Host Calcium Channels and Pumps in Viral Infections


Abstract
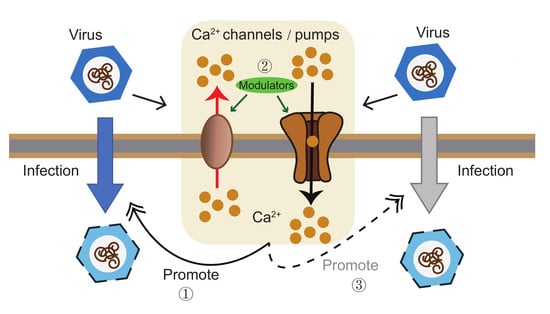
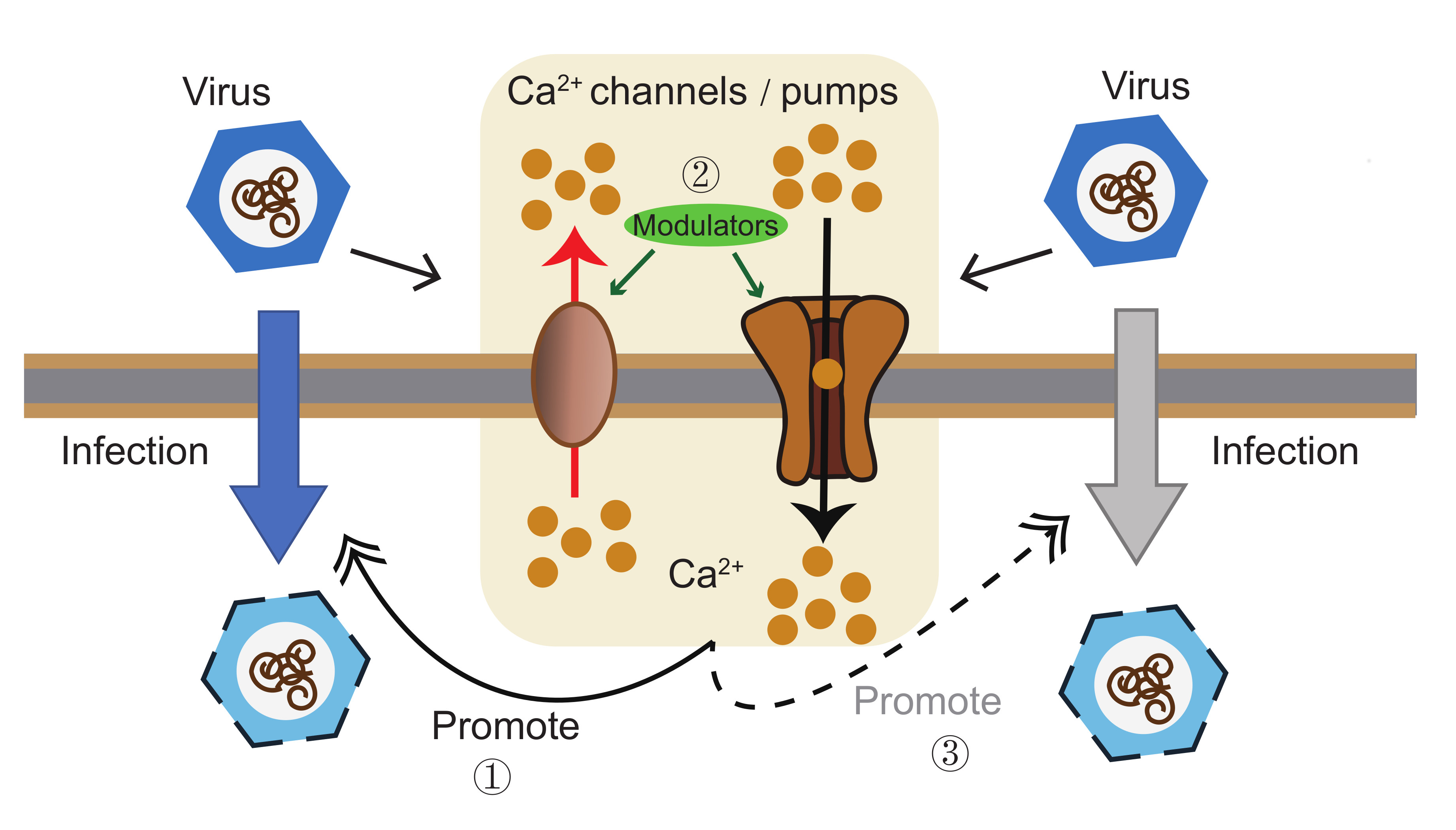
1. Introduction
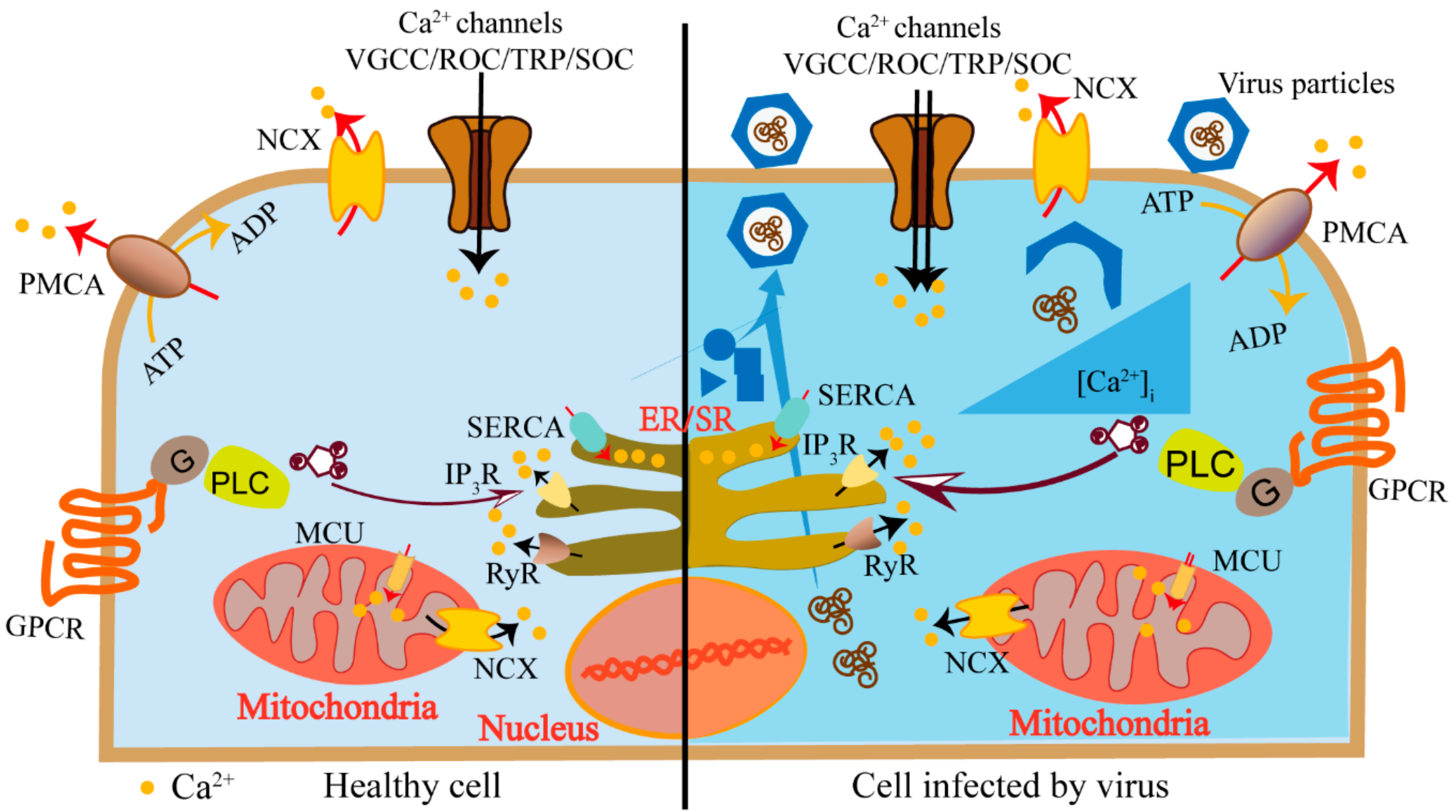
2. Calcium Channels and Pumps in Host Ca2+ Homeostasis
3. Viruses Control Host Voltage-Gated Calcium Channels (VGCCs) and Two-Pore Channels (TPCs)
4. Store-Operated Calcium (SOC) Channel in Viral Assembly and Egress
5. Host Transient Receptor Potential (TRP) Channels and Receptor-Operated Calcium (ROC) Channels
5.1. TRP Channels
5.2. N-Methyl-D-Aspartate (NMDA) Receptors
5.3. IP3 Receptors
6. Calcium Pumps
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Martinez de Victoria, E. Calcium, essential for health. Nutr. Hosp. 2016, 33 (Suppl. S4), 341. [Google Scholar]
- Berridge, M.J.; Bootman, M.D.; Lipp, P. Calcium—A life and death signal. Nature 1998, 395, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Olivier, M. Modulation of host cell intracellular Ca2+. Parasitol. Today 1996, 12, 145–150. [Google Scholar] [CrossRef]
- Clark, K.B.; Eisenstein, E.M. Targeting host store-operated Ca2+ release to attenuate viral infections. Curr. Top. Med. Chem. 2013, 13, 1916–1932. [Google Scholar] [CrossRef] [PubMed]
- Gonzales-van Horn, S.R.; Sarnow, P. Making the Mark: The Role of Adenosine Modifications in the Life Cycle of RNA Viruses. Cell Host Microbe 2017, 21, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Atlas, D. Voltage-gated calcium channels function as Ca2+-activated signaling receptors. Trends Biochem. Sci. 2014, 39, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Nugent, K.M.; Shanley, J.D. Verapamil inhibits influenza A virus replication. Arch. Virol. 1984, 81, 163–170. [Google Scholar] [CrossRef]
- Fujioka, Y.; Tsuda, M.; Nanbo, A.; Hattori, T.; Sasaki, J.; Sasaki, T.; Miyazaki, T.; Ohba, Y. A Ca2+-dependent signalling circuit regulates influenza a virus internalization and infection. Nat. Commun. 2013, 4, 2763. [Google Scholar] [CrossRef]
- Fujioka, Y.; Nishide, S.; Ose, T.; Suzuki, T.; Kato, I.; Fukuhara, H.; Fujioka, M.; Horiuchi, K.; Satoh, A.O.; Nepal, P. A Sialylated Voltage-Dependent Ca2+ Channel Binds Hemagglutinin and Mediates Influenza A Virus Entry into Mammalian Cells. Cell Host Microbe 2018, 23, 809–818. [Google Scholar] [CrossRef]
