PPARγ-Independent Side Effects of Thiazolidinediones on Mitochondrial Redox State in Rat Isolated Hearts

 ,
,  ,
, 
Abstract
1. Introduction
2. Material and Methods
2.1. Animals
2.2. Heart Isolation
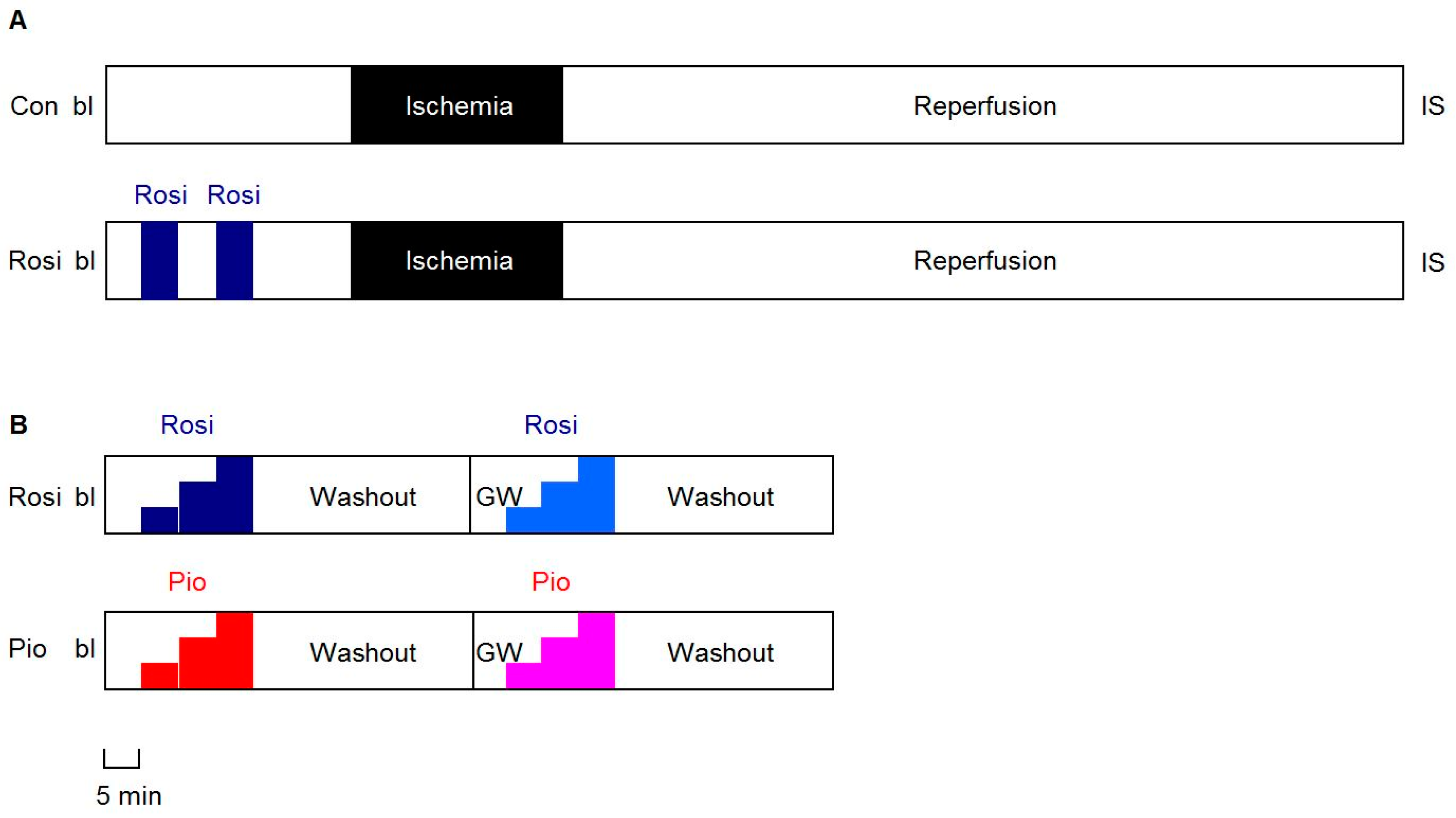
2.3. Protocols
2.3.1. IR Experiments
2.3.2. Dose-Response Experiments
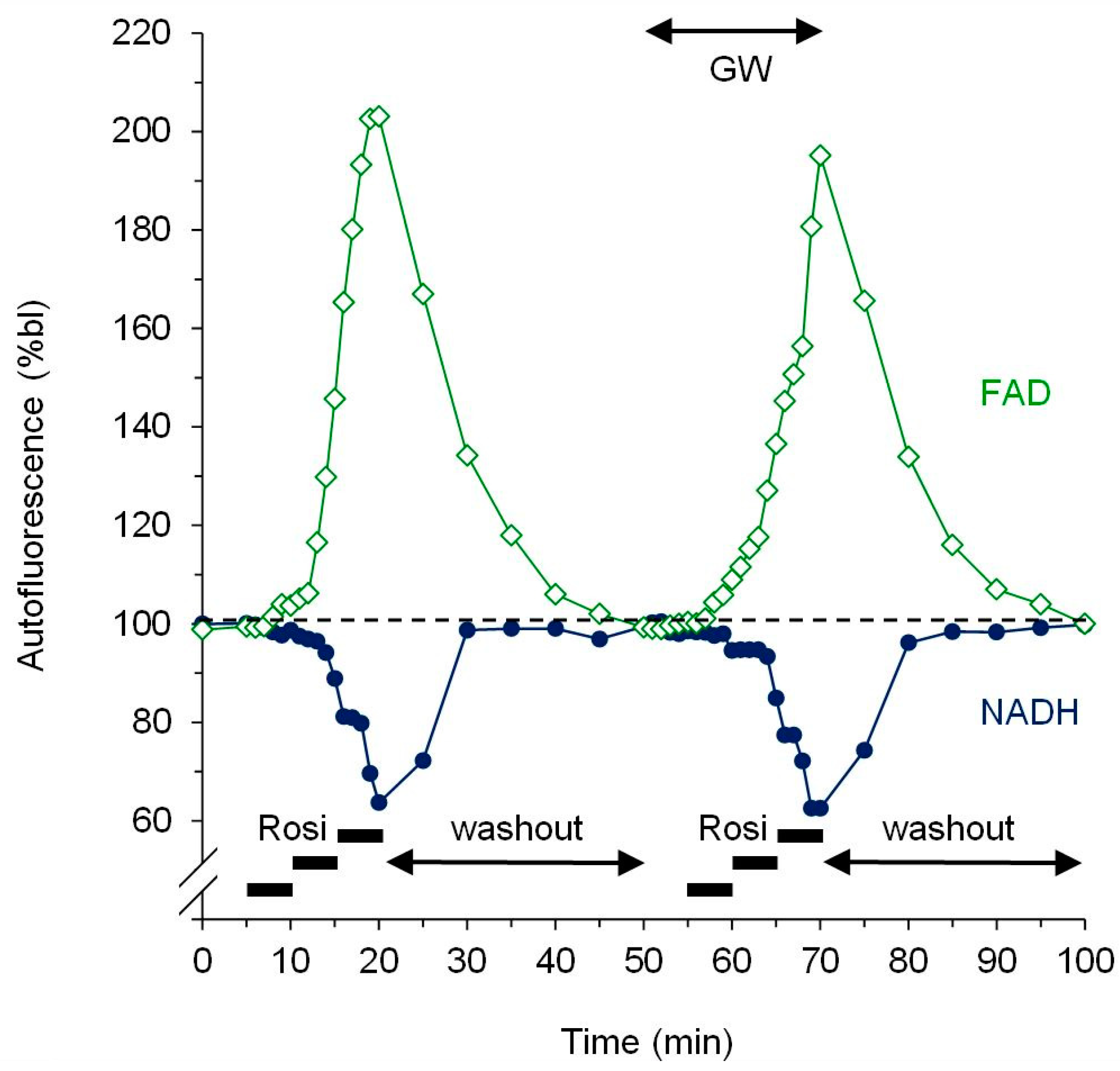
2.4. Fluorescence Measurement of Mitochondrial Redox State
2.5. Statistical Analysis
3. Results
3.1. IR Experiments
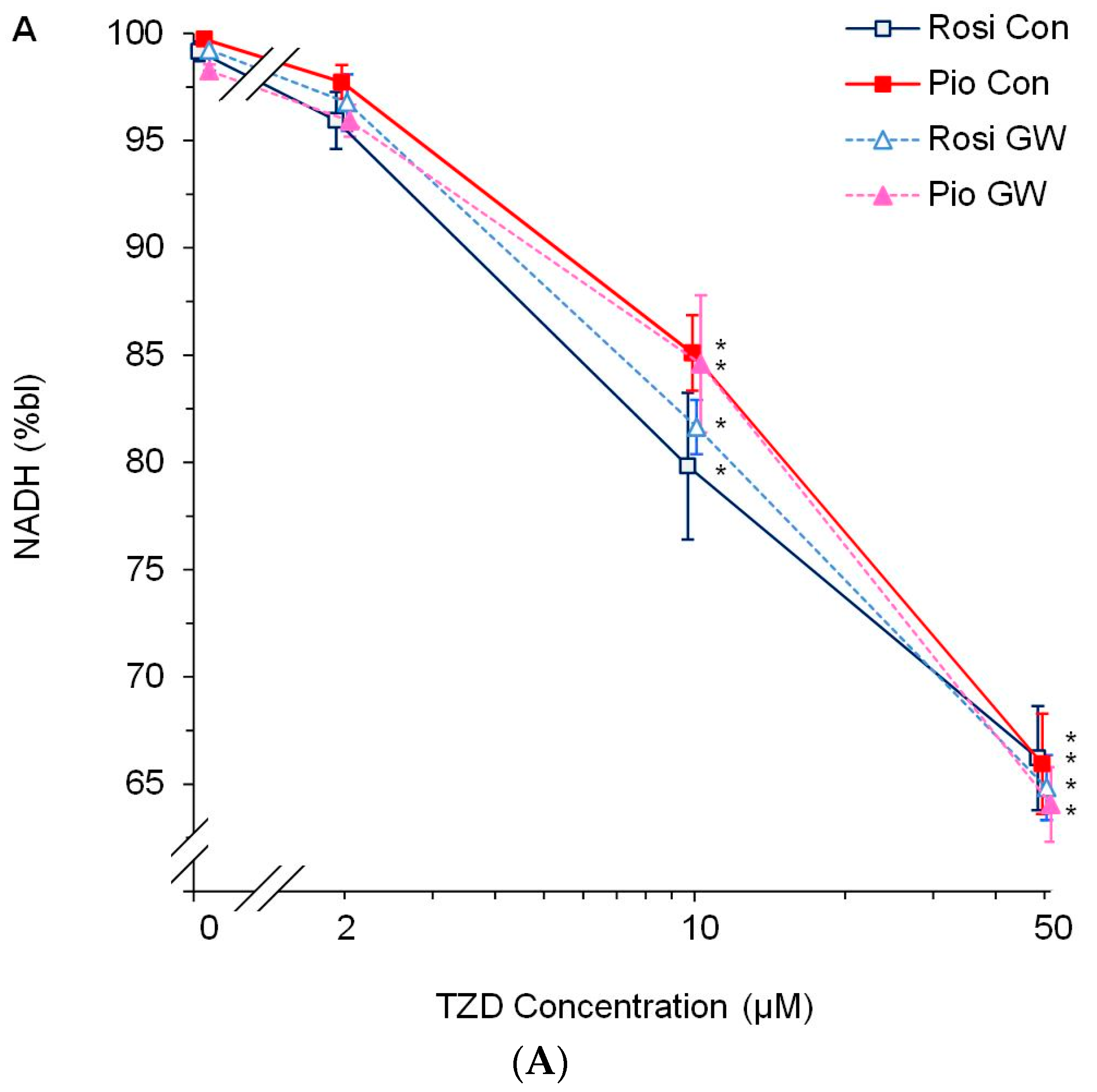
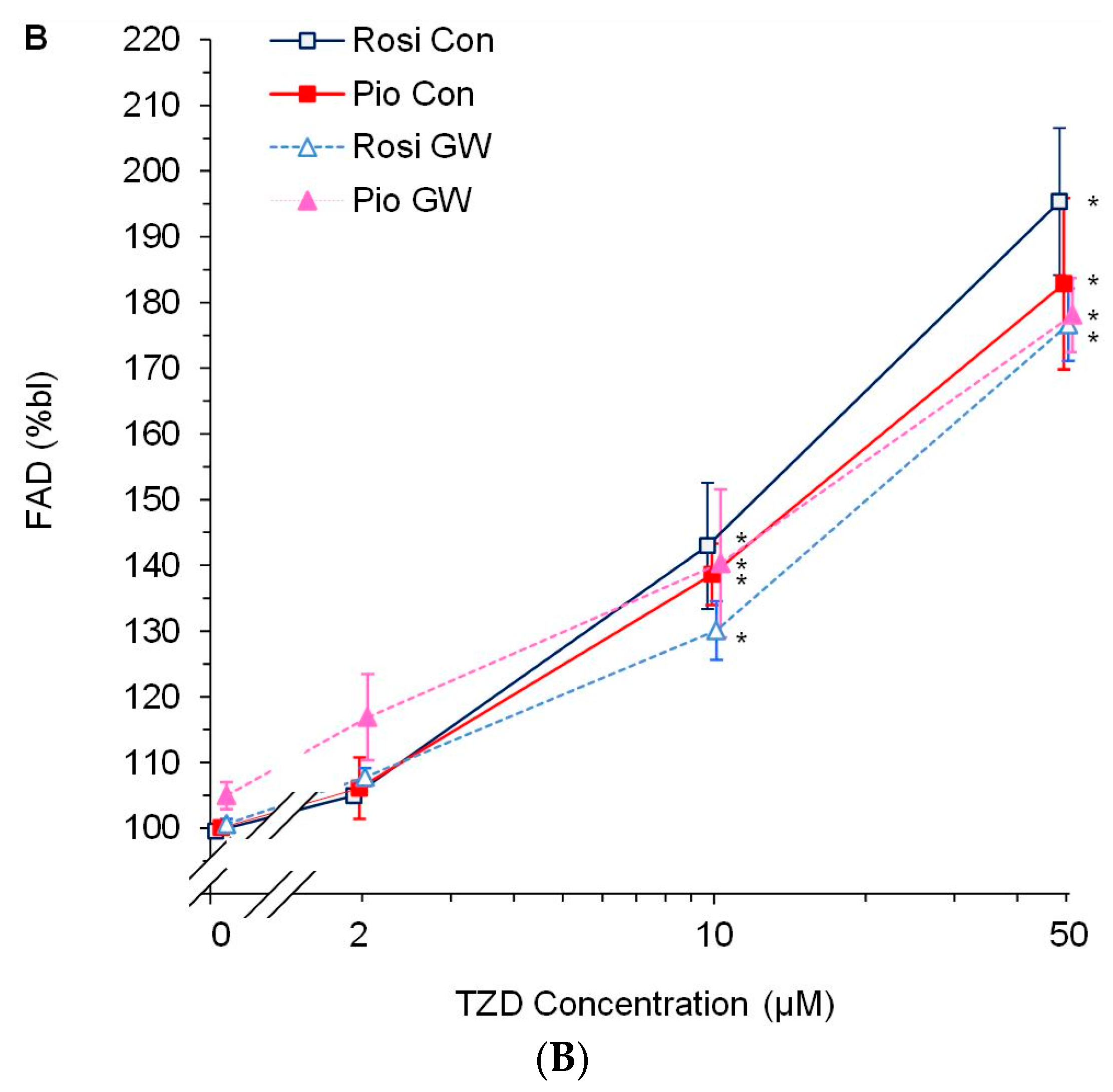
3.2. Dose-Response Experiments
4. Discussion
4.1. PPARγ Activation and Myocardial Protection: Friend or Foe?
4.2. Specificity of TZDs for PPARγ Activation
4.3. TZDs Affect Mitochondrial Function
4.4. Study Limitations and Summary
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Eldor, R.; DeFronzo, R.A.; Abdul-Ghani, M. In vivo actions of peroxisome proliferator-activated receptors: Glycemic control, insulin sensitivity, and insulin secretion. Diabetes Care 2013, 36, S162–S174. [Google Scholar] [CrossRef]
- Kobayashi, N.; Ohno, T.; Yoshida, K.; Fukushima, H.; Mamada, Y.; Nomura, M.; Hirata, H.; Machida, Y.; Shinoda, M.; Suzuki, N.; et al. Cardioprotective mechanism of telmisartan via PPAR-gamma-eNOS pathway in dahl salt-sensitive hypertensive rats. Am. J. Hypertens. 2008, 21, 576–581. [Google Scholar] [CrossRef]
