c-Jun N-Terminal Kinase as a Therapeutic Target in Experimental Autoimmune Encephalomyelitis

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. In Vivo and In Vitro Treatments
2.2. Active Myelin Oligodendrocyte Glycoprotein (MOG35-55) Experimental Autoimmune Encephalomyelitis (MOG35-55 EAE)
2.3. Apoptosis Assay
2.4. Proliferation Assay
2.5. RNA Isolation
2.6. Quantitative Real-Time Reverse Transcription Polymerase Chain Reaction (qRT-PCR) and Analysis
2.7. Protein Analysis
2.8. Histology
2.9. Mass Spectrometry
2.10. Statistical Analysis
3. Results
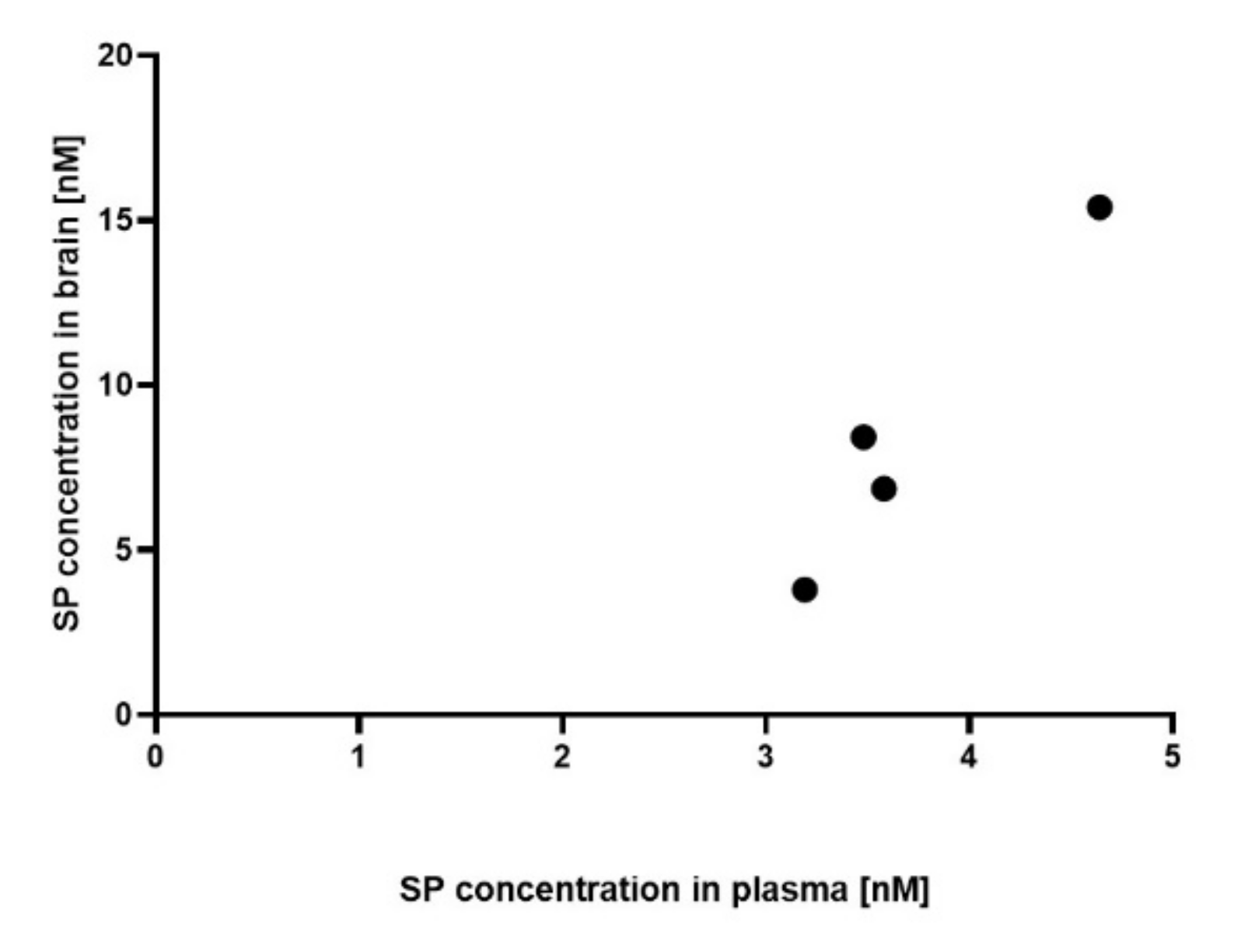
3.1. Plasma and Brain SP Concentrations of SP 30 mg/kg/day-Treated Acute MOG35-55 EAE-Diseased Mice Positively Correlate
3.2. Effects of Pharmacological Pan-JNK Inhibition on MOG35-55 EAE Disease Course
3.3. JNK Inhibition Increases Spinal Cord INF-γ and TNF-α mRNA Expression during the Acute Phase of MOG35-55 EAE
3.4. JNK Inhibition Reduces Spinal Cord Cell-Infiltration during the Acute Phase of MOG35-55 EAE
3.5. JNK inhibition Exerts Anti-Apoptotic and Anti-Proliferative Effects in Murine and Human CD3+ T Cells
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A






References
- Goldenberg, M.M. Multiple sclerosis review. A Peer-Rev. J. Formul. Manag. 2012, 37, 175–184. [Google Scholar]
- Davis, R.J. Signal transduction by the JNK group of MAP kinases. Cell 2000, 103, 239–252. [Google Scholar] [CrossRef]
- Hua, L.L.; Zhao, M.L.; Cosenza, M.; Kim, M.O.; Huang, H.; Tanowitz, H.B.; Brosnan, C.F.; Lee, S.C. Role of mitogen-activated protein kinases in inducible nitric oxide synthase and TNFalpha expression in human fetal astrocytes. J. Neuroimmunol. 2002, 126, 180–189. [Google Scholar] [CrossRef]
- Cobb, A.S.K.A.M.H. MAP Kinase Pathways: Functions and Modulation. Available online: https://pdfs.semanticscholar.org/e817/98ebf70c233741ada55e0666ea96927e5b4a.pdf (accessed on 21 September 2020).