- Chandrasekaran, A.; Srinivasan, A.; Raman, R.; Viswanathan, K.; Raguram, S.; Tumpey, T.M.; Sasisekharan, V.; Sasisekharan, R. Glycan topology determines human adaptation of avian H5N1 virus hemagglutinin. Nat. Biotechnol. 2008, 26, 107–113. [Google Scholar] [CrossRef]
- Lazniewska, J.; Weiss, N. Glycosylation of voltage-gated calcium channels in health and disease. Biochim. Biophys. Acta Biomembr. 2017, 1859, 662–668. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhang, L.K.; Li, S.F.; Zhang, S.F.; Wan, W.W.; Zhang, Y.L.; Xin, Q.L.; Dai, K.; Hu, Y.Y.; Wang, Z.B. Calcium channel blockers reduce severe fever with thrombocytopenia syndrome virus (SFTSV) related fatality. Cell Res. 2019, 29, 739–753. [Google Scholar] [CrossRef] [PubMed]
- Lavanya, M.; Cuevas, C.D.; Thomas, M.; Cherry, S.; Ross, S.R. siRNA screen for genes that affect Junin virus entry uncovers voltage-gated calcium channels as a therapeutic target. Sci. Transl. Med. 2013, 5, 204ra131. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Liu, Y.; Guo, J.; Wang, P.; Zhang, L.; Xiao, G.; Wang, W. Screening of FDA-Approved Drugs for Inhibitors of Japanese Encephalitis Virus Infection. J. Virol. 2017, 91, e01055-17. [Google Scholar] [CrossRef]
- Dreyer, E.B.; Kaiser, P.K.; Offermann, J.T.; Lipton, S.A. HIV-1 coat protein neurotoxicity prevented by calcium channel antagonists. Science 1990, 248, 364–367. [Google Scholar] [CrossRef]
- Haughey, N.J.; Mattson, M.P. Calcium dysregulation and neuronal apoptosis by the HIV-1 proteins Tat and gp120. J. Acquir. Immune. Defic. Syndr. 2002, 31 (Suppl. S2), S55–S61. [Google Scholar] [CrossRef]
- Hu, X.T. HIV-1 Tat-Mediated Calcium Dysregulation and Neuronal Dysfunction in Vulnerable Brain Regions. Curr. Drug Targets 2016, 17, 4–14. [Google Scholar] [CrossRef]
- Kruman, I.I.; Nath, A.; Mattson, M.P. HIV-1 protein Tat induces apoptosis of hippocampal neurons by a mechanism involving caspase activation, calcium overload, and oxidative stress. Exp. Neurol. 1998, 154, 276–288. [Google Scholar] [CrossRef]
- Zhang, Q.; Hsia, S.C.; Martin-Caraballo, M. Regulation of T-type Ca2+ channel expression by herpes simplex virus-1 infection in sensory-like ND7 cells. J. Neurovirol. 2017, 23, 657–670. [Google Scholar] [CrossRef]
- Zhang, Q.; Hsia, S.C.; Martin-Caraballo, M. Regulation of T-type Ca2+ channel expression by interleukin-6 in sensory-like ND7/23 cells post-herpes simplex virus (HSV-1) infection. J. Neurochem. 2019, 151, 238–254. [Google Scholar] [CrossRef]