- Yasuda, S.; Kobayashi, H.; Iwasa, M.; Kawamura, I.; Sumi, S.; Narentuoya, B.; Yamaki, T.; Ushikoshi, H.; Nishigaki, K.; Nagashima, K.; et al. Antidiabetic drug pioglitazone protects the heart via activation of PPAR-gamma receptors, PI3-kinase, Akt, and eNOS pathway in a rabbit model of myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H1558–H1565. [Google Scholar] [CrossRef]
- Zhu, P.; Lu, L.; Xu, Y.; Schwartz, G.G. Troglitazone improves recovery of left ventricular function after regional ischemia in pigs. Circulation 2000, 101, 1165–1171. [Google Scholar] [CrossRef] [PubMed]
- Yue, T.L.; Chen, J.; Bao, W.; Narayanan, P.K.; Bril, A.; Jiang, W.; Lysko, P.G.; Gu, J.L.; Boyce, R.; Zimmerman, D.M.; et al. In vivo myocardial protection from ischemia/reperfusion injury by the peroxisome proliferator-activated receptor-gamma agonist rosiglitazone. Circulation 2001, 104, 2588–2594. [Google Scholar] [CrossRef] [PubMed]
- Khandoudi, N.; Delerive, P.; Berrebi-Bertrand, I.; Buckingham, R.E.; Staels, B.; Bril, A. Rosiglitazone, a peroxisome proliferator-activated receptor-gamma, inhibits the Jun NH(2)-terminal kinase/activating protein 1 pathway and protects the heart from ischemia/reperfusion injury. Diabetes 2002, 51, 1507–1514. [Google Scholar] [CrossRef] [PubMed]
- Wayman, N.S.; Hattori, Y.; McDonald, M.C.; Mota-Filipe, H.; Cuzzocrea, S.; Pisano, B.; Chatterjee, P.K.; Thiemermann, C. Ligands of the peroxisome proliferator-activated receptors (PPAR-gamma and PPAR-alpha) reduce myocardial infarct size. FASEB J. 2002, 16, 1027–1040. [Google Scholar] [CrossRef]
