Neuron-Glia Interactions in Neurodevelopmental Disorders

Abstract
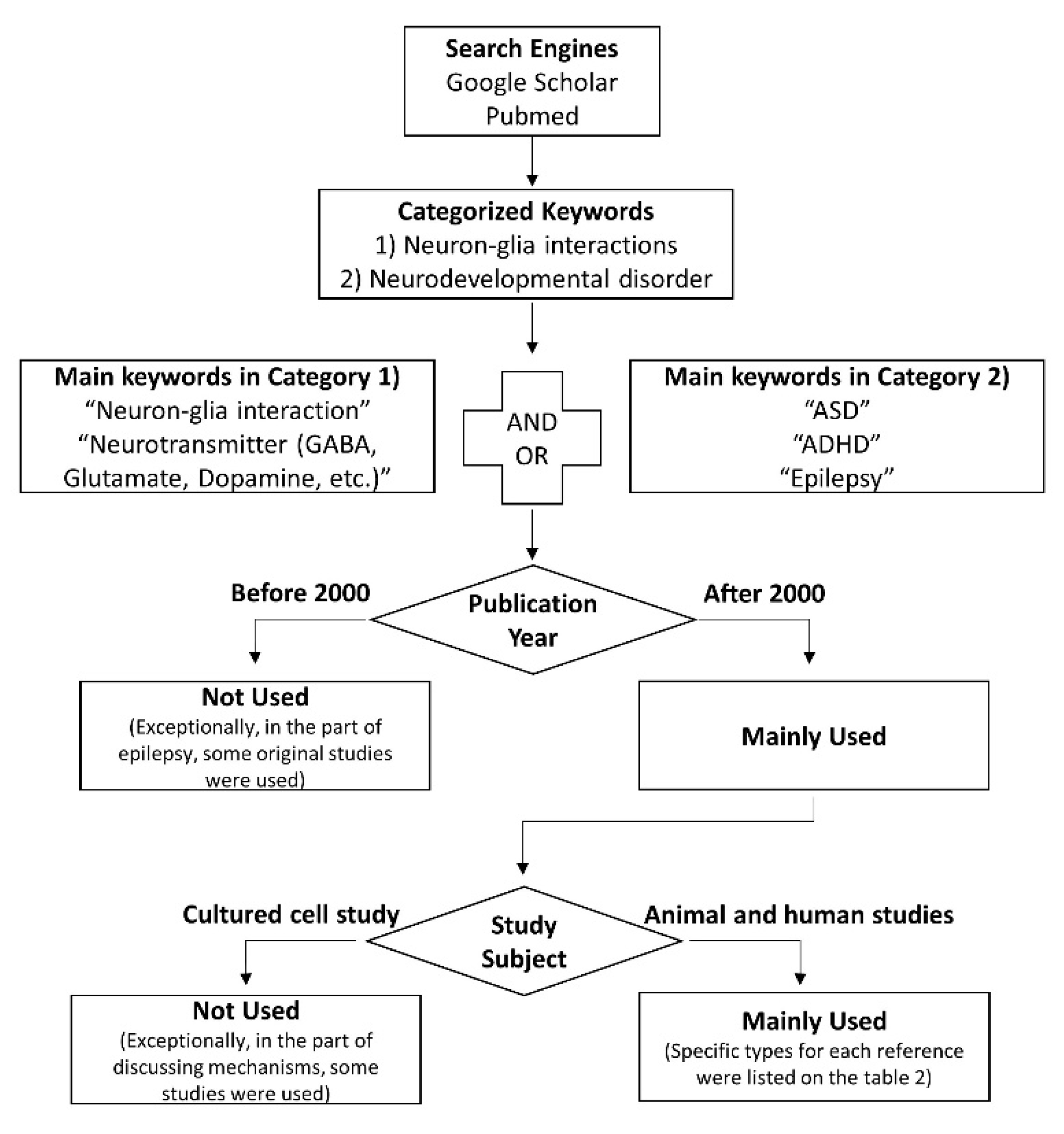
1. Introduction
2. ASD (Autism Spectrum Disorder) and ADHD (Attention-Deficit/Hyperactivity Disorder)
2.1. Neuroinflammation in ASD and ADHD
2.2. Impaired E/I Balance in Patients with ASD and ADHD
3. Epilepsy
3.1. Neuroinflammation in Epilepsy
3.2. Impaired E/I Balance in Epilepsy
4. Conclusions
Funding
Conflicts of Interest
References
- Alyagor, I.; Berkun, V.; Keren-Shaul, H.; Marmor-Kollet, N.; David, E.; Mayseless, O.; Issman-Zecharya, N.; Amit, I.; Schuldiner, O. Combining Developmental and Perturbation-Seq Uncovers Transcriptional Modules Orchestrating Neuronal Remodeling. Dev. Cell 2018, 47, 38–52.e6. [Google Scholar] [CrossRef]
- Puro, D.G.; Nirenberg, M. On the specificity of synapse formation. Proc. Natl. Acad. Sci. USA 1976, 73, 3544–3548. [Google Scholar] [CrossRef] [PubMed]
- Zaki, Y.; Cai, D.J. Creating Space for Synaptic Formation-A New Role for Microglia in Synaptic Plasticity. Cell 2020, 182, 265–267. [Google Scholar] [CrossRef] [PubMed]
- Simhal, A.K.; Zuo, Y.; Perez, M.M.; Madison, D.V.; Sapiro, G.; Micheva, K.D. Multifaceted Changes in Synaptic Composition and Astrocytic Involvement in a Mouse Model of Fragile X Syndrome. Sci. Rep. 2019, 9, 13855. [Google Scholar] [CrossRef]
- Pannasch, U.; Freche, D.; Dallérac, G.; Ghézali, G.; Escartin, C.; Ezan, P.; Cohen-Salmon, M.; Benchenane, K.; Abudara, V.; Dufour, A. Connexin 30 sets synaptic strength by controlling astroglial synapse invasion. Nat. Neurosci. 2014, 17, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Livne-Bar, I.; Lam, S.; Chan, D.; Guo, X.; Askar, I.; Nahirnyj, A.; Flanagan, J.G.; Sivak, J.M. Pharmacologic inhibition of reactive gliosis blocks TNF-α-mediated neuronal apoptosis. Cell Death Dis. 2016, 7, e2386. [Google Scholar] [CrossRef]
- Deverman, B.E.; Patterson, P.H. Cytokines and CNS development. Neuron 2009, 64, 61–78. [Google Scholar] [CrossRef]
- Alvarez-Maubecin, V.; García-Hernández, F.; Williams, J.T.; Van Bockstaele, E.J. Functional coupling between neurons and glia. J. Neurosci. 2000, 20, 4091–4098. [Google Scholar] [CrossRef]
- Parpura, V.; Basarsky, T.A.; Liu, F.; Jeftinija, K.; Jeftinija, S.; Haydon, P.G. Glutamate-mediated astrocyte–neuron signalling. Nature 1994, 369, 744–747. [Google Scholar] [CrossRef]
- Jo, S.; Yarishkin, O.; Hwang, Y.J.; Chun, Y.E.; Park, M.; Woo, D.H.; Bae, J.Y.; Kim, T.; Lee, J.; Chum, H. GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat. Med. 2014, 20, 886–896. [Google Scholar] [CrossRef]
- Heo, J.Y.; Nam, M.H.; Yoon, H.H.; Kim, J.; Hwang, Y.J.; Won, W.; Woo, D.H.; Lee, J.A.; Park, H.J.; Jo, S. Aberrant Tonic Inhibition of Dopaminergic Neuronal Activity Causes Motor Symptoms in Animal Models of Parkinson’s Disease. Curr. Biol. 2020, 30, 276–291. [Google Scholar] [CrossRef] [PubMed]
- Pandit, S.; Neupane, C.; Woo, J.; Sharma, R.; Nam, M.H.; Lee, G.S.; Yi, M.H.; Shin, N.; Kim, D.W.; Cho, H. Bestrophin1-mediated tonic GABA release from reactive astrocytes prevents the development of seizure-prone network in kainate-injected hippocampi. Glia 2020, 68, 1065–1080. [Google Scholar] [CrossRef] [PubMed]
- Eftekharian, M.M.; Ghafouri-Fard, S.; Noroozi, R.; Omrani, M.D.; Arsang-Jang, S.; Ganji, M.; Gharzi, V.; Noroozi, H.; Komaki, A.; Mazdeh, M. Cytokine profile in autistic patients. Cytokine 2018, 108, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Patterson, S.L. Immune dysregulation and cognitive vulnerability in the aging brain: Interactions of microglia, IL-1β, BDNF and synaptic plasticity. Neuropharmacology 2015, 96, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Liimatainen, S.; Fallah, M.; Kharazmi, E.; Peltola, M.; Peltola, J. Interleukin-6 levels are increased in temporal lobe epilepsy but not in extra-temporal lobe epilepsy. J. Neurol. 2009, 256, 796–802. [Google Scholar] [CrossRef] [PubMed]
- Van Vliet, E.A.; da Costa Araujo, S.; Redeker, S.; Van Schaik, R.; Aronica, E.; Gorter, J.A. Blood–brain barrier leakage may lead to progression of temporal lobe epilepsy. Brain 2007, 130, 521–534. [Google Scholar] [CrossRef]
- Elkabes, S.; DiCicco-Bloom, E.M.; Black, I.B. Brain microglia/macrophages express neurotrophins that selectively regulate microglial proliferation and function. J. Neurosci. 1996, 16, 2508–2521. [Google Scholar] [CrossRef]
- Nakajima, K.; Honda, S.; Tohyama, Y.; Imai, Y.; Kohsaka, S.; Kurihara, T. Neurotrophin secretion from cultured microglia. J. Neurosci. Res. 2001, 65, 322–331. [Google Scholar] [CrossRef]
- Jankowsky, J.L.; Patterson, P.H. The role of cytokines and growth factors in seizures and their sequelae. Prog. Neurobiol. 2001, 63, 125–149. [Google Scholar] [CrossRef]
- Zhang, C.; Tabatabaei, M.; Bélanger, S.; Girouard, H.; Moeini, M.; Lu, X.; Lesage, F. Astrocytic endfoot Ca2+ correlates with parenchymal vessel responses during 4-AP induced epilepsy: An in vivo two-photon lifetime microscopy study. J. Cereb. Blood Flow Metab. 2019, 39, 260–271. [Google Scholar] [CrossRef]
- Delorme, R.; Ey, E.; Toro, R.; Leboyer, M.; Gillberg, C.; Bourgeron, T. Progress toward treatments for synaptic defects in autism. Nat. Med. 2013, 19, 685–694. [Google Scholar] [CrossRef]