- Kolokoltsov, A.A.; Saeed, M.F.; Freiberg, A.N.; Holbrook, M.R.; Davey, R.A. Identification of novel cellular targets for therapeutic intervention against Ebola virus infection by siRNA screening. Drug Dev. Res. 2009, 70, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Gehring, G.; Rohrmann, K.; Atenchong, N.; Mittler, E.; Becker, S.; Dahlmann, F.; Pohlmann, S.; Vondran, F.W.; David, S.; Manns, M.P. The clinically approved drugs amiodarone, dronedarone and verapamil inhibit filovirus cell entry. J. Antimicrob. Chemother. 2014, 69, 2123–2131. [Google Scholar] [CrossRef] [PubMed]
- Madrid, P.B.; Chopra, S.; Manger, I.D.; Gilfillan, L.; Keepers, T.R.; Shurtleff, A.C.; Green, C.E.; Iyer, L.V.; Dilks, H.H.; Davey, R.A. A systematic screen of FDA-approved drugs for inhibitors of biological threat agents. PLoS ONE 2013, 8, e60579. [Google Scholar] [CrossRef] [PubMed]
- Calcraft, P.J.; Ruas, M.; Pan, Z.; Cheng, X.; Arredouani, A.; Hao, X.; Tang, J.; Rietdorf, K.; Teboul, L.; Chuang, K.T. NAADP mobilizes calcium from acidic organelles through two-pore channels. Nature 2009, 459, 596–600. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.X.; Ma, J.; Parrington, J.; Calcraft, P.J.; Galione, A.; Evans, A.M. Calcium signaling via two-pore channels: Local or global, that is the question. Am. J. Physiol. Cell Physiol. 2010, 298, C430–C441. [Google Scholar] [CrossRef] [PubMed]
- Morgan, A.J. Ca2+ dialogue between acidic vesicles and ER. Biochem. Soc. Trans. 2016, 44, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, Y.; Kolokoltsov, A.A.; Chen, C.C.; Tidwell, M.W.; Bauta, W.E.; Klugbauer, N.; Grimm, C.; Wahl-Schott, C.; Biel, M.; Davey, R.A. Two-pore channels control Ebola virus host cell entry and are drug targets for disease treatment. Science 2015, 347, 995–998. [Google Scholar] [CrossRef]
- Kintzer, A.F.; Stroud, R.M. Structure, inhibition and regulation of two-pore channel TPC1 from Arabidopsis thaliana. Nature 2016, 531, 258–262. [Google Scholar] [CrossRef]
- Hogan, P.G.; Lewis, R.S.; Rao, A. Molecular basis of calcium signaling in lymphocytes: STIM and ORAI. Annu. Rev. Immunol. 2010, 28, 491–533. [Google Scholar] [CrossRef]
- Nwokonko, R.M.; Cai, X.; Loktionova, N.A.; Wang, Y.; Zhou, Y.; Gill, D.L. The STIM-Orai Pathway: Conformational Coupling between STIM and Orai in the Activation of Store-Operated Ca2+ Entry. Adv. Exp. Med. Biol. 2017, 993, 83–98. [Google Scholar]