- Wynne, A.M.; Mocanu, M.M.; Yellon, D.M. Pioglitazone mimics preconditioning in the isolated perfused rat heart: A role for the prosurvival kinases PI3K and P42/44MAPK. J. Cardiovasc. Pharmacol. 2005, 46, 817–822. [Google Scholar] [CrossRef]
- Li, J.; Lang, M.J.; Mao, X.B.; Tian, L.; Feng, Y.B. Antiapoptosis and mitochondrial effect of pioglitazone preconditioning in the ischemic/reperfused heart of rat. Cardiovasc. Drugs Ther. 2008, 22, 283–291. [Google Scholar] [CrossRef]
- Hu, Q.; Chen, J.; Jiang, C.; Liu, H.F. Effect of peroxisome proliferator-activated receptor gamma agonist on heart of rabbits with acute myocardial ischemia/reperfusion injury. Asian Pac. J. Trop. Med. 2014, 7, 271–275. [Google Scholar] [CrossRef]
- Nabor, D.; ElOrbany, R.; Cheng, Q.; Kersten, J.; Stowe, D.; Riess, M. PPARγ Mediates Endogenous and Exogenous Cardioprotection Associated with Rat Chromosome 6. Anesth. Analg. 2013, 116, S93. [Google Scholar]
- Lotz, C.; Lange, M.; Redel, A.; Stumpner, J.; Schmidt, J.; Tischer-Zeitz, T.; Roewer, N.; Kehl, F. Peroxisome-proliferator-activated receptor gamma mediates the second window of anaesthetic-induced preconditioning. Exp. Physiol. 2011, 96, 317–324. [Google Scholar] [CrossRef]
- Xu, Y.; Lu, L.; Greyson, C.; Lee, J.; Gen, M.; Kinugawa, K.; Long, C.S.; Schwartz, G.G. Deleterious effects of acute treatment with a peroxisome proliferator-activated receptor-gamma activator in myocardial ischemia and reperfusion in pigs. Diabetes 2003, 52, 1187–1194. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Gen, M.; Lu, L.; Fox, J.; Weiss, S.O.; Brown, R.D.; Perlov, D.; Ahmad, H.; Zhu, P.; Greyson, C.; et al. PPAR-gamma activation fails to provide myocardial protection in ischemia and reperfusion in pigs. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H1314–H1323. [Google Scholar] [CrossRef] [PubMed]
- Palee, S.; Weerateerangkul, P.; Surinkeaw, S.; Chattipakorn, S.; Chattipakorn, N. Effect of rosiglitazone on cardiac electrophysiology, infarct size and mitochondrial function in ischaemia and reperfusion of swine and rat heart. Exp. Physiol. 2011, 96, 778–789. [Google Scholar] [CrossRef]
- Sarraf, M.; Lu, L.; Ye, S.; Reiter, M.J.; Greyson, C.R.; Schwartz, G.G. Thiazolidinedione drugs promote onset, alter characteristics, and increase mortality of ischemic ventricular fibrillation in pigs. Cardiovasc. Drugs Ther. 2012, 26, 195–204. [Google Scholar] [CrossRef][Green Version]
- Palee, S.; Weerateerangkul, P.; Chinda, K.; Chattipakorn, S.C.; Chattipakorn, N. Mechanisms responsible for beneficial and adverse effects of rosiglitazone in a rat model of acute cardiac ischaemia-reperfusion. Exp. Physiol. 2013, 98, 1028–1037. [Google Scholar] [CrossRef]
- Nissen, S.E.; Wolski, K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N. Engl. J. Med. 2007, 356, 2457–2471. [Google Scholar] [CrossRef]
- Chen, X.; Yang, L.; Zhai, S.D. Risk of cardiovascular disease and all-cause mortality among diabetic patients prescribed rosiglitazone or pioglitazone: A meta-analysis of retrospective cohort studies. Chin. Med. J. 2012, 125, 4301–4306. [Google Scholar]