- Reisinger, S.; Khan, D.; Kong, E.; Berger, A.; Pollak, A.; Pollak, D.D. The poly(I:C)-induced maternal immune activation model in preclinical neuropsychiatric drug discovery. Pharmacol. Ther. 2015, 149, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Neal, A.P.; Guilarte, T.R. Molecular Neurobiology of Lead (Pb2+): Effects on Synaptic Function. Mol. Neurobiol. 2010, 42, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Sloan, S.A.; Barres, B.A. Mechanisms of astrocyte development and their contributions to neurodevelopmental disorders. Curr. Opin. Neurobiol. 2014, 27, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Südhof, T.C. Synaptic neurexin complexes: A molecular code for the logic of neural circuits. Cell 2017, 171, 745–769. [Google Scholar] [CrossRef]
- Loe, I.M.; Feldman, H.M. Academic and educational outcomes of children with ADHD. J. Pediatr. Psychol. 2007, 32, 643–654. [Google Scholar] [CrossRef]
- Pringsheim, T.; Steeves, T. Pharmacological treatment for Attention Deficit Hyperactivity Disorder (ADHD) in children with comorbid tic disorders. Cochrane Database Syst. Rev. 2011. [Google Scholar] [CrossRef]
- Estes, M.L.; McAllister, A.K. Immune mediators in the brain and peripheral tissues in autism spectrum disorder. Nat. Rev. Neurosci. 2015, 16, 469–486. [Google Scholar] [CrossRef]
- Aloisi, F. Immune function of microglia. Glia 2001, 36, 165–179. [Google Scholar] [CrossRef]
- Sofroniew, M.V. Astrocyte barriers to neurotoxic inflammation. Nat. Rev. Neurosci. 2015, 16, 249–263. [Google Scholar] [CrossRef]
- Greter, M.; Merad, M. Regulation of microglia development and homeostasis. Glia 2013, 61, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Hanisch, U.K.; Kettenmann, H. Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 2007, 10, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Russell, V.A. Overview of animal models of attention deficit hyperactivity disorder (ADHD). Curr. Protoc. Neurosci. 2011, 54, 9–35. [Google Scholar] [CrossRef] [PubMed]
- Pajarillo, E.; Rizor, A.; Lee, J.; Aschner, M.; Lee, E. The role of astrocytic glutamate transporters GLT-1 and GLAST in neurological disorders: Potential targets for neurotherapeutics. Neuropharmacology 2019, 161, 107559. [Google Scholar] [CrossRef] [PubMed]
- McBean, G.J. Cerebral cystine uptake: A tale of two transporters. Trends Pharmacol. Sci. 2002, 23, 299–302. [Google Scholar] [CrossRef]
- Schmidt-Wilcke, T.; Fuchs, E.; Funke, K.; Vlachos, A.; Müller-Dahlhaus, F.; Puts, N.A.J.; Harris, R.E.; Edden, R.A.E. GABA—From inhibition to cognition: Emerging concepts. Neuroscientist 2018, 24, 501–515. [Google Scholar] [CrossRef]
- Roux, L.; Buzsáki, G. Tasks for inhibitory interneurons in intact brain circuits. Neuropharmacology 2015, 88, 10–23. [Google Scholar] [CrossRef]
- Pizzarelli, R.; Cherubini, E. Alterations of GABAergic signaling inautism spectrum disorders. Neural Plast. 2011, 297153. [Google Scholar] [CrossRef]
- Purkayastha, P.; Malapati, A.; Yogeeswari, P.; Sriram, D. A review on GABA/glutamate pathway for therapeutic intervention of ASD and ADHD. Curr. Med. Chem. 2015, 22, 1850–1859. [Google Scholar] [CrossRef]
- Hubbard, J.A.; Hsu, M.S.; Fiacco, T.A.; Binder, D.K. Glial cell changes in epilepsy: Overview of the clinical problem and therapeutic opportunities. Neurochem. Int. 2013, 63, 638–651. [Google Scholar] [CrossRef]
- Anderson, C.M.; Swanson, R.A. Astrocyte glutamate transport: Review of properties, regulation, and physiological functions. Glia 2000, 32, 1–14. [Google Scholar] [CrossRef]
- Chapman, A.G. Glutamate and epilepsy. J. Nutr. 2000, 130, 1043S–1045S. [Google Scholar] [CrossRef]
- Treiman, D.M. GABAergic mechanisms in epilepsy. Epilepsia 2001, 42, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Eroglu, C.; Barres, B.A. Regulation of synaptic connectivity by glia. Nature 2010, 468, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Coulter, D.A.; Eid, T. Astrocytic regulation of glutamate homeostasis in epilepsy. Glia 2012, 60, 1215–1226. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A. Anti-inflammatory drugs in epilepsy: Does it impact epileptogenesis? Expert Opin. Drug Saf. 2015, 14, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A.; Moneta, D.; Richichi, C.; Perego, C.; De Simoni, M.G. Functional role of proinflammatory and anti-inflammatory cytokines in seizures. Adv. Exp. Med. Biol. 2004, 548, 123–133. [Google Scholar] [CrossRef]
- Vezzani, A.; Ravizza, T.; Balosso, S.; Aronica, E. Glia as a source of cytokines: Implications for neuronal excitability and survival. Epilepsia 2008, 49, 24–32. [Google Scholar] [CrossRef]
- Hanisch, U.K. Microglia as a source and target of cytokines. Glia 2002, 40, 140–155. [Google Scholar] [CrossRef]
- Santello, M.; Volterra, A. TNFα in synaptic function: Switching gears. Trends Neurosci. 2012, 35, 638–647. [Google Scholar] [CrossRef]
- Suzumura, A.; Takeuchi, H.; Zhang, G.; Kuno, R.; Mizuno, T. Roles of glia-derived cytokines on neuronal degeneration and regeneration. Ann. N. Y. Acad. Sci. 2006, 1088, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Bernardino, L.; Ferreira, R.; Cristovao, A.J.; Sales, F.; Malva, J.O. Inflammation and neurogenesis in temporal lobe epilepsy. Curr. Drug Targets CNS Neurol. Disord. 2005, 4, 349–360. [Google Scholar] [CrossRef] [PubMed]
- MacVicar, B.A.; Newman, E.A. Astrocyte regulation of blood flow in the brain. Cold Spring Harbor Perspect. Biol. 2015, 7, 1290–1300. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, U.; Kaufer, D.; Friedman, A. Blood-brain barrier dysfunction, TGFβ signaling, and astrocyte dysfunction in epilepsy. Glia 2012, 60, 1251–1257. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Qin, Z.H. Molecular and cellular mechanisms of excitotoxic neuronal death. Apoptosis 2010, 15, 1382–1402. [Google Scholar] [CrossRef]
- Barreto, G.E.; Gonzalez, J.; Torres, Y.; Morales, L. Astrocytic-neuronal crosstalk: Implications for neuroprotection from brain injury. Neurosci. Res. 2011, 71, 107–113. [Google Scholar] [CrossRef]
