Depolarization-Associated CircRNA Regulate Neural Gene Expression and in Some Cases May Function as Templates for Translation

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Differentiation
2.2. Depolarization
2.3. RNA Extraction and Quality Control
2.4. RNA-Seq Library Generation and Sequencing
2.5. Prediction and Quantification of CircRNAs
2.6. mRNA and miRNA Expression Analysis
2.7. cDNA Synthesis and Quantitative PCR
2.8. Functional Enrichment Analysis
2.9. MiRNA Binding Site Prediction and circRNA–miRNA–mRNA Network Construction
2.10. circRNA Knockdown
2.11. Ribosome Profiling
2.12. Analysis of Ribosome Profiling Data
2.13. Annotation of ribo-circRNAs and Prediction of Coding Sequence and Domains
3. Results
3.1. Annotation of circRNAs in Differentiated Neuroblastoma
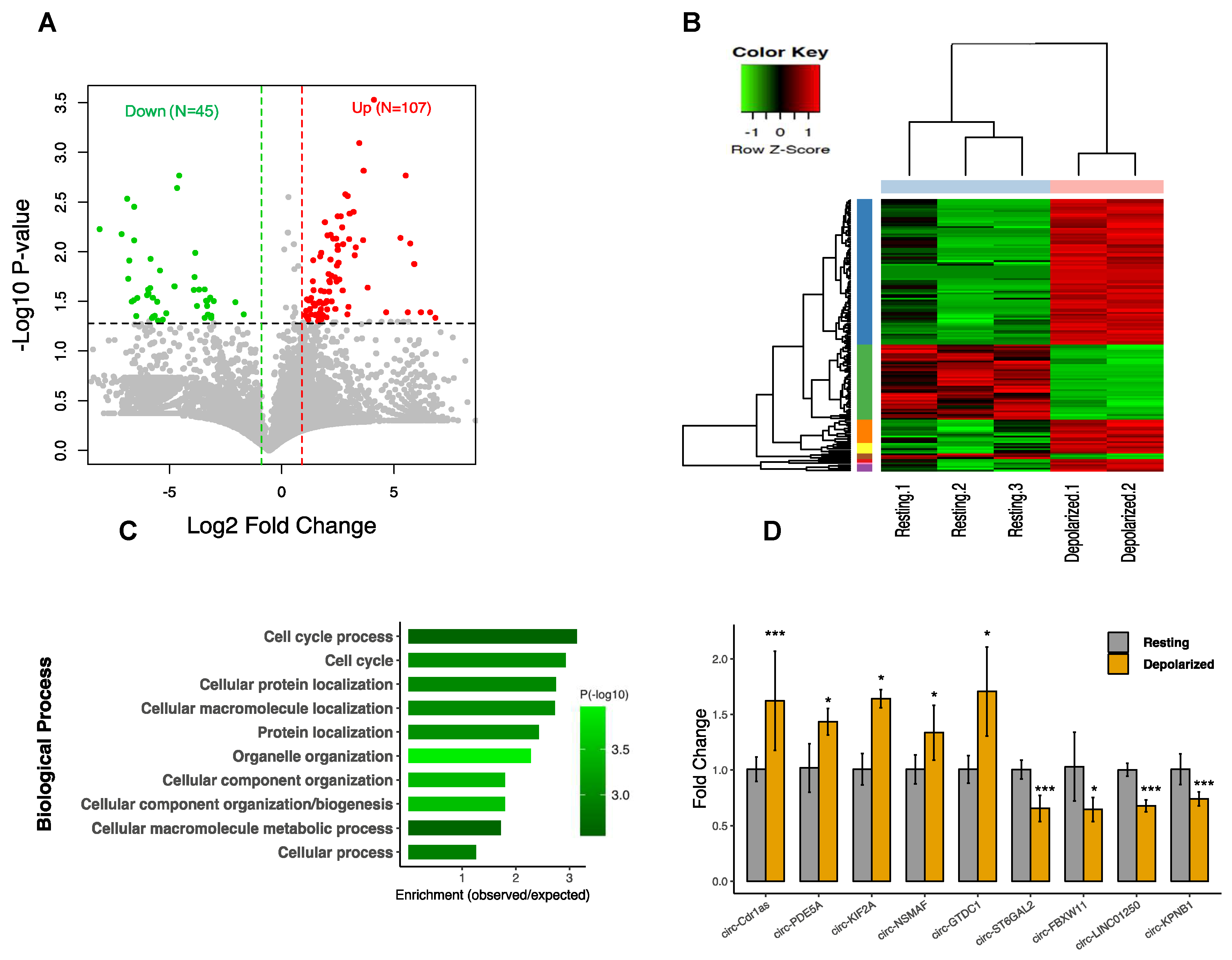
3.2. CircRNAs are Differentially Expressed in Neuronal Depolarization
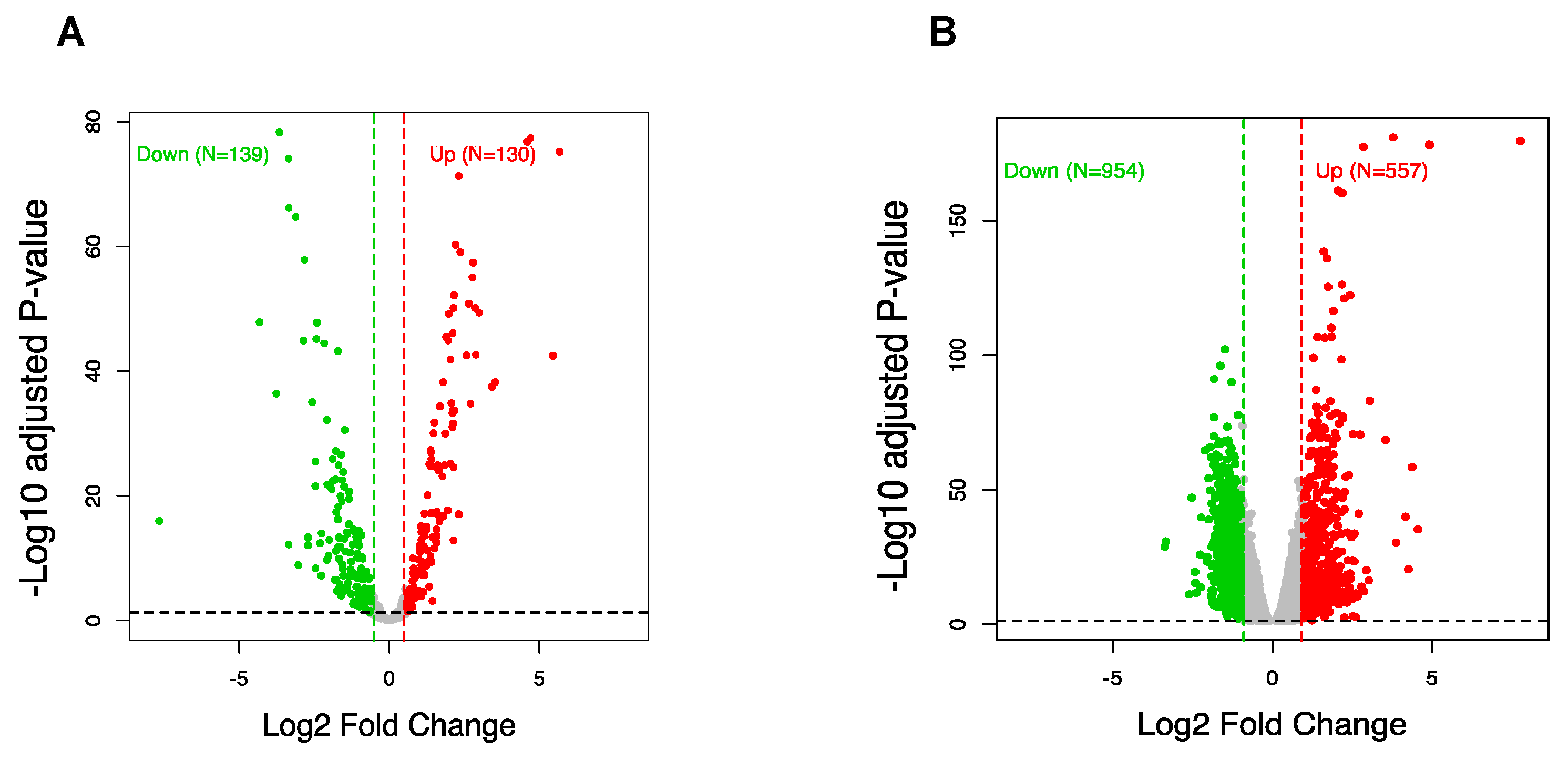
3.3. CircRNA Regulation Coincides with Global Alteration of miRNA and mRNA
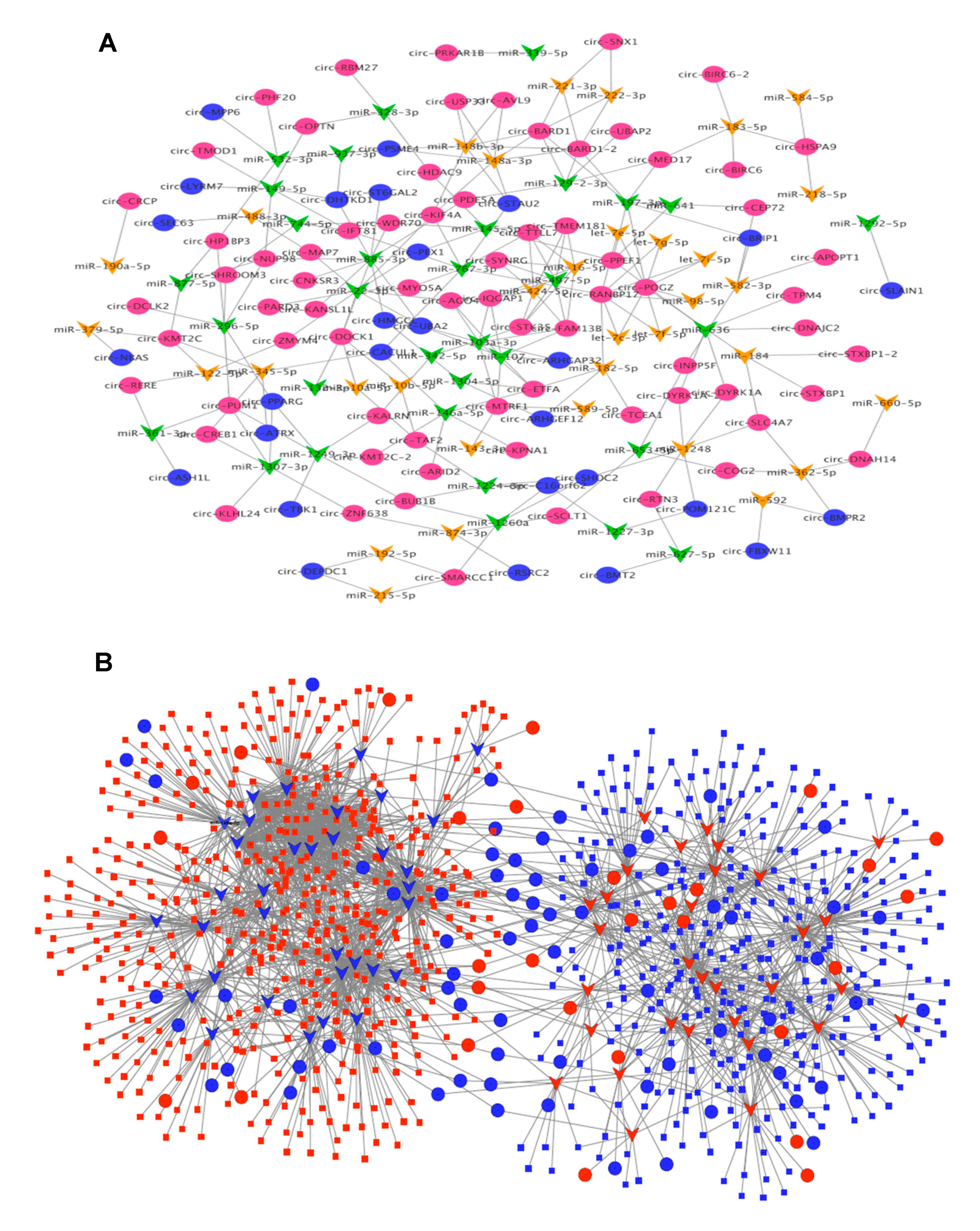
3.4. CircRNA–miRNA–mRNA Association Network
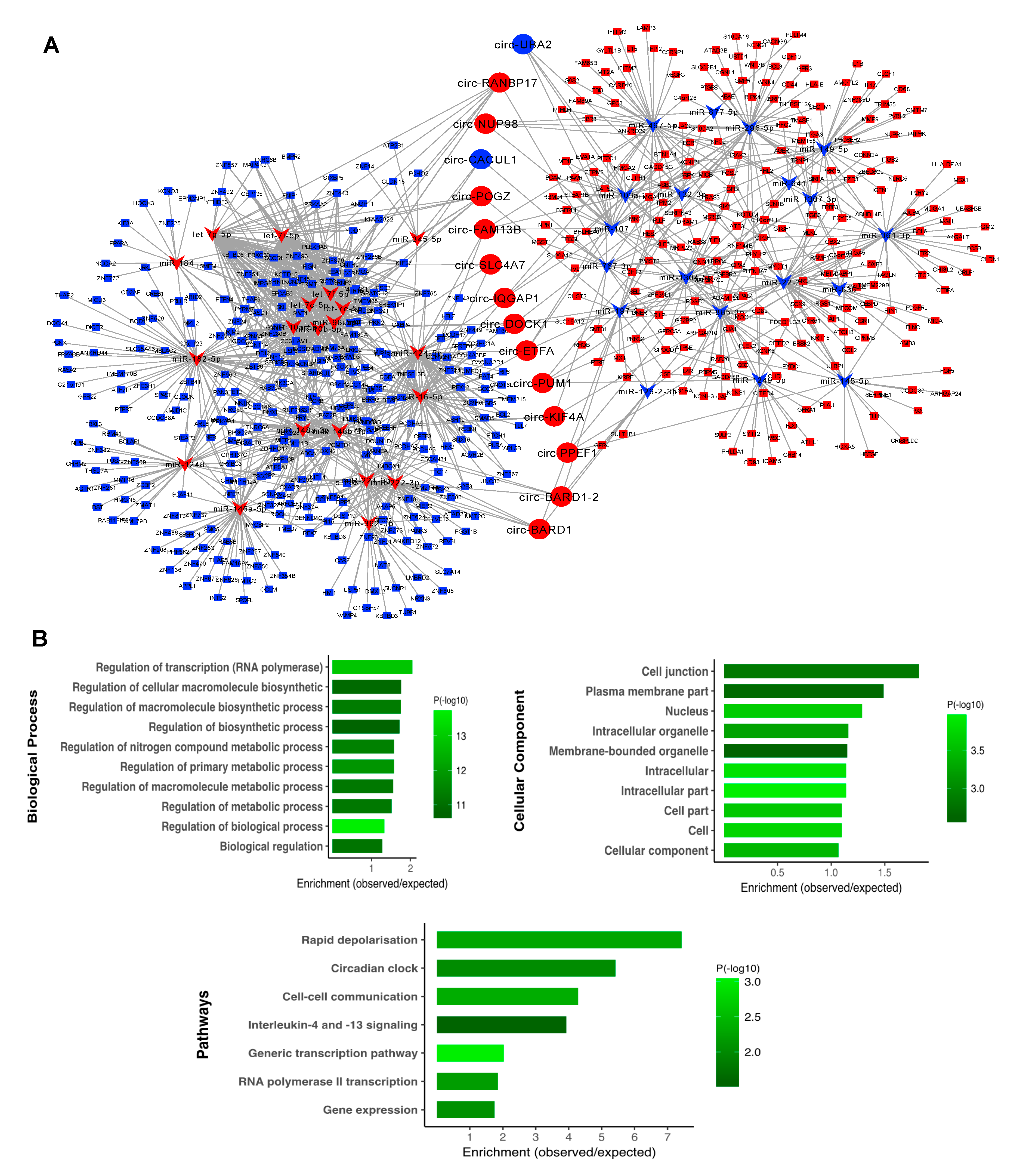
3.5. Subnetwork Analysis and Functional Regulatory Modules
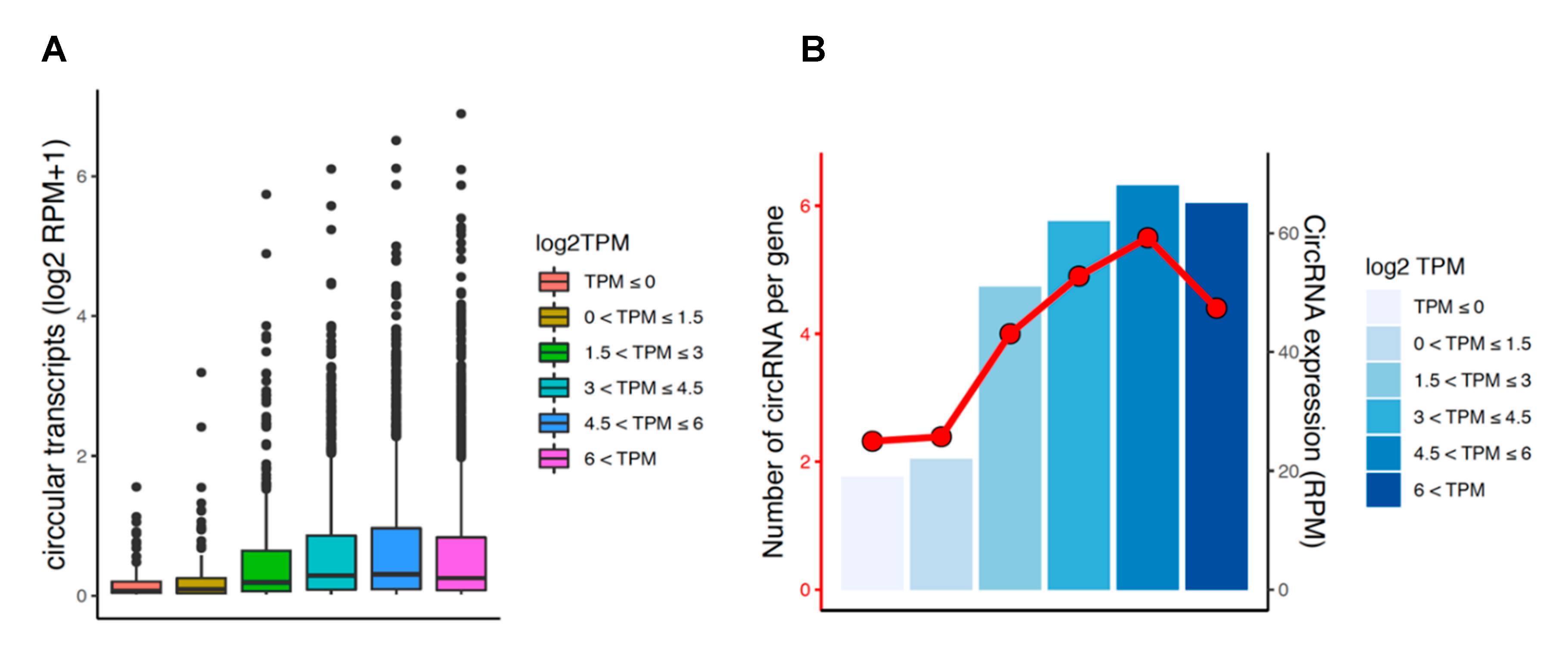
3.6. Circular and Linear Transcripts Expression are Associated in Neuronal Biology
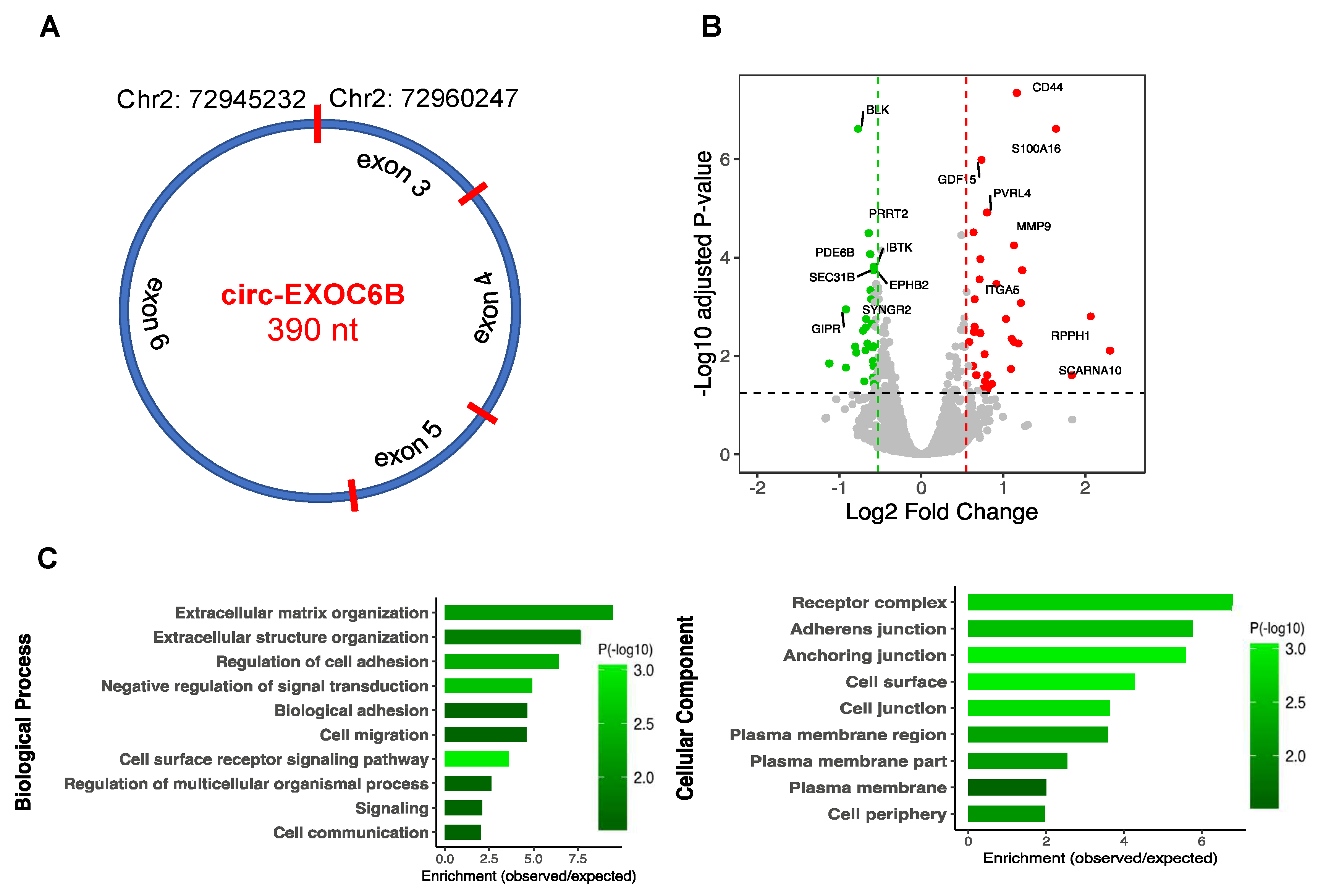
3.7. CircRNA Loss-of-Function Regulates Specific Gene Expression
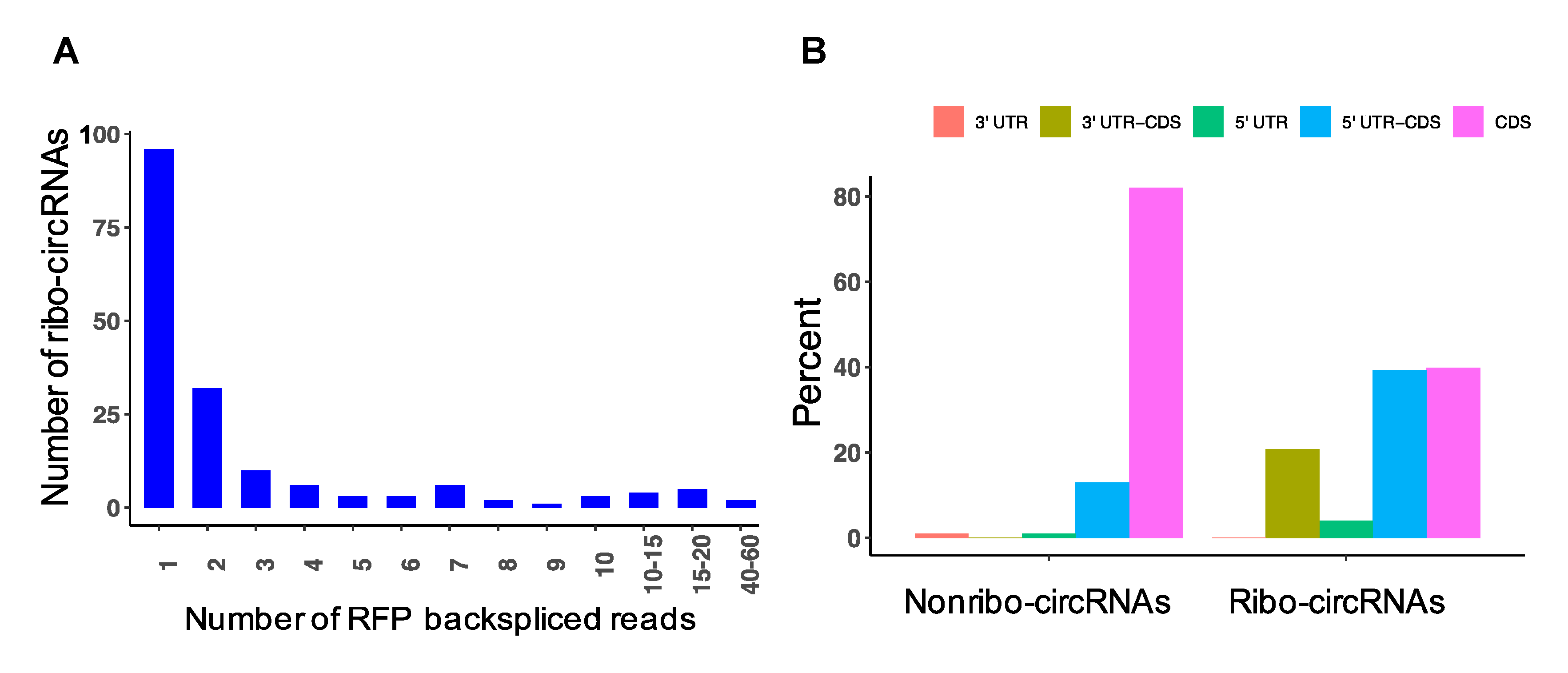
3.8. Detection of circRNAs with Protein-Coding Potential
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef]