- Nissen, S.E. Rosiglitazone: A case of regulatory hubris. BMJ 2013, 347, f7428. [Google Scholar] [CrossRef]
- Hickson, R.P.; Cole, A.L.; Dusetzina, S.B. Implications of Removing Rosiglitazone’s Black Box Warning and Restricted Access Program on the Uptake of Thiazolidinediones and Dipeptidyl Peptidase-4 Inhibitors Among Patients with Type 2 Diabetes. J. Manag. Care Spec. Pharm. 2019, 25, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Lincoff, A.M.; Wolski, K.; Nicholls, S.J.; Nissen, S.E. Pioglitazone and risk of cardiovascular events in patients with type 2 diabetes mellitus: A meta-analysis of randomized trials. JAMA 2007, 298, 1180–1188. [Google Scholar] [CrossRef] [PubMed]
- Colmers, I.N.; Bowker, S.L.; Majumdar, S.R.; Johnson, J.A. Use of thiazolidinediones and the risk of bladder cancer among people with type 2 diabetes: A meta-analysis. CMAJ 2012, 184, E675–E683. [Google Scholar] [CrossRef] [PubMed]
- Turner, R.M.; Kwok, C.S.; Chen-Turner, C.; Maduakor, C.A.; Singh, S.; Loke, Y.K. Thiazolidinediones and associated risk of bladder cancer: A systematic review and meta-analysis. Br. J. Clin. Pharmacol. 2014, 78, 258–273. [Google Scholar] [CrossRef]
- Tang, H.; Shi, W.; Fu, S.; Wang, T.; Zhai, S.; Song, Y.; Han, J. Pioglitazone and bladder cancer risk: A systematic review and meta-analysis. Cancer Med. 2018, 7, 1070–1080. [Google Scholar] [CrossRef]