- De Simoni, M.G.; Imeri, L. Cytokine-neurotransmitter interactions in the brain. Neurosignals Recept 1998, 7, 33–44. [Google Scholar] [CrossRef]
- Vesce, S.; Rossi, D.; Brambilla, L.; Volterra, A. Glutamate release from astrocytes in physiological conditions and in neurodegenerative disorders characterized by neuroinflammation. Int. Rev. Neurobiol. 2007, 82, 57–71. [Google Scholar] [CrossRef]
- Choi, D.W.; Rothman, S.M. The role of glutamate neurotoxicity in hypoxic-ischemic neuronal death. Annu. Rev. Neurosci. 1990, 13, 171–182. [Google Scholar] [CrossRef]
- Hertz, L.; Dringen, R.; Schousboe, A.; Robinson, S.R. Astrocytes: Glutamate producers for neurons. J. Neurosci. Res. 1999, 57, 417–428. [Google Scholar] [CrossRef]
- Tilleux, S.; Hermans, E. Neuroinflammation and regulation of glial glutamate uptake in neurological disorders. J. Neurosci. Res. 2007, 85, 2059–2070. [Google Scholar] [CrossRef] [PubMed]
- Kreiser, N.L.; White, S.W. ASD in females: Are we overstating the gender difference in diagnosis? Clin. Child Fam. Psychol. Rev. 2014, 17, 67–84. [Google Scholar] [CrossRef] [PubMed]
- Horváth, G.; Otrokocsi, L.; Beko, K.; Baranyi, M.; Kittel, Á.; Fritz-Ruenes, P.A.; Sperlágh, B. P2X7 Receptors Drive Poly(I:C) Induced Autism-like Behavior in Mice. J. Neurosci. 2019, 39, 2542–2561. [Google Scholar] [CrossRef] [PubMed]
- Tsai, P.T.; Hull, C.; Chu, Y.; Greene-Colozzi, E.; Sadowski, A.R.; Leech, J.M.; Steinberg, J.; Crawley, J.N.; Regehr, W.G.; Sahin, M. Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature 2012, 488, 647–651. [Google Scholar] [CrossRef]
- Chao, H.T.; Chen, H.; Samaco, R.C.; Xue, M.; Chahrour, M.; Yoo, J.; Neul, J.L.; Gong, S.; Lu, H.; Heintz, N. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature 2010, 468, 263–269. [Google Scholar] [CrossRef]
- Han, S.; Tai, C.; Westenbroek, R.E.; Yu, F.H.; Cheah, C.S.; Potter, G.B.; Rubenstein, J.L.; Scheuer, T.; De la Iglesia, H.O.; Catterall, W.A. Autistic-like behaviour in Scn1a+/−mice and rescue by enhanced GABA-mediated neurotransmission. Nature 2012, 489, 385–390. [Google Scholar] [CrossRef]
- Hammer, M.; Krueger-Burg, D.; Tuffy, L.P.; Cooper, B.H.; Taschenberger, H.; Goswami, S.P.; Ehrenreich, H.; Jonas, P.; Variqueaux, F.; Rhee, J.S. Perturbed hippocampal synaptic inhibition and γ-oscillations in a neuroligin-4 knockout mouse model of autism. Cell Rep. 2015, 13, 516–523. [Google Scholar] [CrossRef]
- Won, H.; Mah, W.; Kim, E.; Kim, J.W.; Hahm, E.K.; Kim, M.H.; Cho, S.; Kim, J.; Jang, H.; Cho, S.C. GIT1 is associated with ADHD in humans and ADHD-like behaviors in mice. Nat. Med. 2011, 17, 566–572. [Google Scholar] [CrossRef]
- Moore, A.H.; Wu, M.; Shaftel, S.S.; Graham, K.A.; O’Banion, M.K. Sustained expression of interleukin-1β in mouse hippocampus impairs spatial memory. Neuroscience 2009, 164, 1484–1495. [Google Scholar] [CrossRef]
- Gogolla, N.; Takesian, A.E.; Feng, G.; Fagiolini, M.; Hensch, T.K. Sensory integration in mouse insular cortex reflects GABA circuit maturation. Neuron 2014, 83, 894–905. [Google Scholar] [CrossRef]
- Gumusoglu, S.B.; Fine, R.S.; Murray, S.J.; Bittle, J.L.; Stevens, H.E. The role of IL-6 in neurodevelopment after prenatal stress. Brain Behav. Immun. 2017, 65, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Rivero, O.; Selten, M.M.; Sich, S.; Popp, S.; Bacmeister, L.; Amendola, E.; Negwer, M.; Schuber, D.; Proft, F.; Kiser, D. Cadherin-13, a risk gene for ADHD and comorbid disorders, impacts GABAergic function in hippocampus and cognition. Transl. Psychiatry 2015, 5, e655. [Google Scholar] [CrossRef] [PubMed]
- Kozłowska, A.; Wojtacha, P.; Równiak, M.; Kolenkiewicz, M.; Huang, A.C.W. ADHD pathogenesis in the immune, endocrine and nervous systems of juvenile and maturating SHR and WKY rats. Psychopharmacology 2019, 236, 2937–2958. [Google Scholar] [CrossRef] [PubMed]
- Higashimori, H.; Schin, C.S.; Chiang, M.S.R.; Morel, L.; Shoneye, T.A.; Nelson, D.L.; Yang, Y. Selective deletion of astroglial FMRP dysregulates glutamate transporter GLT1 and contributes to fragile X syndrome phenotypes in vivo. J. Neurosci. 2016, 36, 7079–7094. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, R.M.; Tanaka, K.; Heilig, M.; Holmes, A. Loss of glial glutamate and aspartate transporter (excitatory amino acid transporter 1) causes locomotor hyperactivity and exaggerated responses to psychotomimetics: Rescue by haloperidol and metabotropic glutamate 2/3 agonist. Biol. Psychiatry 2008, 64, 810–814. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.; Yang, Y.; Rothstein, J.D.; Robinson, M.B. Nuclear factor-κB contributes to neuron-dependent induction of glutamate transporter-1 expression in astrocytes. J. Neurosci. 2011, 31, 9159–9169. [Google Scholar] [CrossRef] [PubMed]
- Omrani, A.; Melone, M.; Bellesi, M.; Safiulina, V.; Aida, T.; Tanaka, K.; Cherubin, E.; Conti, F. Up-regulation of GLT-1 severely impairs LTD at mossy fibre—CA3 synapses. J. Physiol. 2009, 587 Pt 19, 4575–4588. [Google Scholar] [CrossRef]
- Qin, S.; Colin, C.; Hinners, I.; Gervais, A.; Cheret, C.; Mallat, M. System xc− and apolipoprotein E expressed by microglia have opposite effects on the neurotoxicity of amyloid-β peptide 1–40. J. Neurosci. 2006, 26, 3345–3356. [Google Scholar] [CrossRef]
- Ribak, C.E.; Tong, W.M.; Brecha, N.C. GABA plasma membrane transporters, GAT-1 and GAT-3, display different distributions in the rat hippocampus. J. Comp. Neurol. 1996, 367, 595–606. [Google Scholar] [CrossRef]