- Tibbles, L.A.; Woodgett, J.R. The stress-activated protein kinase pathways. Cell. Mol. Life Sci. 1999, 55, 1230–1254. [Google Scholar] [CrossRef] [PubMed]
- Zeke, A.; Misheva, M.; Remenyi, A.; Bogoyevitch, M.A. JNK Signaling: Regulation and Functions Based on Complex Protein-Protein Partnerships. Microbiol. Mol. Biol. Rev. 2016, 80, 793–835. [Google Scholar] [CrossRef] [PubMed]
- Johnson, G.L.; Nakamura, K. The c-jun kinase/stress-activated pathway: Regulation, function and role in human disease. Biochim. Biophys. Acta 2007, 1773, 1341–1348. [Google Scholar] [CrossRef]
- Cui, J.; Zhang, M.; Zhang, Y.Q.; Xu, Z.H. JNK pathway: Diseases and therapeutic potential. Acta Pharmacol. Sin. 2007, 28, 601–608. [Google Scholar] [CrossRef]
- Roy, P.K.; Rashid, F.; Bragg, J.; Ibdah, J.A. Role of the JNK signal transduction pathway in inflammatory bowel disease. World J. Gastroenterol. 2008, 14, 200–202. [Google Scholar] [CrossRef]
- Ferrandi, C.; Richard, F.; Tavano, P.; Hauben, E.; Barbie, V.; Gotteland, J.P.; Greco, B.; Fortunato, M.; Mariani, M.F.; Furlan, R.; et al. Characterization of immune cell subsets during the active phase of multiple sclerosis reveals disease and c-Jun N-terminal kinase pathway biomarkers. Mult. Scler. 2011, 17, 43–56. [Google Scholar] [CrossRef]
- Shin, T.; Ahn, M.; Jung, K.; Heo, S.; Kim, D.; Jee, Y.; Lim, Y.K.; Yeo, E.J. Activation of mitogen-activated protein kinases in experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2003, 140, 118–125. [Google Scholar] [CrossRef]
- Offen, D.; Gilgun-Sherki, Y.; Barhum, Y.; Benhar, M.; Grinberg, L.; Reich, R.; Melamed, E.; Atlas, D. A low molecular weight copper chelator crosses the blood-brain barrier and attenuates experimental autoimmune encephalomyelitis. J. Neurochem. 2004, 89, 1241–1251. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Qi, Y.; Yuan, Y.; Cai, L.; Xu, H.; Zhang, L.; Su, B.; Nie, H. Paeoniflorin Ameliorates Experimental Autoimmune Encephalomyelitis via Inhibition of Dendritic Cell Function and Th17 Cell Differentiation. Sci. Rep. 2017, 7, 41887. [Google Scholar] [CrossRef] [PubMed]
