The Roles of CD38 and CD157 in the Solid Tumor Microenvironment and Cancer Immunotherapy


Abstract
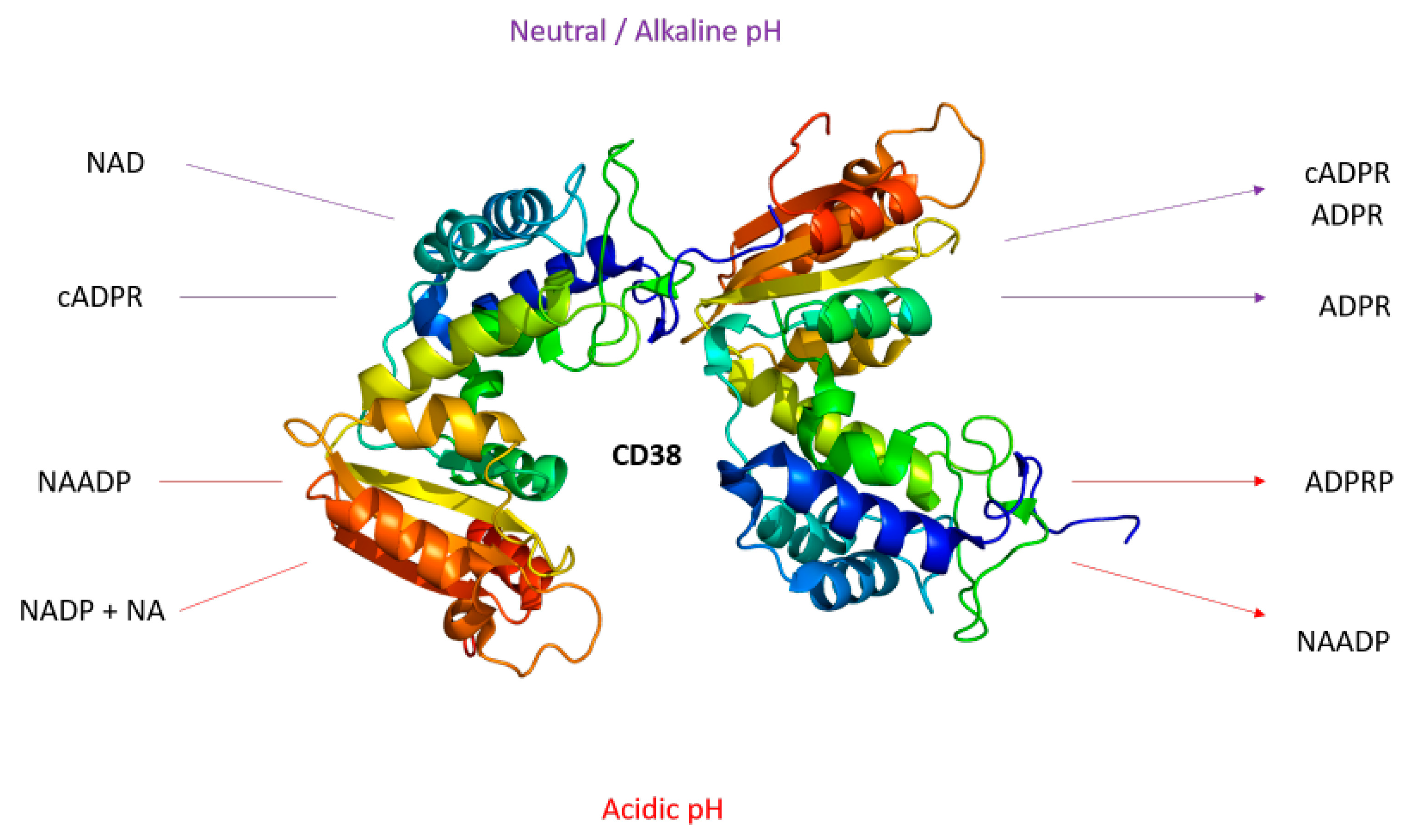
1. Background
1.1. The Role of CD38 in the TME
1.2. The Role of CD157 in the TME
2. CD38 and CD157 as Targets for Cancer Immunotherapy
2.1. CD38 in the Solid Tumor Microenvironment
2.1.1. CD38 in Hepatocellular Carcinoma
2.1.2. CD38 in Non-Small Cell Lung Cancer
2.1.3. CD38 in Melanoma
2.1.4. CD38 in Pancreatic Ductal Adenocarcinoma
2.1.5. CD38 in Glioma
2.1.6. CD38 in Breast Cancer
2.2. CD157 in the Solid Tumor Microenvironment
2.2.1. CD157 in Ovarian Cancer
2.2.2. CD157 in Pleural Mesothelioma
2.2.3. Potential Functions of CD157 in Other Types of Tumor
3. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Sheng, Z.; Zhu, X.; Sun, Y.; Zhang, Y. The efficacy of anti-PD-1/PD-L1 therapy and its comparison with EGFR-TKIs for advanced non-small-cell lung cancer. Oncotarget 2017, 8, 57826–57835. [Google Scholar] [CrossRef] [PubMed]
- Pai-Scherf, L.; Blumenthal, G.M.; Li, H.; Subramaniam, S.; Mishra-Kalyani, P.S.; He, K.; Zhao, H.; Yu, J.; Paciga, M.; Goldberg, K.B.; et al. FDA Approval Summary: Pembrolizumab for Treatment of Metastatic Non-Small Cell Lung Cancer: First-Line Therapy and Beyond. Oncologist 2017, 22, 1392–1399. [Google Scholar] [CrossRef] [PubMed]
- Teixido, C.; Vilarino, N.; Reyes, R.; Reguart, N. PD-L1 expression testing in non-small cell lung cancer. Ther. Adv. Med. Oncol. 2018, 10, 1758835918763493. [Google Scholar] [CrossRef] [PubMed]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-γ–related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef] [PubMed]
- Roma-Rodrigues, C.; Mendes, R.; Baptista, P.V.; Fernandes, A.R. Targeting Tumor Microenvironment for Cancer Therapy. Int. J. Mol. Sci. 2019, 20, 840. [Google Scholar] [CrossRef]
- Goh, S.; Ng, H.H.M.; Chew, V.; Sim, X.N.; Li, H.; Lim, S.; Lim, J.C.T.; Loh, J.J.H.; Sabai, K.; Ong, C.C.H.; et al. CD38 is a good predictor of anti-PD-1 immunotherapy responsiveness in hepatocellular carcinoma. BioXiv 2019, 638981. [Google Scholar] [CrossRef]
- Di Blasi, D.; Boldanova, T.; Mori, L.; Terracciano, L.; Heim, M.H.; De Libero, G. Unique T-Cell Populations Define Immune-Inflamed Hepatocellular Carcinoma. Cell. Mol. Gastroenterol. Hepatol. 2019. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer—Immune set point. Nature 2017, 541, 321. [Google Scholar] [CrossRef]
- Wang, M.; Zhao, J.; Zhang, L.; Wei, F.; Lian, Y.; Wu, Y.; Gong, Z.; Zhang, S.; Zhou, J.; Cao, K.; et al. Role of tumor microenvironment in tumorigenesis. J. Cancer 2017, 8, 761–773. [Google Scholar] [CrossRef]
- Malavasi, F.; Deaglio, S.; Funaro, A.; Ferrero, E.; Horenstein, A.L.; Ortolan, E.; Vaisitti, T.; Aydin, S. Evolution and Function of the ADP Ribosyl Cyclase/CD38 Gene Family in Physiology and Pathology. Physiol. Rev. 2008, 88, 841–886. [Google Scholar] [CrossRef]
- Lee, H.C. Structure and enzymatic functions of human CD38. Mol. Med. 2006, 12, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Ortolan, E.; Augeri, S.; Fissolo, G.; Musso, I.; Funaro, A. CD157: From immunoregulatory protein to potential therapeutic target. Immun. Lett. 2019, 205, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Van de Donk, N.W.C.J.; Janmaat, M.L.; Mutis, T.; Lammerts van Bueren, J.J.; Ahmadi, T.; Sasser, A.K.; Lokhorst, H.M.; Parren, P.W.H.I. Monoclonal antibodies targeting CD38 in hematological malignancies and beyond. Immun. Rev. 2016, 270, 95–112. [Google Scholar] [CrossRef] [PubMed]