- Rybak-Wolf, A.; Stottmeister, C.; Glazar, P.; Jens, M.; Pino, N.; Giusti, S.; Hanan, M.; Behm, M.; Bartok, O.; Ashwal-Fluss, R.; et al. Circular RNAs in the Mammalian Brain Are Highly Abundant, Conserved, and Dynamically Expressed. Mol. Cell 2015, 58, 870–885. [Google Scholar] [CrossRef] [PubMed]
- Westholm, J.O.; Miura, P.; Olson, S.; Shenker, S.; Joseph, B.; Sanfilippo, P.; Celniker, S.E.; Graveley, B.R.; Lai, E.C. Genome-wide analysis of drosophila circular RNAs reveals their structural and sequence properties and age-dependent neural accumulation. Cell Rep. 2014, 9, 1966–1980. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Cui, L.; Zhou, Y.; Zhu, C.; Fan, D.; Gong, H.; Zhao, Q.; Zhou, C.; Zhao, Y.; Lu, D.; et al. Transcriptome-wide investigation of circular RNAs in rice. RNA 2015, 21, 2076–2087. [Google Scholar] [CrossRef] [PubMed]
- Haque, S.; Harries, L.W. Circular RNAs (circRNAs) in Health and Disease. Genes 2017, 8, 353. [Google Scholar] [CrossRef]
- Fischer, J.W.; Leung, A.K. CircRNAs: A regulator of cellular stress. Crit. Rev. Biochem Mol. Biol. 2017, 52, 220–233. [Google Scholar] [CrossRef]
- Li, J.; Lin, H.; Sun, Z.; Kong, G.; Yan, X.; Wang, Y.; Wang, X.; Wen, Y.; Liu, X.; Zheng, H.; et al. High-Throughput Data of Circular RNA Profiles in Human Temporal Cortex Tissue Reveals Novel Insights into Temporal Lobe Epilepsy. Cell Physiol. Biochem. 2018, 45, 677–691. [Google Scholar] [CrossRef]
- Lukiw, W.J. Circular RNA (circRNA) in Alzheimer’s disease (AD). Front. Genet. 2013, 4, 307. [Google Scholar] [CrossRef]