- Ichiyama, K.; Sekiya, T.; Inoue, N.; Tamiya, T.; Kashiwagi, I.; Kimura, A.; Morita, R.; Muto, G.; Shichita, T.; Takahashi, R.; et al. Transcription factor Smad-independent T helper 17 cell induction by transforming-growth factor-β is mediated by suppression of eomesodermin. Immunity 2011, 34, 741–754. [Google Scholar] [CrossRef] [PubMed]
- Nicolson, K.; Freland, S.; Weir, C.; Delahunt, B.; Flavell, R.A.; Backstrom, B.T. Induction of experimental autoimmune encephalomyelitis in the absence of c-Jun N-terminal kinase 2. Int. Immunol. 2002, 14, 849–856. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tran, E.H.; Azuma, Y.T.; Chen, M.; Weston, C.; Davis, R.J.; Flavell, R.A. Inactivation of JNK1 enhances innate IL-10 production and dampens autoimmune inflammation in the brain. Proc. Natl. Acad. Sci. USA 2006, 103, 13451–13456. [Google Scholar] [CrossRef] [PubMed]
- Bennett, B.L.; Sasaki, D.T.; Murray, B.W.; O’Leary, E.C.; Sakata, S.T.; Xu, W.; Leisten, J.C.; Motiwala, A.; Pierce, S.; Satoh, Y.; et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc. Natl. Acad. Sci. USA 2001, 98, 13681–13686. [Google Scholar] [CrossRef] [PubMed]
- Hoepner, R.; Bagnoud, M.; Pistor, M.; Salmen, A.; Briner, M.; Synn, H.; Schrewe, L.; Guse, K.; Ahmadi, F.; Demir, S.; et al. Vitamin D increases glucocorticoid efficacy via inhibition of mTORC1 in experimental models of multiple sclerosis. Acta Neuropathol. 2019, 138, 443–456. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, W.P.; Ness, D.K.; van Lier, R.B. Analysis of rodent growth data in toxicology studies. Toxicol. Sci. 2002, 66, 313–319. [Google Scholar] [CrossRef] [PubMed]
- ThermoFischer. Available online: https://www.thermofisher.com/ch/en/home/life-science/pcr/real-time-pcr/real-time-pcr-learning-center/real-time-pcr-basics/efficiency-real-time-pcr-qpcr.html (accessed on 21 September 2020).