- Boddum, K.; Jensen, T.P.; Magloire, V.; Kristiansen, U.; Rusakov, D.A.; Pavlov, I.; Walker, M.C. Astrocytic GABA transporter activity modulates excitatory neurotransmission. Nat. Commun. 2016, 7, 1–10. [Google Scholar] [CrossRef]
- Yoon, B.E.; Woo, J.; Chun, Y.E.; Chun, H.; Jo, S.; Bae, J.Y.; Lee, C.J. Glial GABA, synthesized by monoamine oxidase B, mediates tonic inhibition. J. Physiol. 2014, 592, 4951–4968. [Google Scholar] [CrossRef] [PubMed]
- Silverman, J.L.; Pride, M.C.; Hayes, J.E.; Puhger, K.R.; Butler-Struben, H.M.; Baker, S.; Crawley, J.N. GABA B receptor agonist R-baclofen reverses social deficits and reduces repetitive behavior in Two mouse models of autism. Neuropsychopharmacology 2015, 40, 2228–2239. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Woo, J.; Lee, C.J.; Yoon, B.E. Decreased glial GABA and tonic inhibition in cerebellum of mouse model for attention-deficit/hyperactivity disorder (ADHD). Exp. Neurobiol. 2017, 26, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Sterley, T.L.; Howells, F.M.; Russell, V.A. Evidence for reduced tonic levels of GABA in the hippocampus of an animal model of ADHD, the spontaneously hypertensive rat. Brain Res. 2013, 1541, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Yang, X.; Zhou, X.; Wang, C.; Gong, X.; Chen, B.; Chen, Y. Hyperactivity and impaired attention in Gamma aminobutyric acid transporter subtype 1 gene knockout mice. Acta Neuropsychiatr. 2015, 27, 368–374. [Google Scholar] [CrossRef]
- Nagai, J.; Rajbhandari, A.K.; Gangwani, M.R.; Hachisuka, A.; Coppola, G.; Masmanidis, S.C.; Fanselow, M.S.; Khakh, B.S. Hyperactivity with disrupted attention by activation of an astrocyte synaptogenic cue. Cell 2019, 177, 1280–1292. [Google Scholar] [CrossRef]
- Lee, S.; Yoon, B.E.; Berglund, K.; Oh, S.J.; Park, H.; Shin, H.S.; Augustine, G.J.; Lee, C.J. Channel-mediated tonic GABA release from glia. Science 2010, 330, 790–796. [Google Scholar] [CrossRef]
- Buckingham, S.C.; Campbell, S.L.; Haas, B.R.; Montana, V.; Robel, S.; Ogunrinu, T.; Sontheimer, H. Glutamate release by primary brain tumors induces epileptic activity. Nat. Med. 2011, 17, 1269–1274. [Google Scholar] [CrossRef]
- Lahuerta, M.; Gonzalez, D.; Aguado, C.; Fathinajafabadi, A.; García-Giménez, J.L.; Moreno-Estellés, M.; Romá-Mateo, C.; Knecht, E.; Pallardó, F.V.; Sanz, P. Reactive Glia-Derived Neuroinflammation: A Novel Hallmark in Lafora Progressive Myoclonus Epilepsy That Progresses with Age. Mol. Neurobiol. 2020, 57, 1607–1621. [Google Scholar] [CrossRef]
- Rubio-Villena, C.; Viana, R.; Bonet, J.; Garcia-Gimeno, M.A.; Casado, M.; Heredia, M.; Sanz, P. Astrocytes: New players in progressive myoclonus epilepsy of Lafora type. Hum. Mol. Genet. 2018, 27, 1290–1300. [Google Scholar] [CrossRef]
- Stellwagen, D.; Malenka, R.C. Synaptic scaling mediated by glial TNF-α. Nature 2006, 440, 1054–1059. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.H.; Bero, A.W.; Zhang, B.; Holtzman, D.M.; Wong, M. Modulation of astrocyte glutamate transporters decreases seizures in a mouse model of Tuberous Sclerosis Complex. Neurobiol. Dis. 2010, 37, 764–771. [Google Scholar] [CrossRef] [PubMed]
- Truccolo, W.; Donoghue, J.A.; Hochberg, L.R.; Eskandar, E.N.; Madsen, J.R.; Anderson, W.S.; Brown, E.N.; Halgren, E.; Cash, S.S. Single-neuron dynamics in human focal epilepsy. Nat. Neurosci. 2011, 14, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Iyer, A.; Zurolo, E.; Prabowo, A.; Fluiter, K.; Spliet, W.G.; van Rijen, P.C.; Gorter, J.A.; Aronica, E. MicroRNA-146a: A key regulator of astrocyte-mediated inflammatory response. PLoS ONE 2012, 7, e44789. [Google Scholar] [CrossRef]
- Héja, L.; Barabás, P.; Nyitrai, G.; Kékesi, K.A.; Lasztóczi, B.; Toke, O.; Tárkányi, G.; Madsen, K.; Schousboe, A.; Dobolyi, A.; et al. Glutamate uptake triggers transporter-mediated GABA release from astrocytes. PLoS ONE 2009, 4, e7153. [Google Scholar] [CrossRef]
- Hu, S.; Sheng, W.S.; Ehrlich, L.C.; Peterson, P.K.; Chao, C.C. Cytokine effects on glutamate uptake by human astrocytes. Neuroimmunomodulation 2000, 7, 153–159. [Google Scholar] [CrossRef]
- Yao, C.; He, Z.; Nakano, T.; Shuai, J. Spiking patterns of a neuron model to stimulus: Rich dynamics and oxygen’s role. Chaos 2018, 28, 1–8. [Google Scholar] [CrossRef]
- Savin, C.; Triesch, J.; Meyer-Hermann, M. Epileptogenesis due to glia-mediated synaptic scaling. J. R. Soc. Interface 2009, 6, 655–668. [Google Scholar] [CrossRef]
- Hsiao, E.Y.; McBride, S.W.; Hsien, S.; Sharon, G.; Hyde, E.R.; McCue, T.; Codelli, J.A.; Chow, J.; Reisman, S.E.; Petrosino, J.F. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 2013, 155, 1451–1463. [Google Scholar] [CrossRef]
- Choi, G.B.; Yim, Y.S.; Wong, H.; Kim, S.; Kim, H.; Kim, S.V.; Hoeffer, C.A.; Littman, D.R.; Huh, J.R. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science 2016, 351, 933–939. [Google Scholar] [CrossRef]
- Shi, L.; Smith, S.E.; Malkova, N.; Tse, D.; Su, Y.; Patterson, P.H. Activation of the maternal immune system alters cerebellar development in the offspring. Brain Behav. Immun. 2009, 23, 116–123. [Google Scholar] [CrossRef]
- Lombardo, M.V.; Moon, H.M.; Su, J.; Palmer, T.D.; Courchesne, E.; Pramparo, T. Maternal immune activation dysregulation of the fetal brain transcriptome and relevance to the pathophysiology of autism spectrum disorder. Mol. Psychiatry 2018, 23, 1001–1013. [Google Scholar] [CrossRef]
- Wei, H.; Chadman, K.K.; McCloskey, D.P.; Sheikh, A.M.; Malik, M.; Brown, W.T.; Li, X. Brain IL-6 elevation causes neuronal circuitry imbalances and mediates autism-like behaviors. Biochim. Biophys. Acta 2012, 1822, 831–842. [Google Scholar] [CrossRef]