- Li, J.; Yang, J.; Zhou, P.; Le, Y.; Zhou, C.; Wang, S.; Xu, D.; Lin, H.K.; Gong, Z. Circular RNAs in cancer: Novel insights into origins, properties, functions and implications. Am. J. Cancer Res. 2015, 5, 472–480. [Google Scholar]
- Mahmoudi, E.; Fitzsimmons, C.; Geaghan, M.P.; Shannon Weickert, C.; Atkins, J.R.; Wang, X.; Cairns, M.J. Circular RNA biogenesis is decreased in postmortem cortical gray matter in schizophrenia and may alter the bioavailability of associated miRNA. Neuropsychopharmacology 2019, 44, 1043–1054. [Google Scholar] [CrossRef]
- Junn, E.; Lee, K.W.; Jeong, B.S.; Chan, T.W.; Im, J.Y.; Mouradian, M.M. Repression of alpha-synuclein expression and toxicity by microRNA-7. Proc. Natl. Acad. Sci. USA 2009, 106, 13052–13057. [Google Scholar] [CrossRef] [PubMed]
- de Chevigny, A.; Core, N.; Follert, P.; Gaudin, M.; Barbry, P.; Beclin, C.; Cremer, H. miR-7a regulation of Pax6 controls spatial origin of forebrain dopaminergic neurons. Nat. Neurosci. 2012, 15, 1120–1126. [Google Scholar] [CrossRef] [PubMed]
- Pollock, A.; Bian, S.; Zhang, C.; Chen, Z.; Sun, T. Growth of the developing cerebral cortex is controlled by microRNA-7 through the p53 pathway. Cell Rep. 2014, 7, 1184–1196. [Google Scholar] [CrossRef] [PubMed]
- Piwecka, M.; Glazar, P.; Hernandez-Miranda, L.R.; Memczak, S.; Wolf, S.A.; Rybak-Wolf, A.; Filipchyk, A.; Klironomos, F.; Cerda Jara, C.A.; Fenske, P.; et al. Loss of a mammalian circular RNA locus causes miRNA deregulation and affects brain function. Science 2017. [Google Scholar] [CrossRef]