- Sepulveda, C.S.; Garcia, C.C.; Damonte, E.B. Determining the Virus Life-Cycle Stage Blocked by an Antiviral. Methods Mol. Biol. 2018, 1604, 371–392. [Google Scholar] [PubMed]
- Hartlieb, B.; Weissenhorn, W. Filovirus assembly and budding. Virology 2006, 344, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.J.; Lamb, R.A. Mechanisms for enveloped virus budding: Can some viruses do without an ESCRT? Virology 2008, 372, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Licata, J.M.; Simpson-Holley, M.; Wright, N.T.; Han, Z.; Paragas, J.; Harty, R.N. Overlapping motifs (PTAP and PPEY) within the Ebola virus VP40 protein function independently as late budding domains: Involvement of host proteins TSG101 and VPS-4. J. Virol. 2003, 77, 1812–1819. [Google Scholar] [CrossRef]
- Han, Z.; Madara, J.J.; Herbert, A.; Prugar, L.I.; Ruthel, G.; Lu, J.; Liu, Y.; Liu, W.; Liu, X.; Wrobel, J.E. Calcium Regulation of Hemorrhagic Fever Virus Budding: Mechanistic Implications for Host-Oriented Therapeutic Intervention. PLoS Pathog. 2015, 11, e1005220. [Google Scholar] [CrossRef]
- Ehrlich, L.S.; Carter, C.A. HIV Assembly and Budding: Ca2+ Signaling and Non-ESCRT Proteins Set the Stage. Mol. Biol. Int. 2012, 2012, 851670. [Google Scholar] [CrossRef]
- Ehrlich, L.S.; Medina, G.N.; Carter, C.A. ESCRT machinery potentiates HIV-1 utilization of the PI (4,5) P (2)-PLC-IP3R-Ca2+ signaling cascade. J. Mol. Biol. 2011, 413, 347–358. [Google Scholar] [CrossRef]
- Dionicio, C.L.; Pena, F.; Constantino-Jonapa, L.A.; Vazquez, C.; Yocupicio-Monroy, M.; Rosales, R.; Zambrano, J.L.; Ruiz, M.C.; Del Angel, R.M.; Ludert, J.E. Dengue virus induced changes in Ca2+ homeostasis in human hepatic cells that favor the viral replicative cycle. Virus Res. 2018, 245, 17–28. [Google Scholar] [CrossRef]
- Cheshenko, N.; Liu, W.; Satlin, L.M.; Herold, B.C. Multiple receptor interactions trigger release of membrane and intracellular calcium stores critical for herpes simplex virus entry. Mol. Biol. Cell 2007, 18, 3119–3130. [Google Scholar] [CrossRef]
- Robinson, L.C.; Marchant, J.S. Enhanced Ca2+ leak from ER Ca2+ stores induced by hepatitis C NS5A protein. Biochem. Biophys. Res. Commun. 2008, 368, 593–599. [Google Scholar] [CrossRef]
