Aquaporin-4 Expression during Toxic and Autoimmune Demyelination

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Groupings
2.2. Cuprizone-Induced Demyelination and Tissue Preparation
2.3. EAE and Disease Scoring
2.4. Tissue Preparation
2.5. Human Samples
2.6. Immunohistochemistry and Immunofluorescence
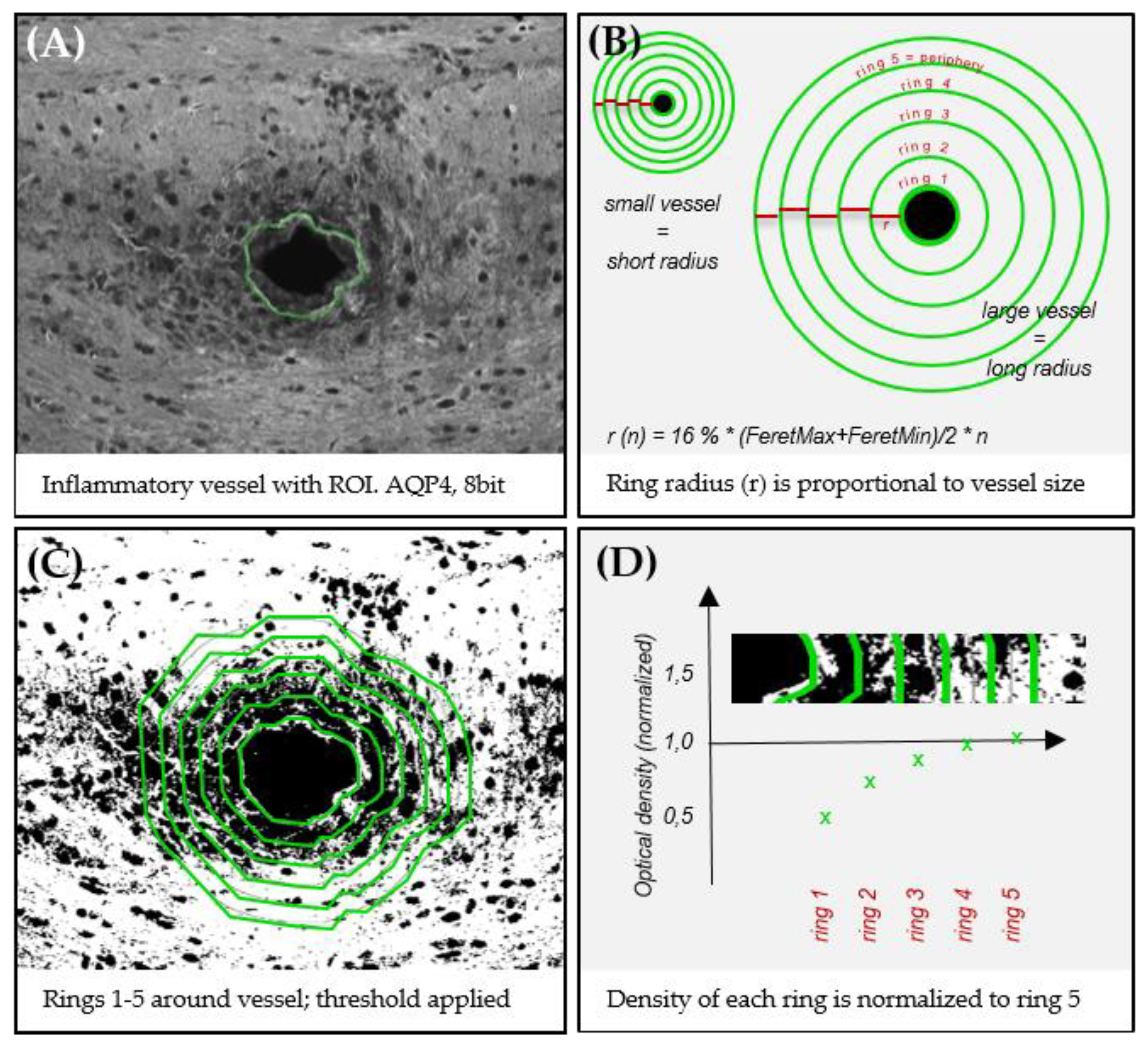
2.7. Digital Image Analysis
2.8. ELISA
2.9. Statistical Analysis
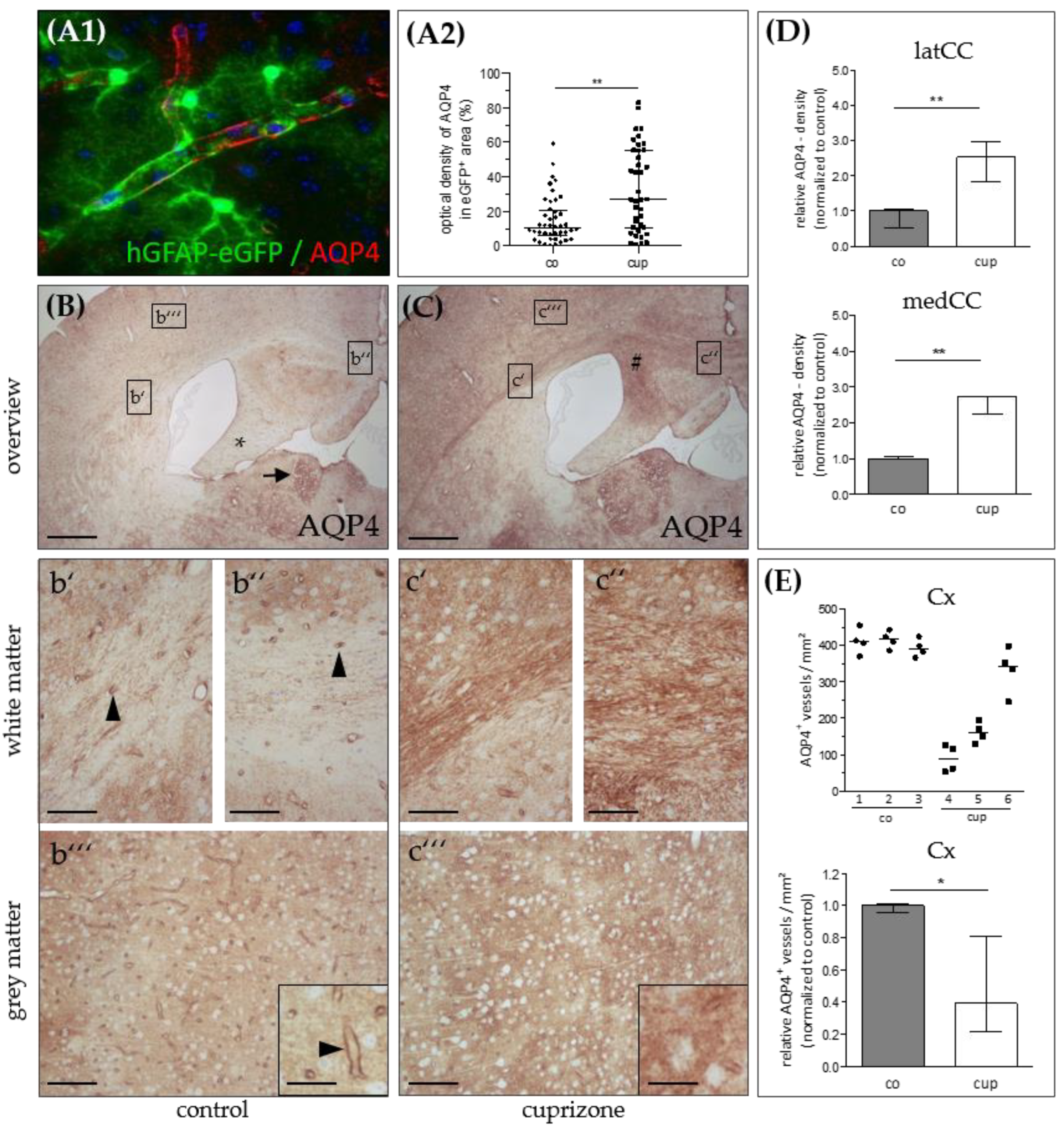
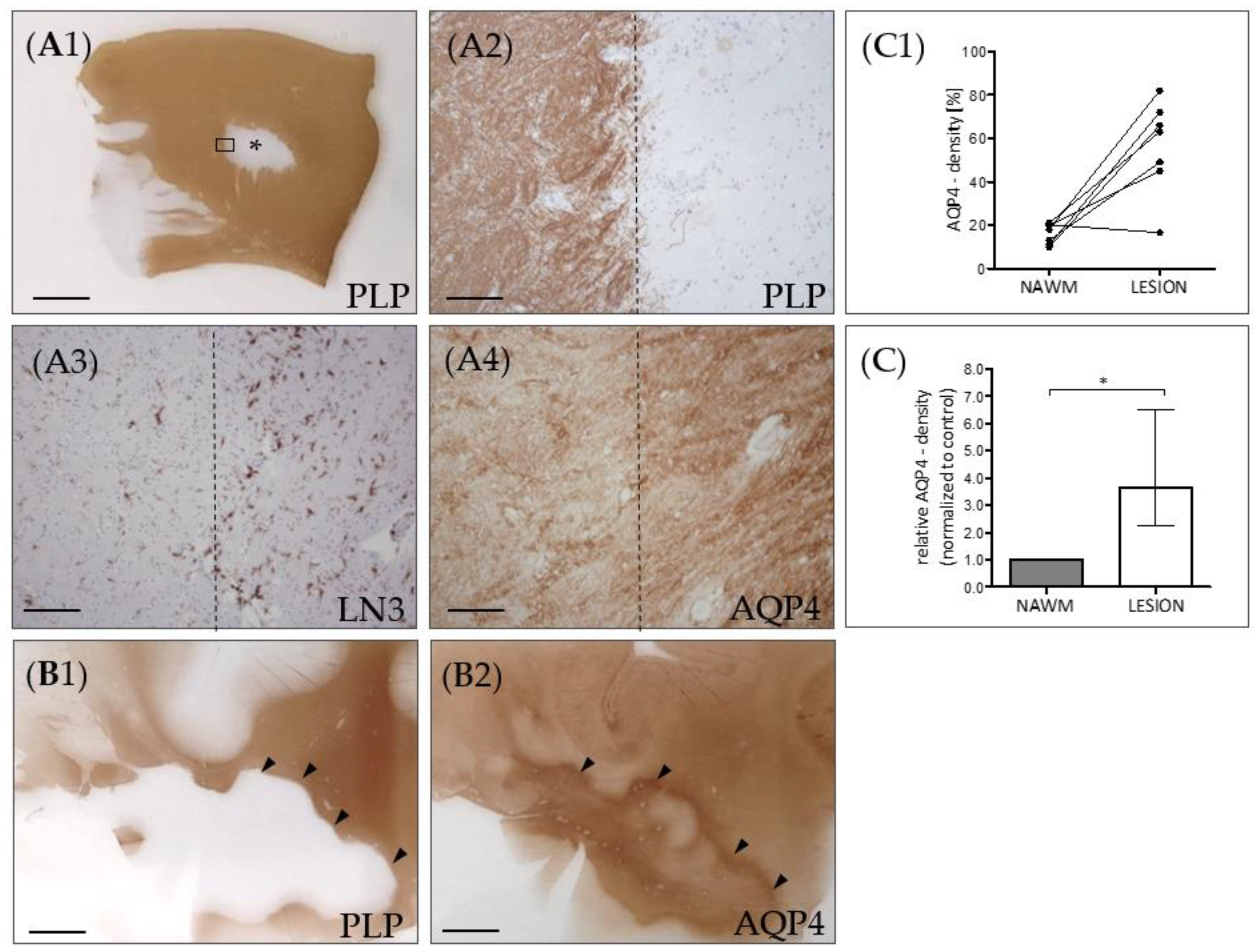
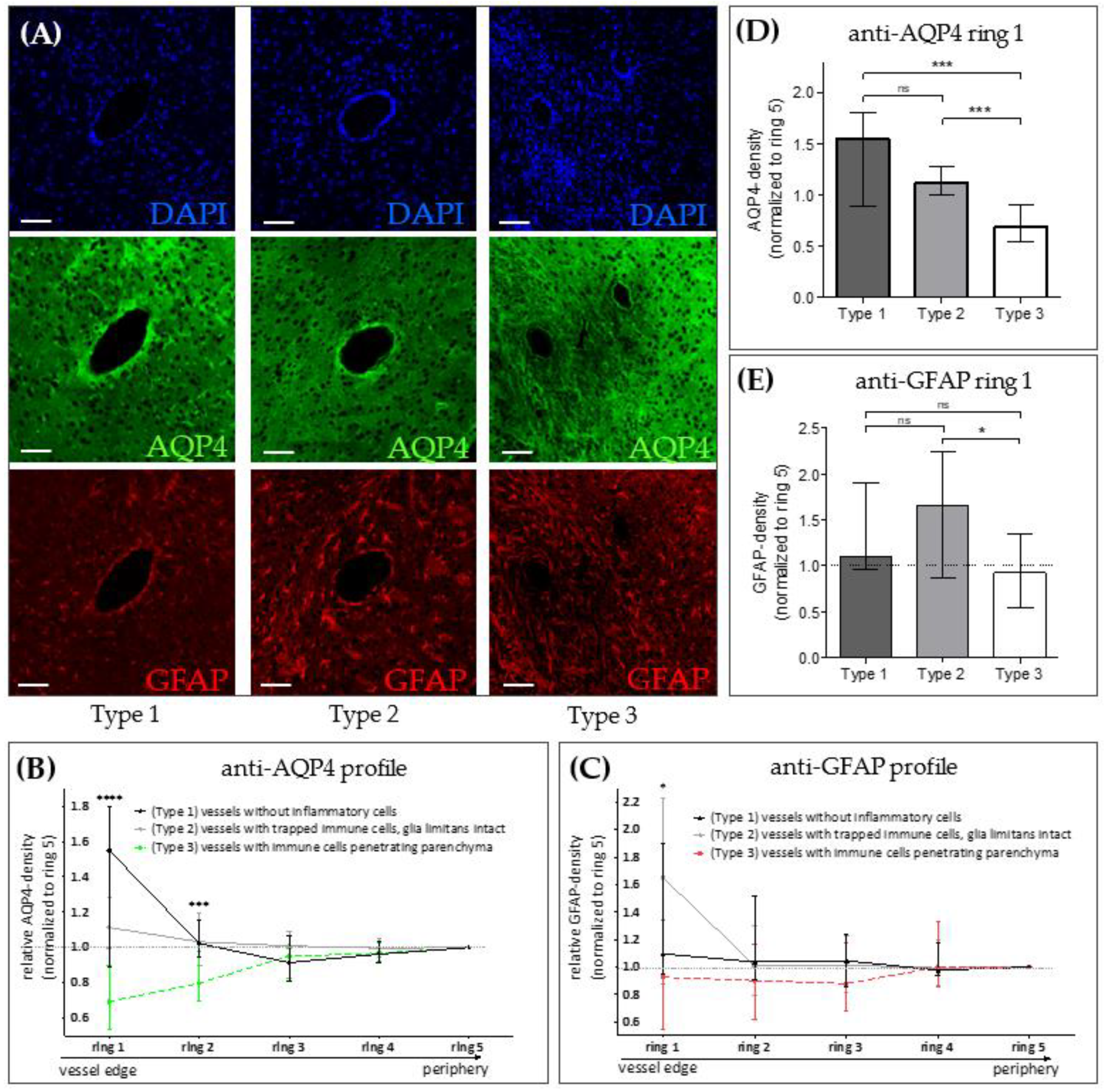
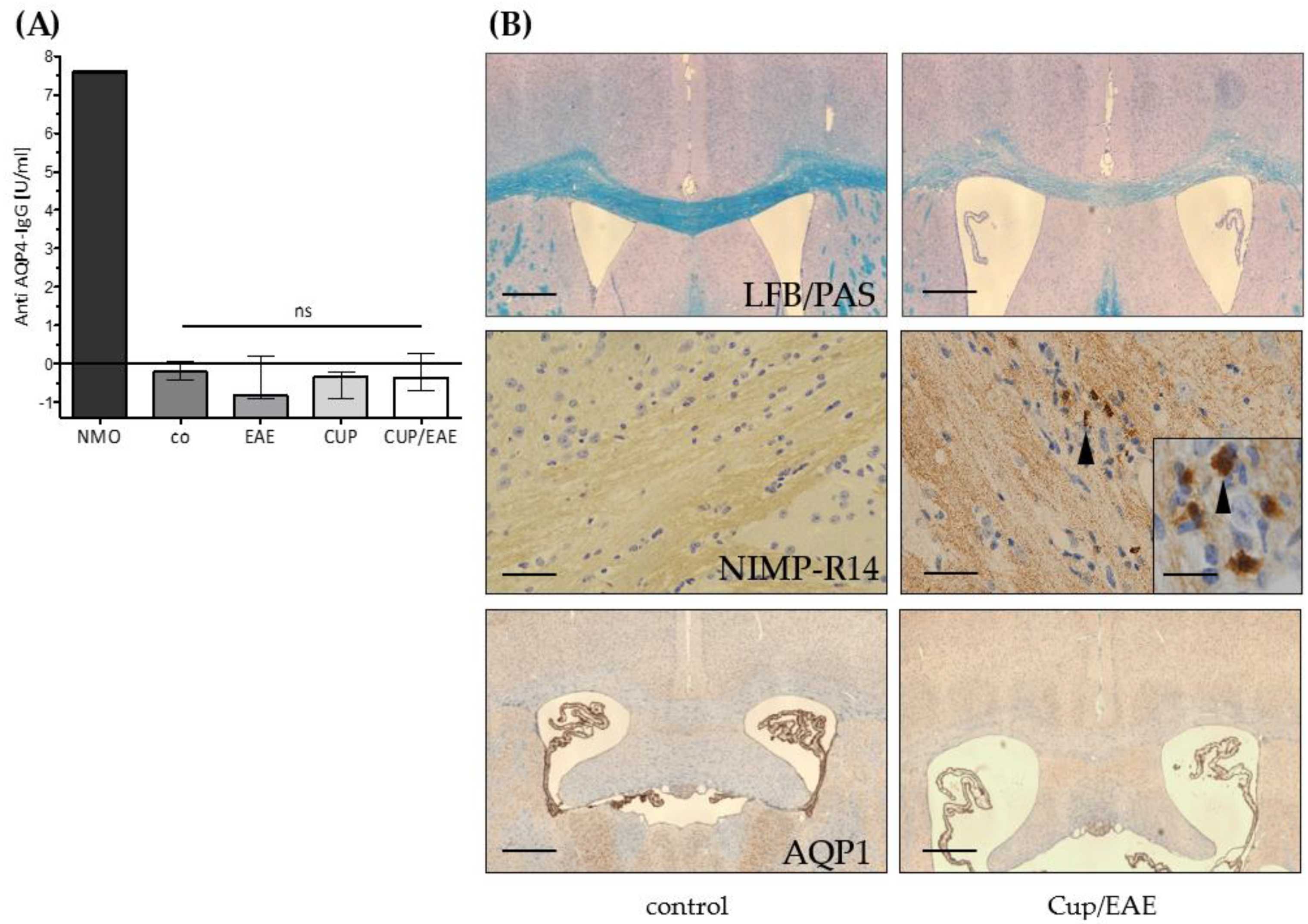
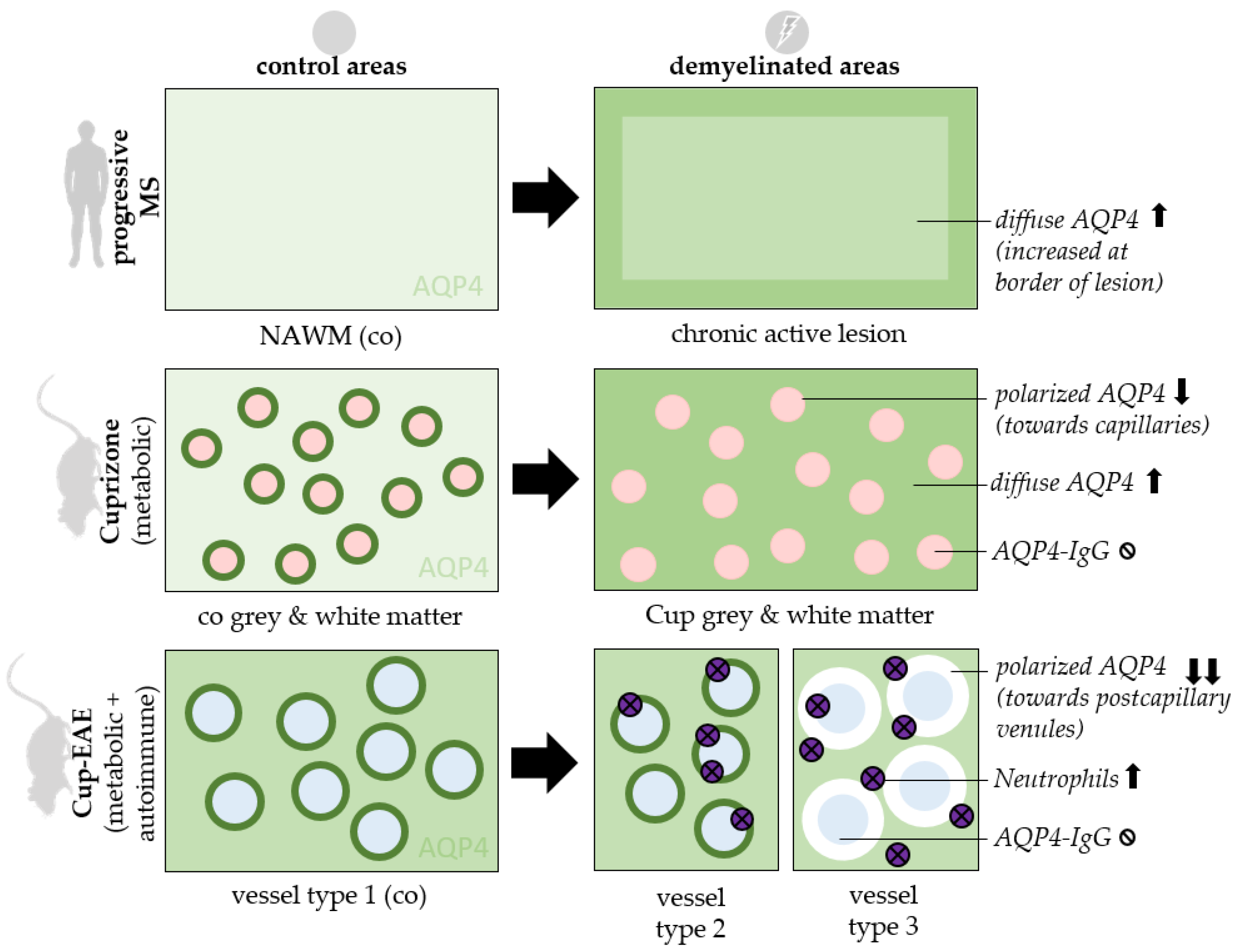
3. Results
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Sex | Age | Time to Fixation | Illness Duration | MS-Type | Cause of Death |
|---|---|---|---|---|---|---|
| 1 | M | 72 | 8:30 | 26 J | MS (PP) | Respiratory insufficiency |
| 2 | F | 50 | 5.35 | 14 J | MS | Pneumonia |
| 3 | F | 41 | 8:25 | 11 J | MS (SP) | Natural death |
| 4 | F | 76 | 6:00 | 22 J | MS (SP) | Unknown |
| 5 | M | 47 | 7:20 | 41 J | MS (PP) | Urosepsis and pneumonia |
| 6 | M | 49 | 8:00 | 24 J | MS (SP) | Pneumonia |
| 7 | F | 77 | 10:00 | 48 J | MS (PP) | Euthanasia |
| co | F | 84 | 4:45 | - | - | Heart failure, lung emphysema, dehydration |
| Use | Primary Antibody | Dilution | Antigen-Unmasking | Normal Serum | Secondary Antibody |
|---|---|---|---|---|---|
| AQP4, IHC mouse | AQP4 (H-80), rabbit, polyclonal (Santa-Cruz sc20812) | 1:1000 | - | Goat | Goat anti-rabbit, (VECTOR BA-1000) |
| AQP4, IHC human | AQP4 (H-80), rabbit, polyclonal (Santa Cruz sc20812) | 1:1000 | - | Goat | Goat anti-rabbit, (VECTOR BA-1000) |
| AQP4, IF mouse | AQP4 (H-80), rabbit, polyclonal (Santa Cruz sc20812) | 1:125 | - | Donkey | AlexaFluor 594 donkey-anti-rabbit (Invitrogen, A21207) |
| AQP1, IHC Mouse | AQP1, mouse monoclonal (Abcam ab9566) | 1:600 | Citrate | Goat | Dako enVision anti-mouse (DAKO 4400) |
| NIMP-R14, IHC mouse | NIMP-R14, rat monoclonal (Abcam ab2557) | 1:300 | - | Goat | Goat anti-rat, (VECTOR BA9400) |
| HLA-DR, IHC human | HLA-DR (LN3), mouse, monoclonal (eBioscience 14-9956) | 1:1000 | Citrate | Goat | Dako enVision anti-mouse (DAKO 4400) |
| PLP, IHC human | PLP, mouse, monoclonal (Serotec MCA839G) | 1:5000 | - | Goat | Goat anti-mouse, (VECTOR BA9200) |
| DAPI, IF mouse | Bisbenzimid (DAPI) (Sigma 14530) | 1:1000 | TRIS | - | - |
| Double-IF for AQP4 and GFAP, mouse | AQP4 (H-80), rabbit, polyclonal (Santa Cruz sc20812) | 1:500 | TRIS | Goat | Alexa Fluor goat anti-rabbit 488 (Life A11008) |
| GFAP, chicken, polyclonal (ab4674) | 1:4000 | TRIS | Goat | Alexa Fluor goat anti-chicken 594 (Life A11042) |

References
- Popescu, B.F.; Pirko, I.; Lucchinetti, C.F. Pathology of multiple sclerosis: Where do we stand? Continuum Minneap Minn 2013, 19, 901–921. [Google Scholar] [CrossRef] [PubMed]