- Krupka, C.; Jansen, A.; Lassmann, I.; Terrett, J.; Aud, D.; Dusek, R.; Bisht, A.; Pombo-Villar, E.; Rohlff, C.; Köhnke, T.; et al. Targeting AML Using an Fc-Engineered BST1/CD157 Monoclonal Antibody. Blood 2014, 124, 987. [Google Scholar] [CrossRef]
- Czura, A.W.; Czura, C.J. CD38 and CD157: Biological observations to clinical therapeutic targets. Mol. Med. 2006, 12, 309–311. [Google Scholar] [CrossRef] [PubMed]
- Petrova, V.; Annicchiarico-Petruzzelli, M.; Melino, G.; Amelio, I. The hypoxic tumour microenvironment. Oncogenesis 2018, 7, 10. [Google Scholar] [CrossRef]
- Kim, Y.; Lin, Q.; Glazer, P.M.; Yun, Z. Hypoxic tumor microenvironment and cancer cell differentiation. Curr. Mol. Med. 2009, 9, 425–434. [Google Scholar] [CrossRef]
- Passarelli, A.; Tucci, M.; Mannavola, F.; Felici, C.; Silvestris, F. The metabolic milieu in melanoma: Role of immune suppression by CD73/adenosine. Tumor Biol. 2019, 41, 1010428319837138. [Google Scholar] [CrossRef]
- Perrot, I.M.H.; Giraudon-Paoli, M.; Augier, S.; Docquier, A.; Gros, L.; Courtois, R.; Déjou, C.; Jecko, D.; Becquart, O.; Rispaud-Blanc, H.; et al. Blocking Antibodies Targeting the CD39/CD73 Immunosuppressive Pathway Unleash Immune Responses in Combination Cancer Therapies. Cell Rep. 2019, 27, 2411–2425. [Google Scholar] [CrossRef]
- Anichini, A.; Molla, A.; Vegetti, C.; Bersani, I.; Zappasodi, R.; Arienti, F.; Ravagnani, F.; Maurichi, A.; Patuzzo, R.; Santinami, M.; et al. Tumor-Reactive CD8 + Early Effector T Cells Identified at Tumor Site in Primary and Metastatic Melanoma. Cancer Res. 2010, 70, 8378–8387. [Google Scholar] [CrossRef]
- Horenstein, A.L.; Chillemi, A.; Zaccarello, G.; Bruzzone, S.; Quarona, V.; Zito, A.; Serra, S.; Malavasi, F. A CD38/CD203a/CD73 ectoenzymatic pathway independent of CD39 drives a novel adenosinergic loop in human T lymphocytes. Oncoimmunology 2013, 2, e26246. [Google Scholar] [CrossRef] [PubMed]
- Horenstein, A.L.; Bracci, C.; Morandi, F.; Malavasi, F. CD38 in Adenosinergic Pathways and Metabolic Re-programming in Human Multiple Myeloma Cells: In-tandem Insights From Basic Science to Therapy. Front. Immunol. 2019, 10, 760. [Google Scholar] [CrossRef] [PubMed]
- Ohta, A. A Metabolic Immune Checkpoint: Adenosine in Tumor Microenvironment. Front. Immunol. 2016, 7, 109. [Google Scholar] [CrossRef] [PubMed]
- Ben Baruch, B.; Blacher, E.; Mantsur, E.; Schwartz, H.; Vaknine, H.; Erez, N.; Stein, R. Stromal CD38 regulates outgrowth of primary melanoma and generation of spontaneous metastasis. Oncotarget 2018, 9, 31797–31811. [Google Scholar] [CrossRef] [PubMed]
- Favia, A.; Desideri, M.; Gambara, G.; D’Alessio, A.; Ruas, M.; Esposito, B.; Del Bufalo, D.; Parrington, J.; Ziparo, E.; Palombi, F.; et al. VEGF-induced neoangiogenesis is mediated by NAADP and two-pore channel-2-dependent Ca2+ signaling. Proc. Natl. Acad. Sci. USA 2014, 111, E4706–E4715. [Google Scholar] [CrossRef] [PubMed]
- Favia, A.; Pafumi, I.; Desideri, M.; Padula, F.; Montesano, C.; Passeri, D.; Nicoletti, C.; Orlandi, A.; Del Bufalo, D.; Sergi, M.; et al. NAADP-Dependent Ca(2+) Signaling Controls Melanoma Progression, Metastatic Dissemination and Neoangiogenesis. Sci. Rep. 2016, 6, 18925. [Google Scholar] [CrossRef] [PubMed]