- Zhadina, M.; Bieniasz, P.D. Functional interchangeability of late domains, late domain cofactors and ubiquitin in viral budding. PLoS Pathog. 2010, 6, e1001153. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, M.C.; Aristimuno, O.C.; Diaz, Y.; Pena, F.; Chemello, M.E.; Rojas, H.; Ludert, J.E.; Michelangeli, F. Intracellular disassembly of infectious rotavirus particles by depletion of Ca2+ sequestered in the endoplasmic reticulum at the end of virus cycle. Virus Res. 2007, 130, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Freedman, B.D.; Harty, R.N. Calcium and filoviruses: A budding relationship. Future Microbiol. 2016, 11, 713–715. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.H.; Liu, Z.J.; Yi, J.H.; Wang, J.; Liu, Y.N. Hepatitis B Virus X Protein Upregulates Intracellular Calcium Signaling by Binding C-terminal of Orail Protein. Curr. Med. Sci. 2018, 38, 26–34. [Google Scholar] [CrossRef]
- Michelangeli, F.; Ruiz, M.C.; del Castillo, J.R.; Ludert, J.E.; Liprandi, F. Effect of rotavirus infection on intracellular calcium homeostasis in cultured cells. Virology 1991, 181, 520–527. [Google Scholar] [CrossRef]
- Pham, T.; Perry, J.L.; Dosey, T.L.; Delcour, A.H.; Hyser, J.M. The Rotavirus NSP4 Viroporin Domain is a Calcium-conducting Ion Channel. Sci. Rep. 2017, 7, 43487. [Google Scholar] [CrossRef]
- Hyser, J.M.; Utama, B.; Crawford, S.E.; Broughman, J.R.; Estes, M.K. Activation of the endoplasmic reticulum calcium sensor STIM1 and store-operated calcium entry by rotavirus requires NSP4 viroporin activity. J. Virol. 2013, 87, 13579–13588. [Google Scholar] [CrossRef]
- Chang-Graham, A.L.; Perry, J.L.; Strtak, A.C.; Ramachandran, N.K.; Criglar, J.M.; Philip, A.A.; Patton, J.T.; Estes, M.K.; Hyser, J.M. Rotavirus Calcium Dysregulation Manifests as Dynamic Calcium Signaling in the Cytoplasm and Endoplasmic Reticulum. Sci. Rep. 2019, 9, 10822. [Google Scholar] [CrossRef]
- Li, J.; Zhang, X.; Song, X.; Liu, R.; Zhang, J.; Li, Z. The structure of TRPC ion channels. Cell Calcium. 2019, 80, 25–28. [Google Scholar] [CrossRef]
- Hicks, G.A. TRP channels as therapeutic targets: Hot property, or time to cool down? Neurogastroenterol. Motil. 2006, 18, 590–594. [Google Scholar] [CrossRef]
- Garcia-Elias, A.; Mrkonjic, S.; Jung, C.; Pardo-Pastor, C.; Vicente, R.; Valverde, M.A. The TRPV4 channel. Handb. Exp. Pharm. 2014, 222, 293–319. [Google Scholar]