- Kipp, M.; Nyamoya, S.; Hochstrasser, T.; Amor, S. Multiple sclerosis animal models: A clinical and histopathological perspective. Brain Pathol. 2017, 27, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Compston, A.; Coles, A. Multiple sclerosis. Lancet 2002, 359, 1221–1231. [Google Scholar] [CrossRef]
- Wheeler, M.A.; Clark, I.C.; Tjon, E.C.; Li, Z.; Zandee, S.E.J.; Couturier, C.P.; Watson, B.R.; Scalisi, G.; Alkwai, S.; Rothhammer, V.; et al. MAFG-driven astrocytes promote CNS inflammation. Nature 2020, 578, 593–599. [Google Scholar] [CrossRef]
- Planas-Fontánez, T.M.; Dreyfus, C.F.; Saitta, K.S. Reactive astrocytes as therapeutic targets for brain degenerative diseases: Roles played by metabotropic glutamate receptors. Neurochem. Res. 2020, 45, 541–550. [Google Scholar] [CrossRef]
- Williams, J.L.; Manivasagam, S.; Smith, B.C.; Sim, J.; Vollmer, L.L.; Daniels, B.P.; Russell, J.H.; Klein, R.S. Astrocyte-T cell crosstalk regulates region-specific neuroinflammation. Glia 2020, 68, 1361–1374. [Google Scholar] [CrossRef]
- Stork, L.; Ellenberger, D.; Ruprecht, K.; Reindl, M.; Beißbarth, T.; Friede, T.; Kümpfel, T.; Gerdes, L.A.; Gloth, M.; Liman, T.; et al. Antibody signatures in patients with histopathologically defined multiple sclerosis patterns. Acta Neuropathol. 2020, 139, 547–564. [Google Scholar] [CrossRef]
- Molina-Gonzalez, I.; Miron, V.E. Astrocytes in myelination and remyelination. Neurosci. Lett. 2019, 713, 134532. [Google Scholar] [CrossRef]
- Brambilla, R.; Persaud, T.; Hu, X.; Karmally, S.; Shestopalov, V.I.; Dvoriantchikova, G.; Ivanov, D.; Nathanson, L.; Barnum, S.R.; Bethea, J.R. Transgenic inhibition of astroglial NF-kappa B improves functional outcome in experimental autoimmune encephalomyelitis by suppressing chronic central nervous system inflammation. J. Immunol. 2009, 182, 2628–2640. [Google Scholar] [CrossRef]
- Draheim, T.; Liessem, A.; Scheld, M.; Wilms, F.; Weißflog, M.; Denecke, B.; Kensler, T.W.; Zendedel, A.; Beyer, C.; Kipp, M.; et al. Activation of the astrocytic Nrf2/ARE system ameliorates the formation of demyelinating lesions in a multiple sclerosis animal model. Glia 2016, 64, 2219–2230. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Misu, T.; Höftberger, R.; Fujihara, K.; Wimmer, I.; Takai, Y.; Nishiyama, S.; Nakashima, I.; Konno, H.; Bradl, M.; Garzuly, F.; et al. Presence of six different lesion types suggests diverse mechanisms of tissue injury in neuromyelitis optica. Acta Neuropathol. 2013, 125, 815–827. [Google Scholar] [CrossRef] [PubMed]
- Misu, T.; Fujihara, K.; Kakita, A.; Konno, H.; Nakashima, I.; Itoyama, Y.; Nakamura, M.; Watanabe, S.; Takahashi, T. Loss of aquaporin 4 in lesions of neuromyelitis optica: Distinction from multiple sclerosis. Brain 2007, 130, 1224–1234. [Google Scholar] [CrossRef] [PubMed]