- Carias, E.; Hamilton, J.; Robison, L.S.; Delis, F.; Eiden, R.; Quattrin, T.; Hadjiargyrou, M.; Komatsu, D.; Thanos, P.K. Chronic oral methylphenidate treatment increases microglial activation in rats. J. Neural Transm. 2018. [Google Scholar] [CrossRef]
- Sadasivan, S.; Pond, B.B.; Pani, A.K.; Qu, C.; Jiao, Y.; Smeyne, R.J. Methylphenidate exposure induces dopamine neuron loss and activation of microglia in the basal ganglia of mice. PLoS ONE 2012, 7, e33693. [Google Scholar] [CrossRef]
- Lucchina, L.; Depino, A.M. Altered peripheral and central inflammatory responses in a mouse model of autism. Autism Res. 2014, 7, 273–289. [Google Scholar] [CrossRef]
- Taguchi, K.; Tamba, M.; Bannai, S.; Sato, H. Induction of cystine/glutamate transporter in bacterial lipopolysaccharide induced endotoxemia in mice. J. Inflamm. 2007, 4, 20. [Google Scholar] [CrossRef]
- Gao, W.; Bi, Y.; Ding, L.; Zhu, W.; Ye, M. SSa ameliorates the Glu uptaking capacity of astrocytes in epilepsy via AP-1/miR-155/GLAST. Biochem. Biophys. Res. Commun. 2017, 493, 1329–1335. [Google Scholar] [CrossRef]
- Schousboe, A.; Larsson, O.M.; Wood, J.D.; Krogsgaard-Larsen, P. Transport and Metabolism of 7-Aminobutyric Acid in Neurons and Glia: Implications for Epilepsy. Epilepsia 1983, 24, 531–538. [Google Scholar] [CrossRef]
- Betti, M.; Minelli, A.; Ambrogini, P.; Ciuffoli, S.; Viola, V.; Galli, F.; Cuppini, R. Dietary supplementation with α-tocopherol reduces neuroinflammation and neuronal degeneration in the rat brain after kainic acid-induced status epilepticus. Free Radic. Res. 2011, 45, 1136–1142. [Google Scholar] [CrossRef]
- Chen, S.; Zeng, X.; Zong, W.; Wang, X.; Chen, L.; Zhou, L.; Li, C.; Huang, Q.; Huang, X.; Zeng, G. Aucubin alleviates seizures activity in Li-Pilocarpine-Induced epileptic mice: Involvement of inhibition of neuroinflammation and regulation of neurotransmission. Neurochem. Res. 2019, 44, 472–484. [Google Scholar] [CrossRef]
- Ambrogini, P.; Minelli, A.; Galati, C.; Betti, M.; Lattanzi, D.; Ciffolilli, S.; Piroddi, M.; Galli, F.; Cuppini, R. Post-seizure α-tocopherol treatment decreases neuroinflammation and neuronal degeneration induced by status epilepticus in rat hippocampus. Mol. Neurobiol. 2014, 50, 246–256. [Google Scholar] [CrossRef]
- Steinmetz, C.C.; Turrigiano, G.G. Tumor necrosis factor-α signaling maintains the ability of cortical synapses to express synaptic scaling. J. Neurosci. 2010, 30, 14685–14690. [Google Scholar] [CrossRef]
- Rappold, P.M.; Lynd-Balta, E.; Joseph, S.A. P2X7 receptor immunoreactive profile confined to resting and activated microglia in the epileptic brain. Brain Res. 2006, 1089, 171–178. [Google Scholar] [CrossRef]
- Zhang, N.; Wei, W.; Mody, I.; Houser, C.R. Altered localization of GABAA receptor subunits on dentate granule cell dendrites influences tonic and phasic inhibition in a mouse model of epilepsy. J. Neurosci. 2007, 27, 7520–7531. [Google Scholar] [CrossRef]
- Labarrera, C.; London, M.; Angelo, K. Tonic inhibition sets the state of excitability in olfactory bulb granule cells. J. Physiol. 2013, 591, 1841–1850. [Google Scholar] [CrossRef]
- Horton, R.W.; Collins, J.F.; Anlezark, G.M.; Meldrum, B.S. Convulsant and anticonvulsant actions in DBA/2 mice of compounds blocking the reuptake of GABA. Eur. J. Pharmacol. 1979, 59, 75–83. [Google Scholar] [CrossRef]
- Deshpande, T.; Li, T.; Henning, L.; Wu, Z.; Müller, J.; Seifert, G.; Steinhäuser, C.; Bedner, P. Constitutive deletion of astrocytic connexins aggravates kainate-induced epilepsy. Glia 2020, 68, 2136–2147. [Google Scholar] [CrossRef]
- Takeuchi, H.; Jin, S.; Wang, J.; Zhang, G.; Kawanokuchi, J.; Kuno, R.; Sonobe, Y.; Mizuno, T.; Suzumura, A. Tumor necrosis factor-α induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J. Biol. Chem. 2006, 281, 21362–21368. [Google Scholar] [CrossRef]
- Okada, M.; Fukuyama, K.; Shiroyama, T.; Ueda, Y. Carbamazepine attenuates astroglial l-glutamate release induced by pro-inflammatory cytokines via chronically activation of adenosine A2A receptor. Int. J. Mol. Sci. 2019, 20, 3727. [Google Scholar] [CrossRef]
- Kong, Q.; Takahashi, K.; Schulte, D.; Stouffer, N.; Lin, Y.; Lin, C.L.G. Increased glial glutamate transporter EAAT2 expression reduces epileptogenic processes following pilocarpine-induced status epilepticus. Neurobiol. Dis. 2012, 47, 145–154. [Google Scholar] [CrossRef]
- Coiret, G.; Ster, J.; Grewe, B.; Wendling, F.; Helmchen, F.; Gerber, U.; Benquet, P. Neuron to astrocyte communication via cannabinoid receptors is necessary for sustained epileptiform activity in rat hippocampus. PLoS ONE 2012, 7, e37320. [Google Scholar] [CrossRef]
- Maenner, M.J.; Shaw, K.A.; Baio, J.; Washington, A.; Patrick, M.; DiRienzo, M.; Christensen, D.L.; Wiggins, L.D.; Pettygrove, S.; Andrews, J.G. Prevalence of autism spectrum disorder among children aged 8 years—Autism and developmental disabilities monitoring network, 11 sites, United States, 2016. MMWR Surveill. Summ. 2020, 69, 1. [Google Scholar] [CrossRef]
- Lee, B.K.; Magnusson, C.; Gardner, R.M.; Blomström, Å.; Newschaffer, C.J.; Burstyn, I.; Karlsson, H.; Dalman, C. Maternal hospitalization with infection during pregnancy and risk of autism spectrum disorders. Brain Behav. Immun. 2015, 44, 100–105. [Google Scholar] [CrossRef]
- Kim, S.M.; Han, D.H.; Lyoo, H.S.; Min, K.J.; Kim, K.H.; Renshaw, P. Exposure to environmental toxins in mothers of children with autism spectrum disorder. Psychiatry Investig. 2010, 7, 122–127. [Google Scholar] [CrossRef]