- Chung, A.S.; Ferrara, N. Developmental and Pathological Angiogenesis. Ann. Rev. Cell Dev. Biol. 2011, 27, 563–584. [Google Scholar] [CrossRef]
- Deaglio, S.; Morra, M.; Mallone, R.; Ausiello, C.M.; Prager, E.; Garbarino, G.; Dianzani, U.; Stockinger, H.; Malavasi, F. Human CD38 (ADP-Ribosyl Cyclase) Is a Counter-Receptor of CD31, an Ig Superfamily Member. J. Immunol. 1998, 160, 395–402. [Google Scholar]
- March, S.; Graupera, M.; Rosa Sarrias, M.; Lozano, F.; Pizcueta, P.; Bosch, J.; Engel, P. Identification and functional characterization of the hepatic stellate cell CD38 cell surface molecule. Am. J. Pathol. 2007, 170, 176–187. [Google Scholar] [CrossRef][Green Version]
- Lam, J.H.; Ng, H.H.M.; Lim, C.J.; Sim, X.N.; Malavasi, F.; Li, H.; Loh, J.J.H.; Sabai, K.; Kim, J.-K.; Ong, C.C.H.; et al. Expression of CD38 on Macrophages Predicts Improved Prognosis in Hepatocellular Carcinoma. Front. Immunol. 2019, 10, 2093. [Google Scholar] [CrossRef]
- Tassi, E.; Grazia, G.; Vegetti, C.; Bersani, I.; Bertolini, G.; Molla, A.; Baldassari, P.; Andriani, F.; Roz, L.; Sozzi, G.; et al. Early Effector T Lymphocytes Coexpress Multiple Inhibitory Receptors in Primary Non–Small Cell Lung Cancer. Cancer Res. 2017, 77, 851–861. [Google Scholar] [CrossRef] [PubMed]
- Kovacsovics-Bankowski, M.; Chisholm, L.; Vercellini, J.; Tucker, C.G.; Montler, R.; Haley, D.; Newell, P.; Ma, J.; Tseng, P.; Wolf, R.; et al. Detailed characterization of tumor infiltrating lymphocytes in two distinct human solid malignancies show phenotypic similarities. J. Immunother. Cancer 2014, 2, 38. [Google Scholar] [CrossRef] [PubMed]
- Dixon, A.R.; Bathany, C.; Tsuei, M.; White, J.; Barald, K.F.; Takayama, S. Recent developments in multiplexing techniques for immunohistochemistry. Expert Rev. Mol. Diagn. 2015, 15, 1171–1186. [Google Scholar] [CrossRef] [PubMed]
- Surace, M.; DaCosta, K.; Huntley, A.; Zhao, W.; Bagnall, C.; Brown, C.; Wang, C.; Roman, K.; Cann, J.; Lewis, A.; et al. Automated Multiplex Immunofluorescence Panel for Immuno-oncology Studies on Formalin-fixed Carcinoma Tissue Specimens. JoVE 2019, 143, e58390. [Google Scholar] [CrossRef]
- Morandi, F.; Morandi, B.; Horenstein, A.L.; Chillemi, A.; Quarona, V.; Zaccarello, G.; Carrega, P.; Ferlazzo, G.; Mingari, M.C.; Moretta, L.; et al. A non-canonical adenosinergic pathway led by CD38 in human melanoma cells induces suppression of T cell proliferation. Oncotarget 2015, 6, 25602–25618. [Google Scholar] [CrossRef]
- Quarona, V.; Zaccarello, G.; Chillemi, A.; Brunetti, E.; Singh, V.K.; Ferrero, E.; Funaro, A.; Horenstein, A.L.; Malavasi, F. CD38 and CD157: A long journey from activation markers to multifunctional molecules. Cytom. Part B Clin. Cytom. 2013, 84B, 207–217. [Google Scholar] [CrossRef]
- Lo Buono, N.M.S.; Giacomino, A.; Parrotta, R.; Ferrero, E.; Malavasi, F.; Ortolan, E.; Funaro, A. CD157 at the intersection between leukocyte trafficking and epithelial ovarian cancer invasion. Front. Biosci. 2014, 19, 366–378. [Google Scholar] [CrossRef]
- Kaisho, T.; Ishikawa, J.; Oritani, K.; Inazawa, J.; Tomizawa, H.; Muraoka, O.; Ochi, T.; Hirano, T. BST-1, a surface molecule of bone marrow stromal cell lines that facilitates pre-B-cell growth. Proc. Natl. Acad. Sci. USA 1994, 91, 5325–5329. [Google Scholar] [CrossRef]