- Herwerth, M.; Kalluri, S.R.; Srivastava, R.; Kleele, T.; Kenet, S.; Illes, Z.; Merkler, D.; Bennett, J.L.; Misgeld, T.; Hemmer, B. In vivo imaging reveals rapid astrocyte depletion and axon damage in a model of neuromyelitis optica-related pathology. Ann. Neurol. 2016, 79, 794–805. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, M.C.; Verkman, A.S. Aquaporin 4 and neuromyelitis optica. Lancet Neurol. 2012, 11, 535–544. [Google Scholar] [CrossRef]
- Jung, J.S.; Bhat, R.V.; Preston, G.M.; Guggino, W.B.; Baraban, J.M.; Agre, P. Molecular characterization of an aquaporin cDNA from brain: Candidate osmoreceptor and regulator of water balance. Proc. Natl. Acad. Sci. USA 1994, 91, 13052–13056. [Google Scholar] [CrossRef]
- Nielsen, S.; Nagelhus, E.A.; Amiry-Moghaddam, M.; Bourque, C.; Agre, P.; Ottersen, O.P. Specialized membrane domains for water transport in glial cells: High-Resolution immunogold cytochemistry of aquaporin-4 in rat brain. J. Neurosci. 1997, 17, 171–180. [Google Scholar] [CrossRef]
- Vizuete, M.; Venero, J.; Vargas, C.; Ilundain, A.; Echevarra, M.; Machado, A.; Cano, J. Differential upregulation of aquaporin-4 mrna expression in reactive astrocytes after brain injury: Potential role in brain edema. Neurobiol. Dis. 1999, 6, 245–258. [Google Scholar] [CrossRef]
- Manley, G.T.; Fujimura, M.; Ma, T.; Noshita, N.; Filiz, F.; Bollen, A.W.; Chan, P.; Verkman, A. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat. Med. 2000, 6, 159–163. [Google Scholar] [CrossRef]
- Solenov, E.; Watanabe, H.; Manley, G.T.; Verkman, A.S. Sevenfold-reduced osmotic water permeability in primary astrocyte cultures from AQP-4-deficient mice, measured by a fluorescence quenching method. Am. J. Physiol. Physiol. 2004, 286, C426–C432. [Google Scholar] [CrossRef]
- Nicchia, G.P.; Frigeri, A.; Liuzzi, G.M.; Svelto, M. Inhibition of AQP4 expression in astrocytes by RNAi determines alterations in cell morphology, growth, and water transport and induces changes in ischemia related genes. FASEB J. 2003, 17, 1508–1510. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, M.C.; Manley, G.T.; Krishna, S.; Verkman, A.S. Aquaporin-4 facilitates reabsorption of excess fluid in vasogenic brain edema. FASEB J. 2004, 18, 1291–1293. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Verkman, A.S. Impaired Hearing in Mice Lacking Aquaporin-4 Water Channels. J. Boil. Chem. 2001, 276, 31233–31237. [Google Scholar] [CrossRef] [PubMed]
