCA-3′ | 62 °C [42] |
| TGFBR2 | 5′-GCAGGCATCAGGACCTCAGTTTGATCC-3′ | 5′-AGAGTGAAGCCGTGGTAGGTGAGCTTG-3′ | 62 °C [42] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ziller, N.; Kotolloshi, R.; Esmaeili, M.; Liebisch, M.; Mrowka, R.; Baniahmad, A.; Liehr, T.; Wolf, G.; Loeffler, I. Sex Differences in Diabetes- and TGF-β1-Induced Renal Damage. Cells 2020, 9, 2236. https://doi.org/10.3390/cells9102236
Ziller N, Kotolloshi R, Esmaeili M, Liebisch M, Mrowka R, Baniahmad A, Liehr T, Wolf G, Loeffler I. Sex Differences in Diabetes- and TGF-β1-Induced Renal Damage. Cells. 2020; 9(10):2236. https://doi.org/10.3390/cells9102236
Chicago/Turabian StyleZiller, Nadja, Roland Kotolloshi, Mohsen Esmaeili, Marita Liebisch, Ralf Mrowka, Aria Baniahmad, Thomas Liehr, Gunter Wolf, and Ivonne Loeffler. 2020. "Sex Differences in Diabetes- and TGF-β1-Induced Renal Damage" Cells 9, no. 10: 2236. https://doi.org/10.3390/cells9102236
APA StyleZiller, N., Kotolloshi, R., Esmaeili, M., Liebisch, M., Mrowka, R., Baniahmad, A., Liehr, T., Wolf, G., & Loeffler, I. (2020). Sex Differences in Diabetes- and TGF-β1-Induced Renal Damage. Cells, 9(10), 2236. https://doi.org/10.3390/cells9102236