- Ortolan, E.; Vacca, P.; Capobianco, A.; Armando, E.; Crivellin, F.; Horenstein, A.; Malavasi, F. CD157, the Janus of CD38 but with a unique personality. Cell Biochem. Funct. 2002, 20, 309–322. [Google Scholar] [CrossRef]
- Ortolan, E.A.R.; Morone, S.; Bovino, P.; Lo-Buono, N.; Nacci, G.; Parrotta, R.; Katsaros, D.; Rapa, I.; Migliaretti, G.; Ferrero, E.; et al. Functional Role and Prognostic Significance of CD157 in Ovarian Carcinoma. J. Natl. Cancer Inst. 2010, 102, 1160–1177. [Google Scholar] [CrossRef]
- Funaro, A.; Ortolan, E.; Ferranti, B.; Gargiulo, L.; Notaro, R.; Luzzatto, L.; Malavasi, F. CD157 is an important mediator of neutrophil adhesion and migration. Blood 2004, 104, 4269–4278. [Google Scholar] [CrossRef] [PubMed]
- Roychowdhury, S.; Chinnaiyan, A.M. Translating Genomics for Precision Cancer Medicine. Ann. Rev. Genom. Hum. Genet. 2014, 15, 395–415. [Google Scholar] [CrossRef]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Diao, L.; Yang, Y.; Yi, X.; Rodriguez, B.L.; Li, Y.; Villalobos, P.A.; Cascone, T.; Liu, X.; Tan, L.; et al. CD38-Mediated Immunosuppression as a Mechanism of Tumor Cell Escape from PD-1/PD-L1 Blockade. Cancer Discov. 2018, 8, 1156–1175. [Google Scholar] [CrossRef]
- Bhat, P.; Leggatt, G.; Waterhouse, N.; Frazer, I.H. Interferon-γ derived from cytotoxic lymphocytes directly enhances their motility and cytotoxicity. Cell Death Dis. 2017, 8, e2836. [Google Scholar] [CrossRef]
- Schoenborn, J.W.; Wilson, C.B. Regulation of Interferon-γ During Innate and Adaptive Immune Responses. Adv. Immunol. 2007, 96, 41–101. [Google Scholar] [CrossRef]
- Ji, T.; Li, G.; Chen, J.; Zhao, J.; Li, X.; Lin, H.; Cai, X.; Cang, Y. Distinct role of interleukin-6 and tumor necrosis factor receptor-1 in oval cell-mediated liver regeneration and inflammation-associated hepatocarcinogenesis. Oncotarget 2016, 7, 66635–66646. [Google Scholar] [CrossRef]
- Zhang, M.Y.J.; Zhou, J.; Gao, W.; Zhang, Y.; Lin, Y.; Wang, H.; Ruan, Z.; Ni, B. Prognostic Values of CD38 + CD101 + PD1 + CD8 + T Cells in Pancreatic Cancer. Immun. Investig. 2019, 48, 1–14. [Google Scholar] [CrossRef]
- Levy, A.; Bercovich-Kinori, A.; Alexandrovich, A.G.; Tsenter, J.; Trembovler, V.; Lund, F.E.; Shohami, E.; Stein, R.; Mayo, L. CD38 facilitates recovery from traumatic brain injury. J. Neurotrauma 2009, 26, 1521–1533. [Google Scholar] [CrossRef]
- Mayo, L.; Jacob-Hirsch, J.; Amariglio, N.; Rechavi, G.; Moutin, M.J.; Lund, F.E.; Stein, R. Dual role of CD38 in microglial activation and activation-induced cell death. J. Immunol. 2008, 181, 92–103. [Google Scholar] [CrossRef]
- Levy, A.; Blacher, E.; Vaknine, H.; Lund, F.E.; Stein, R.; Mayo, L. CD38 deficiency in the tumor microenvironment attenuates glioma progression and modulates features of tumor-associated microglia/macrophages. Neuro Oncol. 2012, 14, 1037–1049. [Google Scholar] [CrossRef] [PubMed]
- Blacher, E.L.A.; Ben Baruch, B.; Green, K.; Garneau-Tsodikova, S.; Berkov, Y.; Stein, R. Targeting CD38 in the tumor microenvironment; a novel approach to treat glioma. Cancer Cell Microenviron. 2015, 2, 2. [Google Scholar]