- Ashwal-Fluss, R.; Meyer, M.; Pamudurti, N.R.; Ivanov, A.; Bartok, O.; Hanan, M.; Evantal, N.; Memczak, S.; Rajewsky, N.; Kadener, S. circRNA biogenesis competes with pre-mRNA splicing. Mol. Cell 2014, 56, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Huang, C.; Bao, C.; Chen, L.; Lin, M.; Wang, X.; Zhong, G.; Yu, B.; Hu, W.; Dai, L.; et al. Corrigendum: Exon-intron circular RNAs regulate transcription in the nucleus. Nat. Struct. Mol. Biol. 2017, 24, 194. [Google Scholar] [CrossRef] [PubMed]
- Abe, N.; Matsumoto, K.; Nishihara, M.; Nakano, Y.; Shibata, A.; Maruyama, H.; Shuto, S.; Matsuda, A.; Yoshida, M.; Ito, Y.; et al. Rolling Circle Translation of Circular RNA in Living Human Cells. Sci. Rep. 2015, 5, 16435. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, Z. Efficient backsplicing produces translatable circular mRNAs. RNA 2015, 21, 172–179. [Google Scholar] [CrossRef]
- Pamudurti, N.R.; Bartok, O.; Jens, M.; Ashwal-Fluss, R.; Stottmeister, C.; Ruhe, L.; Hanan, M.; Wyler, E.; Perez-Hernandez, D.; Ramberger, E.; et al. Translation of CircRNAs. Mol. Cell 2017, 66, 9–21 e27. [Google Scholar] [CrossRef]
- Legnini, I.; Di Timoteo, G.; Rossi, F.; Morlando, M.; Briganti, F.; Sthandier, O.; Fatica, A.; Santini, T.; Andronache, A.; Wade, M.; et al. Circ-ZNF609 Is a Circular RNA that Can Be Translated and Functions in Myogenesis. Mol. Cell 2017, 66, 22–37. e29. [Google Scholar] [CrossRef]
- Zhang, M.; Huang, N.; Yang, X.; Luo, J.; Yan, S.; Xiao, F.; Chen, W.; Gao, X.; Zhao, K.; Zhou, H.; et al. A novel protein encoded by the circular form of the SHPRH gene suppresses glioma tumorigenesis. Oncogene 2018, 37, 1805–1814. [Google Scholar] [CrossRef] [PubMed]