- Fischbach, F.; Nedelcu, J.; Leopold, P.; Zhan, J.; Clarner, T.; Nellessen, L.; Beißel, C.; Van Heuvel, Y.; Goswami, A.; Weis, J.; et al. Cuprizone-induced graded oligodendrocyte vulnerability is regulated by the transcription factor DNA damage-inducible transcript 3. Glia 2018, 67, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Hochstrasser, T.; Jiangshan, Z.; Rühling, S.; Schmitz, C.; Kipp, M. Do pre-clinical multiple sclerosis models allow us to measure neurodegeneration and clinical progression? Expert Rev. Neurother. 2018, 18, 351–353. [Google Scholar] [CrossRef]
- Rühling, S.; Kramer, F.; Schmutz, S.; Amor, S.; Jiangshan, Z.; Schmitz, C.; Kipp, M.; Hochstrasser, T. Visualization of the Breakdown of the Axonal Transport Machinery: A Comparative Ultrastructural and Immunohistochemical Approach. Mol. Neurobiol. 2018, 56, 3984–3998. [Google Scholar] [CrossRef]
- Nack, A.; Brendel, M.; Nedelcu, J.; Daerr, M.; Nyamoya, S.; Beyer, C.; Focke, C.; Deussing, M.; Hoornaert, C.; Ponsaerts, P.; et al. Expression of Translocator Protein and [18F]-GE180 Ligand Uptake in Multiple Sclerosis Animal Models. Cells 2019, 8, 94. [Google Scholar] [CrossRef]
- Höflich, K.M.; Beyer, C.; Clarner, T.; Schmitz, C.; Nyamoya, S.; Kipp, M.; Hochstrasser, T. Acute axonal damage in three different murine models of multiple sclerosis: A comparative approach. Brain Res. 2016, 1650, 125–133. [Google Scholar] [CrossRef]
- Rüther, B.J.; Scheld, M.; Dreymueller, D.; Clarner, T.; Kress, E.; Brandenburg, L.-O.; Swartenbroekx, T.; Hoornaert, C.; Ponsaerts, P.; Fallier-Becker, P.; et al. Combination of cuprizone and experimental autoimmune encephalomyelitis to study inflammatory brain lesion formation and progression. Glia 2017, 65, 1900–1913. [Google Scholar] [CrossRef]
- Scheld, M.; Rüther, B.J.; Große-Veldmann, R.; Ohl, K.; Tenbrock, K.; Dreymüller, D.; Fallier-Becker, P.; Zendedel, A.; Beyer, C.; Clarner, T.; et al. Neurodegeneration Triggers Peripheral Immune Cell Recruitment into the Forebrain. J. Neurosci. 2016, 36, 1410–1415. [Google Scholar] [CrossRef]
- Nyamoya, S.; Leopold, P.; Becker, B.; Beyer, C.; Hustadt, F.; Schmitz, C.; Michel, A.; Kipp, M. G-Protein-Coupled Receptor Gpr17 Expression in Two Multiple Sclerosis Remyelination Models. Mol. Neurobiol. 2018, 56, 1109–1123. [Google Scholar] [CrossRef] [PubMed]
- Nolte, C.; Matyash, M.; Pivneva, T.; Schipke, C.G.; Ohlemeyer, C.; Hanisch, U.K.; Kirchhoff, F.; Kettenmann, H. GFAP promoter-controlled EGFP-expressing transgenic mice: A tool to visualize astrocytes and astrogliosis in living brain tissue. Glia 2001, 33, 72–86. [Google Scholar] [CrossRef]
- Hochstrasser, T.; Exner, G.L.; Nyamoya, S.; Schmitz, C.; Kipp, M. Cuprizone-Containing Pellets are less potent to induce consistent demyelination in the corpus callosum of C57BL/6 mice. J. Mol. Neurosci. 2017, 61, 617–624. [Google Scholar] [CrossRef]