- Badie, B.; Schartner, J. Role of microglia in glioma biology. Microsc. Res. Tech. 2001, 54, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Yang, I.; Han, S.J.; Kaur, G.; Crane, C.; Parsa, A.T. The role of microglia in central nervous system immunity and glioma immunology. J. Clin. Neurosci. 2010, 17, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Yeong, J.; Lim, J.C.T.; Lee, B.; Li, H.; Chia, N.; Ong, C.C.H.; Lye, W.K.; Putti, T.C.; Dent, R.; Lim, E.; et al. High Densities of Tumor-Associated Plasma Cells Predict Improved Prognosis in Triple Negative Breast Cancer. Front. Immunol. 2018, 9, 1209. [Google Scholar] [CrossRef]
- Morone, S.; Lo-Buono, N.; Parrotta, R.; Giacomino, A.; Nacci, G.; Brusco, A.; Larionov, A.; Ostano, P.; Mello-Grand, M.; Chiorino, G.; et al. Overexpression of CD157 contributes to epithelial ovarian cancer progression by promoting mesenchymal differentiation. PLoS ONE 2012, 7, e43649. [Google Scholar] [CrossRef] [PubMed]
- Ortolan, E.; Giacomino, A.; Martinetto, F.; Morone, S.; Lo Buono, N.; Ferrero, E.; Scagliotti, G.; Novello, S.; Orecchia, S.; Ruffini, E.; et al. CD157 enhances malignant pleural mesothelioma aggressiveness and predicts poor clinical outcome. Oncotarget 2014, 5, 6191–6205. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Reig, M.; Sherman, M. Evidence-Based Diagnosis, Staging, and Treatment of Patients With Hepatocellular Carcinoma. Gastroenterology 2016, 150, 835–853. [Google Scholar] [CrossRef]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.-Y.; Choo, S.-P.; Trojan, J.; Welling, T.H., 3rd; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Kalathil, S.G.; Hutson, A.; Barbi, J.; Iyer, R.; Thanavala, Y. Augmentation of IFN-γ + CD8 + T cell responses correlates with survival of HCC patients on sorafenib therapy. JCI Insight 2019, 4, 116. [Google Scholar] [CrossRef]
- Garnelo, M.; Tan, A.; Her, Z.; Yeong, J.; Lim, C.J.; Chen, J.; Lim, K.H.; Weber, A.; Chow, P.; Chung, A.; et al. Interaction between tumour-infiltrating B cells and T cells controls the progression of hepatocellular carcinoma. Gut 2017, 66, 342–351. [Google Scholar] [CrossRef] [PubMed]
- Zilber, M.T.; Gregory, S.; Mallone, R.; Deaglio, S.; Malavasi, F.; Charron, D.; Gelin, C. CD38 expressed on human monocytes: A coaccessory molecule in the superantigen-induced proliferation. Proc. Natl. Acad. Sci. USA 2000, 97, 2840–2845. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Gridelli, C.; Rossi, A.; Carbone, D.P.; Guarize, J.; Karachaliou, N.; Mok, T.; Petrella, F.; Spaggiari, L.; Rosell, R. Non-small-cell lung cancer. Nat. Rev. Dis. Primers 2015, 1, 15009. [Google Scholar] [CrossRef] [PubMed]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the Treatment of Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef]
- Kamphorst, A.O.; Pillai, R.N.; Yang, S.; Nasti, T.H.; Akondy, R.S.; Wieland, A.; Sica, G.L.; Yu, K.; Koenig, L.; Patel, N.T.; et al. Proliferation of PD-1 + CD8 T cells in peripheral blood after PD-1-targeted therapy in lung cancer patients. Proc. Natl. Acad. Sci. USA 2017, 114, 4993–4998. [Google Scholar] [CrossRef]
- Bandarchi, B.; Jabbari, C.A.; Vedadi, A.; Navab, R. Molecular biology of normal melanocytes and melanoma cells. J. Clin. Pathol. 2013, 66, 644–648. [Google Scholar] [CrossRef]
- Zhou, L.; Yang, K.; Andl, T.; Wickett, R.R.; Zhang, Y. Perspective of Targeting Cancer-Associated Fibroblasts in Melanoma. J. Cancer 2015, 6, 717–726. [Google Scholar] [CrossRef]