- Donate-Macian, P.; Jungfleisch, J.; Perez-Vilaro, G.; Rubio-Moscardo, F.; Peralvarez-Marin, A.; Diez, J.; Valverde, M.A. The TRPV4 channel links calcium influx to DDX3X activity and viral infectivity. Nat. Commun. 2018, 9, 2307. [Google Scholar] [CrossRef] [PubMed]
- Ariumi, Y. Multiple functions of DDX3 RNA helicase in gene regulation, tumorigenesis, and viral infection. Front. Genet. 2014, 5, 423. [Google Scholar] [CrossRef] [PubMed]
- Valiente-Echeverria, F.; Hermoso, M.A.; Soto-Rifo, R. RNA helicase DDX3: At the crossroad of viral replication and antiviral immunity. Rev. Med. Virol. 2015, 25, 286–299. [Google Scholar] [CrossRef]
- Fujita, T.; Liu, Y.; Higashitsuji, H.; Itoh, K.; Shibasaki, K.; Fujita, J.; Nishiyama, H. Involvement of TRPV3 and TRPM8 ion channel proteins in induction of mammalian cold-inducible proteins. Biochem. Biophys. Res. Commun. 2018, 495, 935–940. [Google Scholar] [CrossRef]
- Omar, S.; Clarke, R.; Abdullah, H.; Brady, C.; Corry, J.; Winter, H.; Touzelet, O.; Power, U.F.; Lundy, F.; McGarvey, L.P. Respiratory virus infection up-regulates TRPV1, TRPA1 and ASICS3 receptors on airway cells. PLoS ONE 2017, 12, e0171681. [Google Scholar] [CrossRef]
- Abdullah, H.; Heaney, L.G.; Cosby, S.L.; McGarvey, L.P. Rhinovirus upregulates transient receptor potential channels in a human neuronal cell line: Implications for respiratory virus-induced cough reflex sensitivity. Thorax 2014, 69, 46–54. [Google Scholar] [CrossRef]
- Costa, V.V.; Del Sarto, J.L.; Rocha, R.F.; Silva, F.R.; Doria, J.G.; Olmo, I.G.; Marques, R.E.; Queiroz-Junior, C.M.; Foureaux, G.; Araujo, J.M.S. N-Methyl-d-Aspartate (NMDA) Receptor Blockade Prevents Neuronal Death Induced by Zika Virus Infection. MBio 2017, 8, e00543-17. [Google Scholar] [CrossRef]
- Sirohi, D.; Kuhn, R.J. Can an FDA-Approved Alzheimer’s Drug Be Repurposed for Alleviating Neuronal Symptoms of Zika Virus? MBio 2017, 8, e00916-17. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, X.; Ashraf, U.; Zheng, B.; Ye, J.; Zhou, D.; Zhang, H.; Song, Y.; Chen, H.; Zhao, S. Activation of neuronal N-methyl-D-aspartate receptor plays a pivotal role in Japanese encephalitis virus-induced neuronal cell damage. J. Neuroinflammation 2018, 15, 238. [Google Scholar] [CrossRef]