- Croen, L.A.; Grether, J.K.; Yoshida, C.K.; Odouli, R.; Van de Water, J. Maternal autoimmune diseases, asthma and allergies, and childhood autism spectrum disorders: A case-control study. Arch. Pediatr. Adolesc. Med. 2005, 159, 151–157. [Google Scholar] [CrossRef]
- Goines, P.E.; Croen, L.A.; Braunschweig, D.; Yoshida, C.K.; Grether, J.; Hansen, R.; Kharrazi, M.; Ashwood, P.; Van de Water, J. Increased midgestational IFN-γ, IL-4 and IL-5 in women bearing a child with autism: A case-control study. Mol. Autism 2011, 2, 13. [Google Scholar] [CrossRef]
- Jones, K.L.; Croen, L.A.; Yoshida, C.K.; Heuer, L.; Hansen, R.; Zerbo, O.; DeLorenze, G.N.; Kharrazi, M.; Yolken, R.; Ashwood, P. Autism with intellectual disability is associated with increased levels of maternal cytokines and chemokines during gestation. Mol. Psychiatry 2017, 22, 273–279. [Google Scholar] [CrossRef]
- Danielson, M.L.; Bitsko, R.H.; Ghandour, R.M.; Holbrook, J.R.; Kogan, M.D.; Blumberg, S.J. Prevalence of parent-reported ADHD diagnosis and associated treatment among US children and adolescents, 2016. J. Clin. Child Adolesc. Psychol. 2018, 47, 199–212. [Google Scholar] [CrossRef]
- Ashwood, P.; Krakowiak, P.; Hertz-Picciotto, I.; Hansen, R.; Pessah, I.; Van de Water, J. Elevated plasma cytokines in autism spectrum disorders provide evidence of immune dysfunction and are associated with impaired behavioral outcome. Brain Behav. Immun. 2011, 25, 40–45. [Google Scholar] [CrossRef]
- Li, X.; Chauhan, A.; Sheikh, A.M.; Patil, S.; Chauhan, V.; Li, X.M.; Ji, L.; Brown, T.; Malik, M. Elevated immune response in the brain of autistic patients. J. Neuroimmunol. 2009, 207, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Tsilioni, I.; Patel, A.B.; Pantazopoulos, H.; Berretta, S.; Conti, P.; Leeman, S.E.; Theoharides, T.C. IL-37 is increased in brains of children with autism spectrum disorder and inhibits human microglia stimulated by neurotensin. Proc. Natl. Acad. Sci. USA 2019, 116, 21659–21665. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Zou, H.; Sheikh, A.M.; Malik, M.; Dobkin, C.; Brown, W.T.; Li, X. IL-6 is increased in the cerebellum of autistic brain and alters neural cell adhesion, migration and synaptic formation. J. Neuroinflamm. 2011, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Darwish, A.H.; Elgohary, T.M.; Nosair, N.A. Serum Interleukin-6 Level in Children with Attention-Deficit Hyperactivity Disorder (ADHD). J. Child Neurol. 2019, 34, 61–67. [Google Scholar] [CrossRef]
- Khalifa, D.; Shahin, O.; Salem, D.; Raafat, O. Serum glutamate was elevated in children aged 3–10 years with autism spectrum disorders when they were compared with controls. Acta Paediatr. 2019, 108, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, J.D.; Martin, L.J.; Kuncl, R.W. Decreased glutamate transport by the brain and spinal cord in amyotrophic lateral sclerosis. N. Engl. J. Med. 1992, 326, 1464–1468. [Google Scholar] [CrossRef]
- Edden, R.A.; Crocetti, D.; Zhu, H.; Gilbert, D.L.; Mostofsky, S.H. Reduced GABA concentration in attention-deficit/hyperactivity disorder. Arch. Gen. Psychiatry 2012, 69, 750–753. [Google Scholar] [CrossRef]
- Puts, N.A.; Ryan, M.; Oeltzschner, G.; Horska, A.; Edden, R.A.; Mahone, E.M. Reduced striatal GABA in unmedicated children with ADHD at 7T. Pscyhiatry Res. Neuroimaging 2020, 301, 111082. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, Q.; Liu, M.; Wang, H.; Dong, Y.; Ji, T.; Liu, X.; Jiang, Y.; Cai, L.; Wu, Y. Upregulation of HMGB1-TLR4 inflammatory pathway in focal cortical dysplasia type II. J. Neuroinflamm. 2018, 15, 1–11. [Google Scholar] [CrossRef]
- Ravizza, T.; Boer, K.; Redeker, S.; Spliet, W.G.M.; Van Rijen, P.C.; Troost, D.; Vezzani, A.; Aronica, E. The IL-1β system in epilepsy-associated malformations of cortical development. Neurobiol. Dis. 2006, 24, 128–143. [Google Scholar] [CrossRef]
- Chao, C.C.; Hu, S.; Sheng, W.S.; Bu, D.; Bukrinsky, M.I.; Peterson, P.K. Cytokine-stimulated astrocytes damage human neurons via a nitric oxide mechanism. Glia 1996, 16, 276–284. [Google Scholar] [CrossRef]
- Choi, J.; Nordli, D.R.; Alden, T.D.; DiPatri, A.; Laux, L.; Kelley, K.; Rosenow, J.; Schuele, S.U.; Raharam, V.; Koh, S. Cellular injury and neuroinflammation in children with chronic intractable epilepsy. J. Neuroinflamm. 2009, 6, 1–14. [Google Scholar] [CrossRef]
- Lu, J.; Huang, H.; Zeng, Q.; Zhang, X.; Xu, M.; Cai, Y.; Wang, Q.; Huang, Y.; Peng, Q.; Deng, L. Hippocampal neuron loss and astrogliosis in medial temporal lobe epileptic patients with mental disorders. J. Integr. Neurosci. 2019, 18, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Dam, A.M. Epilepsy and neuron loss in the hippocampus. Epilepsia 1980, 21, 617–629. [Google Scholar] [CrossRef]
- Eid, T.; Thomas, M.J.; Spencer, D.D.; Runden-Pran, E.; Lai, J.C.K.; Malthankar, G.V.; Kim, J.H.; Danbolt, N.C.; Ottersen, O.P.; De Lanerolle, N.C. Loss of glutamine synthetase in the human epileptogenic hippocampus: Possible mechanism for raised extracellular glutamate in mesial temporal lobe epilepsy. Lancet 2004, 363, 28–37. [Google Scholar] [CrossRef]
- Bedner, P.; Dupper, A.; Hüttmann, K.; Müller, J.; Herde, M.K.; Dublin, P.; Deshpande, T.; Schramm, J.; Häussler, U.; Hass, C.A. Astrocyte uncoupling as a cause of human temporal lobe epilepsy. Brain 2015, 138, 1208–1222. [Google Scholar] [CrossRef] [PubMed]
- Conlon, O.; Volden, J.; Smith, I.M.; Duku, E.; Zwaigenbaum, L.; Waddell, C.; Szatmari, P.; Mirenda, P.; Vaillancourt, T.; Bennett, T. Gender differences in pragmatic communication in school-aged children with autism spectrum disorder (ASD). J. Autism Dev. Disord. 2019, 49, 1937–1948. [Google Scholar] [CrossRef]