- Erdmann, F.; Lortet-Tieulent, J.; Schüz, J.; Zeeb, H.; Greinert, R.; Breitbart, E.W.; Bray, F. International trends in the incidence of malignant melanoma 1953–2008—Are recent generations at higher or lower risk? Int. J. Cancer 2013, 132, 385–400. [Google Scholar] [CrossRef]
- Callahan, M.K.; Kluger, H.; Postow, M.A.; Segal, N.H.; Lesokhin, A.; Atkins, M.B.; Kirkwood, J.M.; Krishnan, S.; Bhore, R.; Horak, C.; et al. Nivolumab Plus Ipilimumab in Patients With Advanced Melanoma: Updated Survival, Response, and Safety Data in a Phase I Dose-Escalation Study. J. Clin. Oncol. 2018, 36, 391–398. [Google Scholar] [CrossRef]
- Albertini, M.R. The age of enlightenment in melanoma immunotherapy. J. Immun. Ther. Cancer 2018, 6, 80. [Google Scholar] [CrossRef] [PubMed]
- Spinella, F.; Caprara, V.; Cianfrocca, R.; Rosanò, L.; Di Castro, V.; Garrafa, E.; Natali, P.G.; Bagnato, A. The interplay between hypoxia, endothelial and melanoma cells regulates vascularization and cell motility through endothelin-1 and vascular endothelial growth factor. Carcinogenesis 2014, 35, 840–848. [Google Scholar] [CrossRef]
- Bailey, P.; Chang, D.K.; Forget, M.-A.; Lucas, F.A.S.; Alvarez, H.A.; Haymaker, C.; Chattopadhyay, C.; Kim, S.-H.; Ekmekcioglu, S.; Grimm, E.A.; et al. Exploiting the neoantigen landscape for immunotherapy of pancreatic ductal adenocarcinoma. Sci. Rep. 2016, 6, 35848. [Google Scholar] [CrossRef] [PubMed]
- Bazhin, A.V.; Shevchenko, I.; Umansky, V.; Werner, J.; Karakhanova, S. Two immune faces of pancreatic adenocarcinoma: Possible implication for immunotherapy. Cancer Immunol. Immunother. 2014, 63, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Goodenberger, M.L.; Jenkins, R.B. Genetics of adult glioma. Cancer Genet. 2012, 205, 613–621. [Google Scholar] [CrossRef]
- Furnari, F.B.; Fenton, T.; Bachoo, R.M.; Mukasa, A.; Stommel, J.M.; Stegh, A.; Hahn, W.C.; Ligon, K.L.; Louis, D.N.; Brennan, C.; et al. Malignant astrocytic glioma: Genetics, biology, and paths to treatment. Genes Dev. 2007, 21, 2683–2710. [Google Scholar] [CrossRef]
- De Vleeschouwer, S.B.G. Glioblastoma: To Target the Tumor Cell or the Microenvironment? In Glioblastoma; De Vleeschouwer, S., Ed.; Codon Publications: Brisbane, Australia, 2017; pp. 315–340. [Google Scholar]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef]
- Zardavas, D.; Irrthum, A.; Swanton, C.; Piccart, M. Clinical management of breast cancer heterogeneity. Nat. Rev. Clin. Oncol. 2015, 12, 381. [Google Scholar] [CrossRef]
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-Negative Breast Cancer: Clinical Features and Patterns of Recurrence. Clin. Cancer Res. 2007, 13, 4429–4434. [Google Scholar] [CrossRef]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674. [Google Scholar] [CrossRef]
- Bauer, K.R.; Brown, M.; Cress, R.D.; Parise, C.A.; Caggiano, V. Descriptive analysis of estrogen receptor (ER)-negative, progesterone receptor (PR)-negative, and HER2-negative invasive breast cancer, the so-called triple-negative phenotype. Cancer 2007, 109, 1721–1728. [Google Scholar] [CrossRef] [PubMed]
- Jaspers, J.E.; Rottenberg, S.; Jonkers, J. Therapeutic options for triple-negative breast cancers with defective homologous recombination. Biochim. Biophys. Acta 2009, 1796, 266–280. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Carden, C.P.; Kaye, S.B. Beyond chemotherapy: Targeted therapies in ovarian cancer. Nat. Rev. Cancer 2009, 9, 167. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, R.; Kaye, S.B. Ovarian cancer: Strategies for overcoming resistance to chemotherapy. Nat. Rev. Cancer 2003, 3, 502–516. [Google Scholar] [CrossRef]