- Salzman, J.; Gawad, C.; Wang, P.L.; Lacayo, N.; Brown, P.O. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS ONE 2012, 7, e30733. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudi, E.; Cairns, M.J. Circular RNAs are temporospatially regulated throughout development and ageing in the rat. Sci. Rep. 2019, 9, 2564. [Google Scholar] [CrossRef] [PubMed]
- You, X.; Vlatkovic, I.; Babic, A.; Will, T.; Epstein, I.; Tushev, G.; Akbalik, G.; Wang, M.; Glock, C.; Quedenau, C.; et al. Neural circular RNAs are derived from synaptic genes and regulated by development and plasticity. Nat. Neurosci. 2015, 18, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, G.; Denis, S.; Langelier, B.; Denis, I.; Lavialle, M.; Vancassel, S. DHA enhances the noradrenaline release by SH-SY5Y cells. Neurochem. Int. 2010, 56, 94–100. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 1–10. [Google Scholar] [CrossRef]
- Zhang, X.O.; Dong, R.; Zhang, Y.; Zhang, J.L.; Luo, Z.; Zhang, J.; Chen, L.L.; Yang, L. Diverse alternative back-splicing and alternative splicing landscape of circular RNAs. Genome Res. 2016, 26, 1277–1287. [Google Scholar] [CrossRef]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef]
- Kim, D.; Salzberg, S.L. TopHat-Fusion: An algorithm for discovery of novel fusion transcripts. Genome Biol. 2011, 12, R72. [Google Scholar] [CrossRef]