- Yamasaki, T.; Kawasaki, H.; Nishina, H. Diverse Roles of JNK and MKK Pathways in the Brain. J. Signal. Transduct. 2012, 2012, 459265. [Google Scholar] [CrossRef] [PubMed]
- Raphael, I.; Nalawade, S.; Eagar, T.N.; Forsthuber, T.G. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine 2015, 74, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Brehm, M.A.; Daniels, K.A.; Welsh, R.M. Rapid production of TNF-alpha following TCR engagement of naive CD8 T cells. J. Immunol. 2005, 175, 5043–5049. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Hertzog, P.J.; Ravasi, T.; Hume, D.A. Interferon-gamma: An overview of signals, mechanisms and functions. J. Leukoc. Biol. 2004, 75, 163–189. [Google Scholar] [CrossRef] [PubMed]
- Parameswaran, N.; Patial, S. Tumor necrosis factor-α signaling in macrophages. Crit. Rev. Eukaryot. Gene Expr. 2010, 20, 87–103. [Google Scholar] [CrossRef]
- Lau, L.T.; Yu, A.C. Astrocytes produce and release interleukin-1, interleukin-6, tumor necrosis factor alpha and interferon-gamma following traumatic and metabolic injury. J. Neurotrauma 2001, 18, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Haider, S.; Pal, R. Integrated analysis of transcriptomic and proteomic data. Curr Genom. 2013, 14, 91–110. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H.; Bradl, M. Multiple sclerosis: Experimental models and reality. Acta Neuropathol. 2017, 133, 223–244. [Google Scholar] [CrossRef]
- Zamvil, S.S.; Steinman, L. The T lymphocyte in experimental allergic encephalomyelitis. Annu. Rev. Immunol. 1990, 8, 579–621. [Google Scholar] [CrossRef]
- Sosa, R.A.; Murphey, C.; Robinson, R.R.; Forsthuber, T.G. IFN-gamma ameliorates autoimmune encephalomyelitis by limiting myelin lipid peroxidation. Proc. Natl. Acad. Sci. USA 2015, 112, E5038–E5047. [Google Scholar] [CrossRef]
- Arellano, G.; Ottum, P.A.; Reyes, L.I.; Burgos, P.I.; Naves, R. Stage-Specific Role of Interferon-Gamma in Experimental Autoimmune Encephalomyelitis and Multiple Sclerosis. Front. Immunol. 2015, 6, 492. [Google Scholar] [CrossRef]
- Naves, R.; Singh, S.P.; Cashman, K.S.; Rowse, A.L.; Axtell, R.C.; Steinman, L.; Mountz, J.D.; Steele, C.; De Sarno, P.; Raman, C. The interdependent, overlapping, and differential roles of type I and II IFNs in the pathogenesis of experimental autoimmune encephalomyelitis. J. Immunol. 2013, 191, 2967–2977. [Google Scholar] [CrossRef]
- Baker, D.; Butler, D.; Scallon, B.J.; O’Neill, J.K.; Turk, J.L.; Feldmann, M. Control of established experimental allergic encephalomyelitis by inhibition of tumor necrosis factor (TNF) activity within the central nervous system using monoclonal antibodies and TNF receptor-immunoglobulin fusion proteins. Eur. J. Immunol. 1994, 24, 2040–2048. [Google Scholar] [CrossRef] [PubMed]
- Suvannavejh, G.C.; Lee, H.O.; Padilla, J.; Dal Canto, M.C.; Barrett, T.A.; Miller, S.D. Divergent roles for p55 and p75 tumor necrosis factor receptors in the pathogenesis of MOG(35-55)-induced experimental autoimmune encephalomyelitis. Cell Immunol. 2000, 205, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Lenercept Multiple Sclerosis Study Group; The University of British Columbia MS/MRI Analysis Group. TNF neutralization in MS: Results of a randomized, placebo-controlled multicenter study. Neurology 1999, 53, 457–465. [Google Scholar] [CrossRef]