- Auersperg, N.W.; Wong, A.S.; Choi, K.C.; Kang, S.; Leung, P.C.K. Ovarian Surface Epithelium: Biology, Endocrinology, and Pathology. Endocr. Rev. 2001, 22, 255–288. [Google Scholar] [CrossRef]
- Jayson, G.C.K.; Kitchener, H.C.; Ledermann, J. Ovarian cancer. Lancet 2014, 384, 1376–1388. [Google Scholar] [CrossRef]
- Taioli, E.; Wolf, A.S.; Camacho-Rivera, M.; Kaufman, A.; Lee, D.S.; Nicastri, D.; Rosenzweig, K.; Flores, R.M. Determinants of Survival in Malignant Pleural Mesothelioma: A Surveillance, Epidemiology, and End Results (SEER) Study of 14,228 Patients. PLoS ONE 2015, 10, e0145039. [Google Scholar] [CrossRef]
- Bibby, A.C.; Tsim, S.; Kanellakis, N.; Ball, H.; Talbot, D.C.; Blyth, K.G.; Maskell, N.A.; Psallidas, I. Malignant pleural mesothelioma: An update on investigation, diagnosis and treatment. Eur. Respir. Rev. 2016, 25, 472–486. [Google Scholar] [CrossRef]
- Baas, P.F.; Fennell, D.; Kerr, K.M.; Van Schil, P.E.; Peters, S. Malignant pleural mesothelioma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2015, 26, v31–v39. [Google Scholar] [CrossRef]
- Ettinger, D.S.; Wood, D.E.; Akerley, W.; Bazhenova, L.A.; Borghaei, H.; Camidge, D.R.; Cheney, R.T.; Chirieac, L.R.; D’Amico, T.A.; Dilling, T.; et al. NCCN Guidelines Insights: Malignant Pleural Mesothelioma, Version 3.2016. J. Natl. Compr. Cancer Netw. 2016, 14, 825. [Google Scholar] [CrossRef]
- Fassina, A.; Cappellesso, R.; Guzzardo, V.; Dalla Via, L.; Piccolo, S.; Ventura, L.; Fassan, M. Epithelial—Mesenchymal transition in malignant mesothelioma. Mod. Pathol. 2012, 25, 86–99. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Schwarzbauer, J.E. Mammary epithelial cell interactions with fibronectin stimulate epithelial-mesenchymal transition. Oncogene 2014, 33, 1649–1657. [Google Scholar] [CrossRef] [PubMed]
- Bitanihirwe, B.K.Y.; Meerang, M.; Friess, M.; Soltermann, A.; Frischknecht, L.; Thies, S.; Felley-Bosco, E.; Tsao, M.-S.; Allo, G.; de Perrot, M.; et al. PI3K/mTOR Signaling in Mesothelioma Patients Treated with Induction Chemotherapy Followed by Extrapleural Pneumonectomy. J. Thorac. Oncol. 2014, 9, 239–247. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Tumor Type | Cell Type | Function of CD38 |
|---|---|---|
| Hepatocellular carcinoma | Tumor infiltrating lymphocytes (B and T cells) | - Presence of CD38+ TILs within the TME increases effectiveness of cancer immunotherapy [2,7], as activated CD38+ TILs induce inflammation and promote an anti-tumor immune response [7] via the production of cytotoxic compounds and inflammatory cytokines [45,46] |
Tumor cell | - CD38 upregulation on tumor cells suppresses cytotoxic T cell function due to its adenosinergic activity, thus limiting the therapeutic effect of anti-PD-1 immunotherapy [2,44] | |
Macrophages | - CD38+ macrophages in the TME may promote inflammation and exert anti-tumor effects. This is because CD38 expression is associated with M1 macrophages, which produce pro-inflammatory cytokines [47] - Coexpression of CD38 and CD68 on macrophages is correlated with improved prognosis after surgical resection by increasing the M1 to M2 macrophage ratio [30] | |