- Slowik, A.; Schmidt, T.; Beyer, C.; Amor, S.; Clarner, T.; Kipp, M. The sphingosine 1-phosphate receptor agonist FTY720 is neuroprotective after cuprizone-induced CNS demyelination. Br. J. Pharmacol. 2014, 172, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Van Der Valk, P.; De Groot, C.J. Staging of multiple sclerosis (MS) lesions: Pathology of the time frame of MS. Neuropathol. Appl. Neurobiol. 2000, 26, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, J.; Clarner, T.; Beyer, C.; Kipp, M. Anatomical distribution of cuprizone-induced lesions in C57BL6 mice. J. Mol. Neurosci. 2015, 57, 166–175. [Google Scholar] [CrossRef]
- Skripuletz, T.; Lindner, M.; Kotsiari, A.; Garde, N.; Fokuhl, J.; Linsmeier, F.; Trebst, C.; Stangel, M. Cortical demyelination is prominent in the murine cuprizone model and is strain-dependent. Am. J. Pathol. 2008, 172, 1053–1061. [Google Scholar] [CrossRef]
- Aoki-Yoshino, K.; Uchihara, T.; Duyckaerts, C.; Nakamura, A.; Hauw, J.-J.; Wakayama, Y. Enhanced expression of aquaporin 4 in human brain with inflammatory diseases. Acta Neuropathol. 2005, 110, 281–288. [Google Scholar] [CrossRef]
- Roemer, S.F.; Parisi, J.E.; Lennon, V.A.; Benarroch, E.E.; Lassmann, H.; Brück, W.; Mandler, R.N.; Weinshenker, B.G.; Pittock, S.J.; Wingerchuk, D.M.; et al. Pattern-specific loss of aquaporin-4 immunoreactivity distinguishes neuromyelitis optica from multiple sclerosis. Brain 2007, 130 Pt 5, 1194–1205. [Google Scholar] [CrossRef]
- Lennon, V.A.; Kryzer, T.J.; Pittock, S.J.; Verkman, A.; Hinson, S.R. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J. Exp. Med. 2005, 202, 473–477. [Google Scholar] [CrossRef]
- Lucchinetti, C.F.; Mandler, R.N.; McGAVERN, D.B.; Bruck, W.; Gleich, G.; Ransohoff, R.M.; Trebst, C.; Weinshenker, B.; Wingerchuk, D.; Parisi, J.E.; et al. A role for humoral mechanisms in the pathogenesis of Devic’s neuromyelitis optica. Brain 2002, 125 Pt 7, 1450–1461. [Google Scholar] [CrossRef]
- Lehmann, G.; Gradilone, S.A.; Marinelli, R. Aquaporin Water Channels in Central Nervous System. Curr. Neurovascular Res. 2004, 1, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, J.A.; Hsu, M.S.; Seldin, M.M.; Binder, D.K. Expression of the Astrocyte Water Channel Aquaporin-4 in the Mouse Brain. ASN Neuro 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Haj-Yasein, N.N.; Vindedal, G.F.; Eilert-Olsen, M.; Gundersen, G.A.; Skare, Ø.; Laake, P.; Klungland, A.; Thorén, A.E.; Burkhardt, J.M.; Ottersen, O.P.; et al. Glial-conditional deletion of aquaporin-4 (Aqp4) reduces blood-brain water uptake and confers barrier function on perivascular astrocyte endfeet. Proc. Natl. Acad. Sci. USA 2011, 108, 17815–17820. [Google Scholar] [CrossRef] [PubMed]