- Liu, J.; Marino, M.W.; Wong, G.; Grail, D.; Dunn, A.; Bettadapura, J.; Slavin, A.J.; Old, L.; Bernard, C.C. TNF is a potent anti-inflammatory cytokine in autoimmune-mediated demyelination. Nat. Med. 1998, 4, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Eugster, H.P.; Frei, K.; Bachmann, R.; Bluethmann, H.; Lassmann, H.; Fontana, A. Severity of symptoms and demyelination in MOG-induced EAE depends on TNFR1. Eur. J. Immunol. 1999, 29, 626–632. [Google Scholar] [CrossRef]
- Taoufik, E.; Tseveleki, V.; Chu, S.Y.; Tselios, T.; Karin, M.; Lassmann, H.; Szymkowski, D.E.; Probert, L. Transmembrane tumour necrosis factor is neuroprotective and regulates experimental autoimmune encephalomyelitis via neuronal nuclear factor-kappaB. Brain 2011, 134, 2722–2735. [Google Scholar] [CrossRef]
- Palumbo, S.; Pellegrini, S. Experimental In Vivo Models of Multiple Sclerosis: State of the Art. In Multiple Sclerosis: Perspectives in Treatment and Pathogenesis; Zagon, I.S., McLaughlin, P.J., Eds.; Codon Publications: Singapore, 2017. [Google Scholar]
- Dong, C.; Yang, D.D.; Wysk, M.; Whitmarsh, A.J.; Davis, R.J.; Flavell, R.A. Defective T cell differentiation in the absence of Jnk1. Science 1998, 282, 2092–2095. [Google Scholar] [CrossRef]
- Yang, D.D.; Conze, D.; Whitmarsh, A.J.; Barrett, T.; Davis, R.J.; Rincón, M.; Flavell, R.A. Differentiation of CD4+ T cells to Th1 cells requires MAP kinase JNK2. Immunity 1998, 9, 575–585. [Google Scholar] [CrossRef]
- Sabapathy, K.; Kallunki, T.; David, J.P.; Graef, I.; Karin, M.; Wagner, E.F. c-Jun NH2-terminal kinase (JNK)1 and JNK2 have similar and stage-dependent roles in regulating T cell apoptosis and proliferation. J. Exp. Med. 2001, 193, 317–328. [Google Scholar] [CrossRef]
- Weiss, L.; Whitmarsh, A.J.; Yang, D.D.; Rincón, M.; Davis, R.J.; Flavell, R.A. Regulation of c-Jun NH(2)-terminal kinase (Jnk) gene expression during T cell activation. J. Exp. Med. 2000, 191, 139–146. [Google Scholar] [CrossRef]
- Sehgal, V.; Ram, P.T. Network Motifs in JNK Signaling. Genes Cancer 2013, 4, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Melino, M.; Hii, C.S.; McColl, S.R.; Ferrante, A. The effect of the JNK inhibitor, JIP peptide, on human T lymphocyte proliferation and cytokine production. J. Immunol. 2008, 181, 7300–7306. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, J.D.; De, A.K.; Kodys, K.; Puyana, J.C.; Furse, R.K.; Miller-Graziano, C. Relationships between T lymphocyte apoptosis and anergy following trauma. J. Surg. Res. 2000, 88, 200–206. [Google Scholar] [CrossRef] [PubMed]
- DeSilva, D.R.; Feeser, W.S.; Tancula, E.J.; Scherle, P.A. Anergic T cells are defective in both jun NH2-terminal kinase and mitogen-activated protein kinase signaling pathways. J. Exp. Med. 1996, 183, 2017–2023. [Google Scholar] [CrossRef]
- Mocholi, E.; Dowling, S.D.; Botbol, Y.; Gruber, R.C.; Ray, A.K.; Vastert, S.; Shafit-Zagardo, B.; Coffer, P.J.; Macian, F. Autophagy is a Tolerance-Avoidance Mechanism that Modulates TCR-Mediated Signaling and Cell Metabolism to Prevent Induction of T Cell Anergy. Cell Rep. 2018, 24, 1136–1150. [Google Scholar] [CrossRef]
- Assi, K.; Pillai, R.; Gómez-Muñoz, A.; Owen, D.; Salh, B. The specific JNK inhibitor SP600125 targets tumour necrosis factor-alpha production and epithelial cell apoptosis in acute murine colitis. Immunology 2006, 118, 112–121. [Google Scholar] [CrossRef]
- Himes, S.R.; Sester, D.P.; Ravasi, T.; Cronau, S.L.; Sasmono, T.; Hume, D.A. The JNK are important for development and survival of macrophages. J. Immunol. 2006, 176, 2219–2228. [Google Scholar] [CrossRef]
- Zhang, L.; Xie, H.; Cui, L. Activation of astrocytes and expression of inflammatory cytokines in rats with experimental autoimmune encephalomyelitis. Exp. Ther. Med. 2018, 16, 4401–4406. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef]