| Non-small cell lung cancer | Tumor cell | - CD38 upregulation on tumor cell correlates with development of resistance to immunotherapy. This is because CD38 suppresses activity of cytotoxic T cells via the adenosinergic pathway [44] |
T cell | - CD38 is expressed by early effector T cells, hence it serves as a biomarker for the monitoring of the immune response against NSCLC [31] | |
| Melanoma | Tumor microenvironment | - CD38 is a potential immunotherapy target which can be inhibited to restrict growth of primary tumors [24] - CD38 inhibition prevents tumor angiogenesis and metastasis [25,26,27] - CD38 mediates suppression of cytotoxic T cell activity via the adenosinergic pathway [24,35] |
| Pancreatic ductal adenocarcinoma | Tumor infiltrating lymphocytes - B and T cells  | - CD38 and CD101 coexpression on TILs could indicate TIL exhaustion, and hence is a marker for adverse prognosis [48] |
| Glioma | Tumor-associated microglia/macrophages (TMM) | - Regulates TMM activation through cADPR-mediated increase in calcium concentration [49,50], hence serves as a prognostic tool as TMM number is positively correlated with glioma grade and invasiveness [51,52] - Creates an immunosuppressive tumor microenvironment via TMM activation, as TMM promote tumour angiogenesis and metastasis through the secretion of cytokines [52,53,54] |
| Breast cancer | Plasma cell | - High CD38 expression and high CD38+ plasma cell density are correlated with increased rates of disease free-survival and overall survival [55], and hence CD38 plays a prognostic role in triple negative breast cancer |
| Tumor Type | Cell Type | Function of CD157 |
|---|---|---|
| Ovarian cancer | Epithelial | - Promotes tumor cell invasion and migration [37,40], by reducing tumor cell binding to extracellular matrix proteins [40] - Correlated with increased malignancy and greater risk of relapse [37,40], as it promotes tumor cell acquisition of mesenchymal traits and reduces apoptosis [56] |
| Pleural mesothelioma | Mesothelial | - Increases tumor cell proliferation, invasiveness, and dissemination by activating the mTOR pathway [57] - Promotes mesenchymal differentiation [56] by mediating adhesion to extracellular matrix proteins [12] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wo, Y.J.; Gan, A.S.P.; Lim, X.; Tay, I.S.Y.; Lim, S.; Lim, J.C.T.; Yeong, J.P.S. The Roles of CD38 and CD157 in the Solid Tumor Microenvironment and Cancer Immunotherapy. Cells 2020, 9, 26. https://doi.org/10.3390/cells9010026
Wo YJ, Gan ASP, Lim X, Tay ISY, Lim S, Lim JCT, Yeong JPS. The Roles of CD38 and CD157 in the Solid Tumor Microenvironment and Cancer Immunotherapy. Cells. 2020; 9(1):26. https://doi.org/10.3390/cells9010026
Chicago/Turabian StyleWo, Yu Jun, Adelia Shin Ping Gan, Xinru Lim, Isabel Shu Ying Tay, Sherlly Lim, Jeffrey Chun Tatt Lim, and Joe Poh Sheng Yeong. 2020. "The Roles of CD38 and CD157 in the Solid Tumor Microenvironment and Cancer Immunotherapy" Cells 9, no. 1: 26. https://doi.org/10.3390/cells9010026
APA StyleWo, Y. J., Gan, A. S. P., Lim, X., Tay, I. S. Y., Lim, S., Lim, J. C. T., & Yeong, J. P. S. (2020). The Roles of CD38 and CD157 in the Solid Tumor Microenvironment and Cancer Immunotherapy. Cells, 9(1), 26. https://doi.org/10.3390/cells9010026