- Stone, J.C.; Furuya-Kanamori, L.; Barendregt, J.J.; Doi, S.A. Was there really any evidence that rosiglitazone increased the risk of myocardial infarction or death from cardiovascular causes? Pharmacoepidemiol. Drug Saf. 2015, 24, 223–227. [Google Scholar] [CrossRef]
- Loke, Y.K.; Kwok, C.S.; Singh, S. Comparative cardiovascular effects of thiazolidinediones: Systematic review and meta-analysis of observational studies. BMJ 2011, 342, d1309. [Google Scholar] [CrossRef]
- Seferovic, P.M.; Petrie, M.C.; Filippatos, G.S.; Anker, S.D.; Rosano, G.; Bauersachs, J.; Paulus, W.J.; Komajda, M.; Cosentino, F.; de Boer, R.A.; et al. Type 2 diabetes mellitus and heart failure: A position statement from the Heart Failure Association of the European Society of Cardiology. Eur. J. Hear. Fail. 2018, 20, 853–872. [Google Scholar] [CrossRef]
- Camara, A.K.; Bienengraeber, M.; Stowe, D.F. Mitochondrial approaches to protect against cardiac ischemia and reperfusion injury. Front. Physiol. 2011, 2, 13. [Google Scholar] [CrossRef]
- Riess, M.L.; Camara, A.K.S.; Chen, Q.; Novalija, E.; Rhodes, S.S.; Stowe, D.F. Altered NADH and improved function by anesthetic and ischemic preconditioning in guinea pig intact hearts. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H53–H60. [Google Scholar] [CrossRef]