- Chen, S.; Shenk, T.; Nogalski, M.T. P2Y2 purinergic receptor modulates virus yield, calcium homeostasis, and cell motility in human cytomegalovirus-infected cells. Proc. Natl. Acad. Sci. USA 2019, 116, 18971–18982. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, L.S.; Medina, G.N.; Photiadis, S.; Whittredge, P.B.; Watanabe, S.; Taraska, J.W.; Carter, C.A. Tsg101 regulates PI (4,5) P2/Ca2+ signaling for HIV-1 Gag assembly. Front. Microbiol. 2014, 5, 234. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, L.S.; Medina, G.N.; Khan, M.B.; Powell, M.D.; Mikoshiba, K.; Carter, C.A. Activation of the inositol (1,4,5)-triphosphate calcium gate receptor is required for HIV-1 Gag release. J. Virol. 2010, 84, 6438–6451. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Manninen, A.; Saksela, K. HIV-1 Nef interacts with inositol trisphosphate receptor to activate calcium signaling in T cells. J. Exp. Med. 2002, 195, 1023–1032. [Google Scholar] [CrossRef]
- Ding, W.; Albrecht, B.; Kelley, R.E.; Muthusamy, N.; Kim, S.J.; Altschuld, R.A.; Lairmore, M.D. Human T-cell lymphotropic virus type 1 p12 (I) expression increases cytoplasmic calcium to enhance the activation of nuclear factor of activated T cells. J. Virol. 2002, 76, 10374–10382. [Google Scholar] [CrossRef]
- Cheshenko, N.; Del Rosario, B.; Woda, C.; Marcellino, D.; Satlin, L.M.; Herold, B.C. Herpes simplex virus triggers activation of calcium-signaling pathways. J. Cell Biol. 2003, 163, 283–293. [Google Scholar] [CrossRef]
- Strehler, E.E.; Zacharias, D.A. Role of alternative splicing in generating isoform diversity among plasma membrane calcium pumps. Physiol. Rev. 2001, 81, 21–50. [Google Scholar] [CrossRef]
- Brini, M.; Cali, T.; Ottolini, D.; Carafoli, E. Calcium pumps: Why so many? Compr. Physiol. 2012, 2, 1045–1060. [Google Scholar]
- Griffiths, C.; Drews, S.J.; Marchant, D.J. Respiratory Syncytial Virus: Infection, Detection, and New Options for Prevention and Treatment. Clin. Microbiol. Rev. 2017, 30, 277–319. [Google Scholar] [CrossRef]
- Hoffmann, H.H.; Schneider, W.M.; Blomen, V.A.; Scull, M.A.; Hovnanian, A.; Brummelkamp, T.R.; Rice, C.M. Diverse Viruses Require the Calcium Transporter SPCA1 for Maturation and Spread. Cell Host Microbe 2017, 22, 460–470. [Google Scholar] [CrossRef]
- Cervantes-Ortiz, S.L.; Zamorano Cuervo, N.; Grandvaux, N. Respiratory Syncytial Virus and Cellular Stress Responses: Impact on Replication and Physiopathology. Viruses 2016, 8, 124. [Google Scholar] [CrossRef] [PubMed]