- Lopez, R.; Dauvilliers, Y.; Jaussent, I.; Billieux, J.; Bayard, S. A multidimensional approach of impulsivity in adult attention deficit hyperactivity disorder. Psychiatry Res. 2015, 227, 290–295. [Google Scholar] [CrossRef]
- Demontis, D.; Walters, R.K.; Martin, J.; Mattheisen, M.; Als, T.D.; Agerbo, E.; Baldursson, G.; Belliveau, R.; Bybjerg-Grauholm, J.; Bækvad-Hansen, M. Discovery of the first genome-wide significant risk loci for attention deficit/hyperactivity disorder. Nat. Genet. 2019, 51, 63–75. [Google Scholar] [CrossRef]
- Joseph, N.; Zhang-James, Y.; Perl, A.; Faraone, S.V. Oxidative stress and ADHD: A meta-analysis. J. Atten. Disord. 2015, 19, 915–924. [Google Scholar] [CrossRef]
- Bilbo, S.D.; Tsang, V. Enduring consequences of maternal obesity for brain inflammation and behavior of offspring. FASEB J. 2010, 24, 2104–2115. [Google Scholar] [CrossRef]
- Fonfría, E.; Rodríguez-Farré, E.; Suñol, C. Mercury interaction with the GABA(A) receptor modulates the benzodiazepine binding site in primary cultures of mouse cerebellar granule cells. Neuropharmacology 2001, 41, 819–833. [Google Scholar] [CrossRef]
- Allen, J.W.; Mutkus, L.A.; Aschner, M. Mercuric chloride, but not methylmercury, inhibits glutamine synthetase activity in primary cultures of cortical astrocytes. Brain Res. 2001, 891, 148–157. [Google Scholar] [CrossRef]
- Lau, L.T.; Yu, A.C.H. Astrocytes produce and release interleukin-1, interleukin-6, tumor necrosis factor alpha and interferon-gamma following traumatic and metabolic injury. J. Neurotrauma 2001, 18, 351–359. [Google Scholar] [CrossRef] [PubMed]
- D’Ambrosio, R.; Fairbanks, J.P.; Fender, J.S.; Born, D.E.; Doyle, D.L.; Miller, J.W. Post-traumatic epilepsy following fluid percussion injury in the rat. Brain 2004, 127, 304–314. [Google Scholar] [CrossRef] [PubMed]
- Ida, T.; Hara, M.; Nakamura, Y.; Kozaki, S.; Tsunoda, S.; Ihara, H. Cytokine-induced enhancement of calcium-dependent glutamate release from astrocytes mediated by nitric oxide. Neurosci. Lett. 2008, 432, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Mandal, M.; Donnelly, R.; Elkabes, S.; Zhang, P.; Davini, D.; David, B.T.; Ponzio, N.M. Maternal immune stimulation during pregnancy shapes the immunological phenotype of offspring. Brain Behav. Immun. 2013, 33, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Busch, R.M.; Srivastava, S.; Hogue, O.; Frazier, T.W.; Klass, P.; Hardan, A.; Martinez-Agosto, J.A.; Sahin, M.; Eng, C.; Developmental Synaptopathies Consortium; et al. Neurobehavioral phenotype of autism spectrum disorder associated with germline heterozygous mutations in PTEN. Transl. Psychiatry 2019, 9, 253. [Google Scholar] [CrossRef]
- Satterstrom, F.K.; Walters, R.K.; Singh, T.; Wigdor, E.M.; Lescai, F.; Demontis, D.; Kosmicki, J.A.; Grove, J.; Stevens, C.; Bybjerg-Grauholm, J. Autism spectrum disorder and attention deficit hyperactivity disorder have a similar burden of rare protein-truncating variants. Nat. Neurosci. 2019, 22, 1961–1965. [Google Scholar] [CrossRef]
- De Rubeis, S.; He, X.; Goldberg, A.P.; Poultney, C.S.; Samocha, K.; Cicek, A.E.; Kou, Y.; Liu, L.; Fromer, M.; Walker, S. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014, 515, 209–215. [Google Scholar] [CrossRef]
- Iossifov, I.; O’Roak, B.J.; Sanders, S.J.; Ronemus, M.; Krumm, N.; Levy, D.; Stessman, H.A.; Witherspoon, K.T.; Vives, L.; Patterson, K.E. The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014, 515, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Sanders, S.J.; He, X.; Willsey, A.J.; Ercan-Sencicek, A.G.; Samocha, K.E.; Cicek, A.E.; Murtha, M.T.; Bal, V.H.; Bishop, S.L.; Dong, S. Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron 2015, 87, 1215–1233. [Google Scholar] [CrossRef] [PubMed]
- Ohlmeier, M.D.; Peters, K.; Wildt, B.T.T.; Zedler, M.; Ziegenbein, M.; Wiese, B.; Emrich, H.M.; Schneider, U. Comorbidity of alcohol and substance dependence with attention-deficit/hyperactivity disorder (ADHD). Alcohol Alcohol. 2008, 43, 300–304. [Google Scholar] [CrossRef] [PubMed]
- Ohlmeier, M.D.; Peters, K.; Kordon, A.; Seifert, J.; Wildt, B.T.; Wiese, B.; Ziegenbein, M.; Emrich, H.M.; Schneider, U. Nicotine and alcohol dependence in patients with comorbid attention-deficit/hyperactivity disorder (ADHD). Alcohol Alcohol. 2007, 42, 539–543. [Google Scholar] [CrossRef]
- Widera, D.; Mikenberg, I.; Elvers, M.; Kaltschmidt, C.; Kaltschmidt, B. Tumor necrosis factor α triggers proliferation of adult neural stem cells via IKK/NF-κB signaling. BMC Neurosci. 2006, 7, 1–18. [Google Scholar] [CrossRef]
- Flynn, G.; Maru, S.; Loughlin, J.; Romero, I.A.; Male, D. Regulation of chemokine receptor expression in human microglia and astrocytes. J. Neuroimmunol. 2003, 136, 84–93. [Google Scholar] [CrossRef]
- Ende, G.; Cackowski, S.; Van Eijk, J.; Sack, M.; Demirakca, T.; Kleindienst, N.; Bohus, M.; Sobanski, E.; Krause-Tuz, A.; Schmahl, C. Impulsivity and aggression in female BPD and ADHD patients: Association with ACC glutamate and GABA concentrations. Neuropsychopharmacology 2016, 41, 410–418. [Google Scholar] [CrossRef]
- Naaijen, J.; Bralten, J.; Poelmans, G.; Glennon, J.C.; Franke, B.; Buitelaar, J.K. Glutamatergic and GABAergic gene sets in attention-deficit/hyperactivity disorder: Association to overlapping traits in ADHD and autism. Transl. Psychiatry 2017, 7, e999. [Google Scholar] [CrossRef]
- Augé, E.; Pelegrí, C.; Manich, G.; Cabezón, I.; Guinovart, J.J.; Duran, J.; Vilaplana, J. Astrocytes and neurons produce distinct types of polyglucosan bodies in Lafora disease. Glia 2018, 66, 2094–2107. [Google Scholar] [CrossRef]