- Saadoun, S.; Bell, B.A.; Verkman, A.S.; Papadopoulos, M.C. Greatly improved neurological outcome after spinal cord compression injury in AQP4-deficient mice. Brain 2008, 131 Pt 4, 1087–1098. [Google Scholar] [CrossRef]
- Bloch, O.; Papadopoulos, M.C.; Manley, G.T.; Verkman, A.S. Aquaporin-4 gene deletion in mice increases focal edema associated with staphylococcal brain abscess. J. Neurochem. 2005, 95, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Tait, M.J.; Saadoun, S.; Bell, B.A.; Verkman, A.S.; Papadopoulos, M.C. Increased brain edema in aqp4-null mice in an experimental model of subarachnoid hemorrhage. Neuroscience 2010, 167, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.J.; Amini, M.; Hamamura, M.J.; Hsu, M.S.; Seldin, M.M.; Nalcioglu, O.; Binder, D.K. Aquaporin-4-dependent edema clearance following status epilepticus. Epilepsy Res. 2012, 98, 264–268. [Google Scholar] [CrossRef]
- Vertinsky, A.T.; Li, D.; Vavasour, I.M.; Miropolsky, V.; Zhao, G.; Zhao, Y.; Riddehough, A.; Moore, G.W.; Traboulsee, A.; Laule, C. Diffusely Abnormal White Matter, T2 Burden of Disease, and Brain Volume in Relapsing-Remitting Multiple Sclerosis. J. Neuroimaging 2018, 29, 151–159. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, P.; Wang, Q.; Trinkaus, K.; Schmidt, R.E.; Naismith, R.T.; Cross, A.H.; Song, S.-K. Differentiation and quantification of inflammation, demyelination and axon injury or loss in multiple sclerosis. Brain 2015, 138 Pt 5, 1223–1238. [Google Scholar] [CrossRef]
- Zuccoli, G.; Panigrahy, A.; Sreedher, G.; Bailey, A.; Laney, E.J.; La Colla, L.; Alper, G. Vasogenic edema characterizes pediatric acute disseminated encephalomyelitis. Neuroradiology 2014, 56, 679–684. [Google Scholar] [CrossRef] [PubMed]
- Balashov, K.E.; Aung, L.L.; Dhib-Jalbut, S.; Keller, I.A. Acute multiple sclerosis lesion: Conversion of restricted diffusion due to vasogenic edema. J. Neuroimaging 2011, 21, 202–204. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.-W.; Wang, Y.; Sun, P.; Lin, T.-H.; Trinkaus, K.; Cross, A.H.; Song, S.-K. Quantifying white matter tract diffusion parameters in the presence of increased extra-fiber cellularity and vasogenic edema. NeuroImage 2014, 101, 310–319. [Google Scholar] [CrossRef] [PubMed]
- Morrissey, S.P.; Stodal, H.; Zettl, U.; Simonis, C.; Jung, S.; Kiefer, R.; Lassmann, H.; Hartung, H.P.; Haase, A.; Toyka, K.V. In vivo MRI and its histological correlates in acute adoptive transfer experimental allergic encephalomyelitis. Quantification of inflammation and oedema. Brain 1996, 119 Pt 1, 239–248. [Google Scholar] [CrossRef]
- Berghoff, S.A.; Düking, T.; Spieth, L.; Winchenbach, J.; Stumpf, S.K.; Gerndt, N.; Kusch, K.; Ruhwedel, T.; Möbius, W.; Saher, G. Blood-brain barrier hyperpermeability precedes demyelination in the cuprizone model. Acta Neuropathol. Commun. 2017, 5, 94. [Google Scholar] [CrossRef]