- Imai, Y.; Ibata, I.; Ito, D.; Ohsawa, K.; Kohsaka, S. A novel gene iba1 in the major histocompatibility complex class III region encoding an EF hand protein expressed in a monocytic lineage. Biochem. Biophys. Res. Commun. 1996, 224, 855–862. [Google Scholar] [CrossRef]
- Sloka, S.; Metz, L.M.; Hader, W.; Starreveld, Y.; Yong, V.W. Reduction of microglial activity in a model of multiple sclerosis by dipyridamole. J. Neuroinflamm. 2013, 10, 89. [Google Scholar] [CrossRef] [PubMed]
- Waetzig, V.; Czeloth, K.; Hidding, U.; Mielke, K.; Kanzow, M.; Brecht, S.; Goetz, M.; Lucius, R.; Herdegen, T.; Hanisch, U.K. c-Jun N-terminal kinases (JNKs) mediate pro-inflammatory actions of microglia. Glia 2005, 50, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, D.B.; Bhaskaran, A.; Arthur-Farraj, P.; Noon, L.A.; Woodhoo, A.; Lloyd, A.C.; Feltri, M.L.; Wrabetz, L.; Behrens, A.; Mirsky, R.; et al. c-Jun is a negative regulator of myelination. J. Cell Biol. 2008, 181, 625–637. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.D.; Kuan, C.Y.; Whitmarsh, A.J.; Rincón, M.; Zheng, T.S.; Davis, R.J.; Rakic, P.; Flavell, R.A. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature 1997, 389, 865–870. [Google Scholar] [CrossRef]
- Ha, D.; Bing, S.J.; Ahn, G.; Kim, J.; Cho, J.; Kim, A.; Herath, K.H.I.N.M.; Yu, H.S.; Jo, S.A.; Cho, I.-H.; et al. Blocking glutamate carboxypeptidase II inhibits glutamate excitotoxicity and regulates immune responses in experimental autoimmune encephalomyelitis. FEBS J. 2016, 283, 3438–3456. [Google Scholar] [CrossRef]
- Pitt, D.; Werner, P.; Raine, C.S. Glutamate excitotoxicity in a model of multiple sclerosis. Nat. Med. 2000, 6, 67–70. [Google Scholar] [CrossRef]
- Kuan, C.Y.; Whitmarsh, A.J.; Yang, D.D.; Liao, G.; Schloemer, A.J.; Dong, C.; Bao, J.; Banasiak, K.J.; Haddad, G.G.; Flavell, R.A.; et al. A critical role of neural-specific JNK3 for ischemic apoptosis. Proc. Natl. Acad. Sci. USA 2003, 100, 15184–15189. [Google Scholar] [CrossRef]
- Jurewicz, A.; Matysiak, M.; Andrzejak, S.; Selmaj, K. TRAIL-induced death of human adult oligodendrocytes is mediated by JNK pathway. Glia 2006, 53, 158–166. [Google Scholar] [CrossRef]
- Jurewicz, A.; Matysiak, M.; Tybor, K.; Selmaj, K. TNF-induced death of adult human oligodendrocytes is mediated by c-jun NH2-terminal kinase-3. Brain 2003, 126, 1358–1370. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bagnoud, M.; Briner, M.; Remlinger, J.; Meli, I.; Schuetz, S.; Pistor, M.; Salmen, A.; Chan, A.; Hoepner, R. c-Jun N-Terminal Kinase as a Therapeutic Target in Experimental Autoimmune Encephalomyelitis. Cells 2020, 9, 2154. https://doi.org/10.3390/cells9102154
Bagnoud M, Briner M, Remlinger J, Meli I, Schuetz S, Pistor M, Salmen A, Chan A, Hoepner R. c-Jun N-Terminal Kinase as a Therapeutic Target in Experimental Autoimmune Encephalomyelitis. Cells. 2020; 9(10):2154. https://doi.org/10.3390/cells9102154
Chicago/Turabian StyleBagnoud, Maud, Myriam Briner, Jana Remlinger, Ivo Meli, Sara Schuetz, Maximilian Pistor, Anke Salmen, Andrew Chan, and Robert Hoepner. 2020. "c-Jun N-Terminal Kinase as a Therapeutic Target in Experimental Autoimmune Encephalomyelitis" Cells 9, no. 10: 2154. https://doi.org/10.3390/cells9102154
APA StyleBagnoud, M., Briner, M., Remlinger, J., Meli, I., Schuetz, S., Pistor, M., Salmen, A., Chan, A., & Hoepner, R. (2020). c-Jun N-Terminal Kinase as a Therapeutic Target in Experimental Autoimmune Encephalomyelitis. Cells, 9(10), 2154. https://doi.org/10.3390/cells9102154