- Guo, W.; Wang, H.; Watanabe, M.; Shimizu, K.; Zou, S.; LaGraize, S.C.; Wei, F.; Dubner, R.; Ren, K. Glial–cytokine–neuronal interactions underlying the mechanisms of persistent pain. J. Neurosci. 2007, 27, 6006–6018. [Google Scholar] [CrossRef]
- Banks, W.A.; Plotkin, S.R.; Kastin, A.J. Permeability of the blood-brain barrier to soluble cytokine receptors. Neuroimmunomodulation 1995, 2, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Kasari, C.; Shih, W.; Frankel, F.; Whitney, R.; Landa, R.; Lord, C.; Orlich, F.; King, B.; Harwood, R. The peer relationships of girls with ASD at school: Comparison to boys and girls with and without ASD. J. Child Psychol. Psychiatry 2014, 55, 1218–1225. [Google Scholar] [CrossRef]
- Mowlem, F.D.; Rosenqvist, M.A.; Martin, J.; Lichtenstein, P.; Asherson, P.; Larsson, H. Sex differences in predicting ADHD clinical diagnosis and pharmacological treatment. Eur. Child Adolesc. Psychiatry 2019, 28, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Bauermeister, J.J.; Shrout, P.E.; Chávez, L.; Rubio-Stipec, M.; Ramírez, R.; Padilla, L.; Anderson, A.; García, P.; Canino, G. ADHD and gender: Are risks and sequela of ADHD the same for boys and girls? J. Child Psychol. Psychiatry 2007, 48, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Elkins, I.J.; Saunders, G.R.; Malone, S.M.; Keyes, M.A.; McGue, M.; Iacono, W.G. Associations between childhood ADHD, gender, and adolescent alcohol and marijuana involvement: A causally informative design. Drug Alcohol Depend. 2018, 184, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Bauermeister, J.J.; Bird, H.R.; Shrout, P.E.; Chavez, L.; Ramírez, R.; Canino, G. Short-term persistence of DSM-IV ADHD diagnoses: Influence of context, age, and gender. J. Am. Acad. Child Adolesc. Psychiatry 2011, 50, 554–562. [Google Scholar] [CrossRef][Green Version]
- Greenfield, B.; Hechtman, L.; Stehli, A.; Wigal, T. Sexual maturation among youth with ADHD and the impact of stimulant medication. Eur. Child Adolesc. Psychiatry 2014, 23, 835–839. [Google Scholar] [CrossRef]
- Semeijn, E.J.; Comijs, H.C.; De Vet, H.C.W.; Kooij, J.J.S.; Michielsen, M.; Beekman, A.T.F.; Deeg, D.J.H. Lifetime stability of ADHD symptoms in older adults. ADHD Atten. Deficit Hyperact. Disord. 2016, 8, 13–20. [Google Scholar] [CrossRef]
- Signs and Symptoms of Autism Spectrum Disorder. Available online: https://www.cdc.gov/ncbddd/autism/signs.html (accessed on 6 September 2020).
- Symptoms and Diagnosis of ADHD. Available online: https://www.cdc.gov/ncbddd/adhd/diagnosis.html (accessed on 6 September 2020).
- Lee, A.S.; Azmitia, E.C.; Whitaker-Azmitia, P.M. Developmental microglial priming in postmortem autism spectrum disorder temporal cortex. Brain Behav. Immun. 2017, 62, 193–202. [Google Scholar] [CrossRef]
- Bollmann, S.; Ghisleni, C.; Poil, S.S.; Martin, E.; Ball, J.; Eich-Höchli, D.; Edden, R.A.E.; Klaver, P.; Michels, L.; Brandeis, D. Developmental changes in gamma-aminobutyric acid levels in attention-deficit/hyperactivity disorder. Transl. Psychiatry 2015, 5, e589. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Types of Paper | |||||
|---|---|---|---|---|---|
| Review | Research | Others | |||
| Systematic | Animal | Human | Mathematical | ||
| [3,7,14,19,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62] | Genetic | [1,2,4,5,8,9,10,11,12,17,18,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92] | Case | [13,15,16,93,94,95,96] | [97,98] |
| Meta-analysis | Pharmacological | [6,11,12,18,20,63,77,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122] | Cohort | [123,124,125,126,127,128,129,130,131,132,133,134,135,136,137,138,139,140,141,142,143,144,145,146,147,148] | |
| [149,150] | Environmental | [16,71,151,152,153,154,155,156] | Comparative | [157,158,159,160,161,162,163,164,165,166,167,168,169,170,171,172,173,174,175,176,177,178] | |
| ASD | Model |
|---|---|
| Genetic | Shank2 KO [24], Shank3 KO [70], NLGN3 R451C KI [67], NLXN1 KO [24], TSC1 HT [64], BTBR [70], MeCP2 mutant [65], Scn1a HT [64], PTEN mutant [158] |
| Pharmacological | Valproic acid (VPA) [71] |
| Environmental | Methyl mercury [127], maternal immune activation (MIA) [99,100,126], polyinosinic:polycytidylic acid (poly I:C) [63] |
| ADHD | Model |
| Genetic | Dopamine transporter (DAT) mutant [33], NK1R-KO [33], SNAP25 mutant [33], Git1 KO [160,161], Cdk5 KO [72], nicotinic acetylcholine receptor (nAChR) ß2-KO [33] |
| Pharmacological | Ethanol [162], methylazoxymethanol [104,105] |
| Environmental | Neonatal X-radiation, hypoxia, heavy metal exposure (lead, cadmium), oncogenic environmental exposure (polychlorinated biphenyl (PCB)) [33] |
| Epilepsy | Model |
|---|---|
| Genetic | AP (Activator Protein)-1 KO [108], SCN1A KO, DBA/2 KO [109] Lafora disease: Malin KO, Epm2a, Epm2b [89,90] |
| Pharmacological * | Pentylenetetrazol (PTZ), pilocarpine (PA), kainic acid (KA) |
| Environmental | Electrical stimulation, brain injury |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, Y.S.; Choi, J.; Yoon, B.-E. Neuron-Glia Interactions in Neurodevelopmental Disorders. Cells 2020, 9, 2176. https://doi.org/10.3390/cells9102176
Kim YS, Choi J, Yoon B-E. Neuron-Glia Interactions in Neurodevelopmental Disorders. Cells. 2020; 9(10):2176. https://doi.org/10.3390/cells9102176
Chicago/Turabian StyleKim, Yoo Sung, Juwon Choi, and Bo-Eun Yoon. 2020. "Neuron-Glia Interactions in Neurodevelopmental Disorders" Cells 9, no. 10: 2176. https://doi.org/10.3390/cells9102176
APA StyleKim, Y. S., Choi, J., & Yoon, B.-E. (2020). Neuron-Glia Interactions in Neurodevelopmental Disorders. Cells, 9(10), 2176. https://doi.org/10.3390/cells9102176