- Purves, D.; Garrod, I.; Dayan, A. A Comparison of Spongiosis Induced in the Brain by Hexachlorophene, Cuprizone and Triethyl Tin in the Sprague-Dawley Rat. Hum. Exp. Toxicol. 1991, 10, 439–444. [Google Scholar] [CrossRef]
- Wolburg-Buchholz, K.; Mack, A.F.; Steiner, E.; Pfeiffer, F.; Engelhardt, B.; Wolburg, H. Loss of astrocyte polarity marks blood-brain barrier impairment during experimental autoimmune encephalomyelitis. Acta Neuropathol. 2009, 118, 219–233. [Google Scholar] [CrossRef]
- Sun, L.; Weng, H.; Li, Z. Elevation of AQP4 and selective cytokines in experimental autoimmune encephalitis mice provides some potential biomarkers in optic neuritis and demyelinating diseases. Int. J. Clin. Exp. Pathol. 2015, 8, 15749–15758. [Google Scholar]
- Miyamoto, K.; Nagaosa, N.; Motoyama, M.; Kataoka, K.; Kusunoki, S. Upregulation of water channel aquaporin-4 in experimental autoimmune encephalomyeritis. J. Neurol. Sci. 2009, 276, 103–107. [Google Scholar] [CrossRef]
- Zamanian, J.L.; Xu, L.; Foo, L.C.; Nouri, N.; Zhou, L.; Giffard, R.G.; Barres, B.A. Genomic analysis of reactive astrogliosis. J. Neurosci. 2012, 32, 6391–6410. [Google Scholar] [CrossRef]
- Li, L.; Zhang, H.; Verkman, A.S. Greatly attenuated experimental autoimmune encephalomyelitis in aquaporin-4 knockout mice. BMC Neurosci. 2009, 10, 94. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhang, H.; Varrin-Doyer, M.; Zamvil, S.S.; Verkman, A.S. Proinflammatory role of aquaporin-4 in autoimmune neuroinflammation. FASEB J. 2011, 25, 1556–1566. [Google Scholar] [CrossRef] [PubMed]
- Pham, H.; Doerrbecker, J.; Ramp, A.A.; D’Souza, C.S.; Gorasia, D.G.; Purcell, A.W.; Ayers, M.M.; Orian, J.M. Experimental autoimmune encephalomyelitis (EAE) IN C57Bl/6 mice is not associated with astrogliosis. J. Neuroimmunol. 2011, 232, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Kaneyama, T.; Takizawa, S.; Tsugane, S.; Yanagisawa, S.; Takeichi, N.; Ehara, T.; Ichikawa, M.; Koh, C.-S. Downregulation of water channel aquaporin-4 in rats with experimental autoimmune encephalomyeritis induced by myelin basic protein. Cell. Immunol. 2013, 281, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Kawasaki, H.; Masaki, K.; Suzuki, S.O.; Terada, T.; Tsuchida, T.; Tokuyama, T.; Kono, S.; Komori, T.; Baba, S.; et al. An Autopsy Case of the Marburg Variant of Multiple Sclerosis (Acute Multiple Sclerosis). Intern. Med. 2013, 52, 1825–1832. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhou, M.; Ding, H.; Xu, S.; Wang, C.; Chan, P. Myelin oligodendrocyte glycoprotein induces aquaporin-4 autoantibodies in mouse experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2013, 261, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, C.; Kirk, J.; Herron, B.; Fitzgerald, U.; McQuaid, S. Absence of aquaporin-4 expression in lesions of neuromyelitis optica but increased expression in multiple sclerosis lesions and normal-appearing white matter. Acta Neuropathol. 2006, 113, 187–194. [Google Scholar] [CrossRef]