- Zhang, X.O.; Wang, H.B.; Zhang, Y.; Lu, X.; Chen, L.L.; Yang, L. Complementary sequence-mediated exon circularization. Cell 2014, 159, 134–147. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- The Gene Ontology, C. Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Res. 2017, 45, D331–D338. [Google Scholar] [CrossRef]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2018, 46, D649–D655. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Grimson, A.; Farh, K.K.; Johnston, W.K.; Garrett-Engele, P.; Lim, L.P.; Bartel, D.P. MicroRNA targeting specificity in mammals: Determinants beyond seed pairing. Mol. Cell 2007, 27, 91–105. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.H.; Chen, S.H.; Wu, H.H.; Ho, C.W.; Ko, M.T.; Lin, C.Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8 (Suppl. 4), S11. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Ingolia, N.T.; Brar, G.A.; Rouskin, S.; McGeachy, A.M.; Weissman, J.S. The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat. Protoc. 2012, 7, 1534–1550. [Google Scholar] [CrossRef]
- Karolchik, D.; Hinrichs, A.S.; Furey, T.S.; Roskin, K.M.; Sugnet, C.W.; Haussler, D.; Kent, W.J. The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 2004, 32, D493–D496. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.J.; Yang, D.C.; Kong, L.; Hou, M.; Meng, Y.Q.; Wei, L.; Gao, G. CPC2: A fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res. 2017, 45, W12–W16. [Google Scholar] [CrossRef] [PubMed]
- Marchler-Bauer, A.; Derbyshire, M.K.; Gonzales, N.R.; Lu, S.; Chitsaz, F.; Geer, L.Y.; Geer, R.C.; He, J.; Gwadz, M.; Hurwitz, D.I.; et al. CDD: NCBI’s conserved domain database. Nucleic Acids Res. 2015, 43, D222–D226. [Google Scholar] [CrossRef] [PubMed]