- Cui, R.; Wang, Y.; Wang, L.; Li, G.; Lan, K.; Altmeyer, R.; Zou, G. Cyclopiazonic acid, an inhibitor of calcium-dependent ATPases with antiviral activity against human respiratory syncytial virus. Antivir. Res. 2016, 132, 38–45. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Cellular Targets | Virus | Consequences [Ref.] |
|---|---|---|
| VGCCs | IAV | CaV1.2 serves as a host cell surface receptor that binds IAV and is critical for IAV entry [9]. |
| SFTSV | Benidipine hydrochloride, VGCC blocker, inhibits SFTSV infection via impairing virus internalization and genome replication [12]. | |
| NWV | Virus binds to VGCCs and promotes virus entry at the virus–cell fusion step [13]. | |
| Flavivirus (JEV, ZIKV, DENV, and WNV | VGCCs blockers inhibit flavivirus (JEV, ZIKV, DENV and WNV) infection at the stage of replication [14]. | |
| HIV-1 | Tat/gp120 overactivate VGCCs [15,16,17]. | |
| HSV-1 | HSV-1 downregulates the CaV3.2 channel and diminishes the detection of viral infection by host [19,20]. | |
| TPCs | EBOV | Facilitates virus–endosome membrane fusion and releases of virus capsid into the cell cytoplasm [27]. |
| STIM1/ORAI1 | EBOV, MARV, LASV, JUNV, HIV-1, DENV, and HBV | Promote virion assembly and budding [35,36,37,38,44]. |
| TRPV4 | ZIKV | Activation of TRPV4- releases DDX3X and promote the viral RNA metabolism [52]. |
| NMDAr | ZIKV, JEV | NMDAr contributes to ZIKA by triggering the neuronal cell death progress [58,60]. |
| HIV-1 | Increases Ca2+ influx [62]. | |
| IP3R | HIV-1, HSV, HRV, and HCMV | These viral proteins deplete ER Ca2+ store during early stages of viral infection to increase the replication ability of viruses [37,62,63,64,66]. |
| SPCA1 | RSV, ZIKV, DENV and WNV | Trigger to produce functional viral glycoproteins that are essential for virus spread [70] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.; Cao, R.; Zhong, W. Host Calcium Channels and Pumps in Viral Infections. Cells 2020, 9, 94. https://doi.org/10.3390/cells9010094
Chen X, Cao R, Zhong W. Host Calcium Channels and Pumps in Viral Infections. Cells. 2020; 9(1):94. https://doi.org/10.3390/cells9010094
Chicago/Turabian StyleChen, Xingjuan, Ruiyuan Cao, and Wu Zhong. 2020. "Host Calcium Channels and Pumps in Viral Infections" Cells 9, no. 1: 94. https://doi.org/10.3390/cells9010094
APA StyleChen, X., Cao, R., & Zhong, W. (2020). Host Calcium Channels and Pumps in Viral Infections. Cells, 9(1), 94. https://doi.org/10.3390/cells9010094