- Aoki, K.; Uchihara, T.; Tsuchiya, K.; Nakamura, A.; Ikeda, K.; Wakayama, Y. Enhanced expression of aquaporin 4 in human brain with infarction. Acta Neuropathol. 2003, 106, 121–124. [Google Scholar] [CrossRef]
- Dua, R.K.; Devi, B.I.; Yasha, T.C. Increased expression of Aquaporin-4 and its correlation with contrast enhancement and perilesional edema in brain tumors. Br. J. Neurosurg. 2010, 24, 454–459. [Google Scholar] [CrossRef]
- Ng, W.H.; Hy, J.W.; Tan, W.L.; Liew, D.; Lim, T.; Ang, B.T.; Ng, I. Aquaporin-4 expression is increased in edematous meningiomas. J. Clin. Neurosci. 2009, 16, 441–443. [Google Scholar] [CrossRef]
- Walker-Caulfield, M.E.; Guo, Y.; Johnson, R.K.; McCarthy, C.B.; Fitz-Gibbon, P.D.; Lucchinetti, C.F.; Howe, C.L. NFκB signaling drives pro-granulocytic astroglial responses to neuromyelitis optica patient IgG. J. Neuroinflamm. 2015, 12, 185. [Google Scholar] [CrossRef] [PubMed]
- Hamid, S.H.M.; Whittam, D.; Mutch, K.; Linaker, S.; Solomon, T.; Das, K.; Bhojak, M.; Jacob, A. What proportion of AQP4-IgG-negative NMO spectrum disorder patients are MOG-IgG positive? A cross sectional study of 132 patients. J. Neurol. 2017, 264, 2088–2094. [Google Scholar] [CrossRef] [PubMed]
- Marignier, R.; Bernard-Valnet, R.; Giraudon, P.; Collongues, N.; Papeix, C.; Zephir, H.; Cavillon, G.; Rogemond, V.; Casey, R.; Frangoulis, B.; et al. Aquaporin-4 antibody-negative neuromyelitis optica: Distinct assay sensitivity-dependent entity. Neurology 2013, 80, 2194–2200. [Google Scholar] [CrossRef] [PubMed]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rohr, S.O.; Greiner, T.; Joost, S.; Amor, S.; Valk, P.v.d.; Schmitz, C.; Kipp, M. Aquaporin-4 Expression during Toxic and Autoimmune Demyelination. Cells 2020, 9, 2187. https://doi.org/10.3390/cells9102187
Rohr SO, Greiner T, Joost S, Amor S, Valk Pvd, Schmitz C, Kipp M. Aquaporin-4 Expression during Toxic and Autoimmune Demyelination. Cells. 2020; 9(10):2187. https://doi.org/10.3390/cells9102187
Chicago/Turabian StyleRohr, Sven Olaf, Theresa Greiner, Sarah Joost, Sandra Amor, Paul van der Valk, Christoph Schmitz, and Markus Kipp. 2020. "Aquaporin-4 Expression during Toxic and Autoimmune Demyelination" Cells 9, no. 10: 2187. https://doi.org/10.3390/cells9102187
APA StyleRohr, S. O., Greiner, T., Joost, S., Amor, S., Valk, P. v. d., Schmitz, C., & Kipp, M. (2020). Aquaporin-4 Expression during Toxic and Autoimmune Demyelination. Cells, 9(10), 2187. https://doi.org/10.3390/cells9102187