- Glazar, P.; Papavasileiou, P.; Rajewsky, N. circBase: A database for circular RNAs. RNA 2014, 20, 1666–1670. [Google Scholar] [CrossRef]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef]
- Fruhmesser, A.; Blake, J.; Haberlandt, E.; Baying, B.; Raeder, B.; Runz, H.; Spreiz, A.; Fauth, C.; Benes, V.; Utermann, G.; et al. Disruption of EXOC6B in a patient with developmental delay, epilepsy, and a de novo balanced t(2;8) translocation. Eur. J. Hum. Genet. 2013, 21, 1177–1180. [Google Scholar] [CrossRef][Green Version]
- Goldie, B.J.; Dun, M.D.; Lin, M.; Smith, N.D.; Verrills, N.M.; Dayas, C.V.; Cairns, M.J. Activity-associated miRNA are packaged in Map1b-enriched exosomes released from depolarized neurons. Nucleic Acids Res. 2014, 42, 9195–9208. [Google Scholar] [CrossRef]
- Wu, B.; Guo, W. The Exocyst at a Glance. J. Cell Sci. 2015, 128, 2957–2964. [Google Scholar] [CrossRef]
- Mehta, S.Q.; Hiesinger, P.R.; Beronja, S.; Zhai, R.G.; Schulze, K.L.; Verstreken, P.; Cao, Y.; Zhou, Y.; Tepass, U.; Crair, M.C.; et al. Mutations in Drosophila sec15 reveal a function in neuronal targeting for a subset of exocyst components. Neuron 2005, 46, 219–232. [Google Scholar] [CrossRef]
- Jafar-Nejad, H.; Andrews, H.K.; Acar, M.; Bayat, V.; Wirtz-Peitz, F.; Mehta, S.Q.; Knoblich, J.A.; Bellen, H.J. Sec15, a component of the exocyst, promotes notch signaling during the asymmetric division of Drosophila sensory organ precursors. Dev. Cell 2005, 9, 351–363. [Google Scholar] [CrossRef]
- Chatterjee, S.; Pal, J.K. Role of 5’- and 3’-untranslated regions of mRNAs in human diseases. Biol. Cell 2009, 101, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Lipton, J.O.; Sahin, M. The neurology of mTOR. Neuron 2014, 84, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Raab-Graham, K.F.; Haddick, P.C.; Jan, Y.N.; Jan, L.Y. Activity- and mTOR-dependent suppression of Kv1.1 channel mRNA translation in dendrites. Science 2006, 314, 144–148. [Google Scholar] [CrossRef] [PubMed]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahmoudi, E.; Kiltschewskij, D.; Fitzsimmons, C.; Cairns, M.J. Depolarization-Associated CircRNA Regulate Neural Gene Expression and in Some Cases May Function as Templates for Translation. Cells 2020, 9, 25. https://doi.org/10.3390/cells9010025
Mahmoudi E, Kiltschewskij D, Fitzsimmons C, Cairns MJ. Depolarization-Associated CircRNA Regulate Neural Gene Expression and in Some Cases May Function as Templates for Translation. Cells. 2020; 9(1):25. https://doi.org/10.3390/cells9010025
Chicago/Turabian StyleMahmoudi, Ebrahim, Dylan Kiltschewskij, Chantel Fitzsimmons, and Murray J. Cairns. 2020. "Depolarization-Associated CircRNA Regulate Neural Gene Expression and in Some Cases May Function as Templates for Translation" Cells 9, no. 1: 25. https://doi.org/10.3390/cells9010025
APA StyleMahmoudi, E., Kiltschewskij, D., Fitzsimmons, C., & Cairns, M. J. (2020). Depolarization-Associated CircRNA Regulate Neural Gene Expression and in Some Cases May Function as Templates for Translation. Cells, 9(1), 25. https://doi.org/10.3390/cells9010025