- Riess, M.L.; Kevin, L.G.; Camara, A.K.S.; Heisner, J.S.; Stowe, D.F. Dual exposure to sevoflurane improves anesthetic preconditioning in intact hearts. Anesthesiology 2004, 100, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Nabbi, R.; Gadicherla, A.K.; Kersten, J.R.; Stowe, D.F.; Lazar, J.; Riess, M.L. Genetically determined mitochondrial preservation and cardioprotection against myocardial ischemia-reperfusion injury in a consomic rat model. Physiol. Genom. 2014, 46, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Salzman, M.M.; Cheng, Q.; Deklotz, R.J.; Dulai, G.K.; Douglas, H.F.; Dikalova, A.E.; Weihrauch, D.; Barnes, B.M.; Riess, M.L. Lipid emulsion enhances cardiac performance after ischemia-reperfusion in isolated hearts from summer-active arctic ground squirrels. J. Comp. Physiol. B 2017, 187, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.E.; Konorev, E.A.; Gross, G.J.; Chilian, W.M.; Jacob, H.J. Resistance to myocardial ischemia in five rat strains: Is there a genetic component of cardioprotection? Am. J. Physiol. 2000, 278, H1395–H1400. [Google Scholar] [CrossRef] [PubMed]
- Kevin, L.G.; Novalija, E.; Riess, M.L.; Camara, A.K.; Rhodes, S.S.; Stowe, D.F. Sevoflurane exposure generates superoxide but leads to decreased superoxide during ischemia and reperfusion in isolated hearts. Anesth. Analg. 2003, 96, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Kevin, L.G.; Katz, P.; Camara, A.K.; Novalija, E.; Riess, M.L.; Stowe, D.F. Anesthetic preconditioning: Effects on latency to ischemic injury in isolated hearts. Anesthesiology 2003, 99, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, R.; Diaz, R.J.; Mao, G.D.; Wilson, G.J. Ischemic preconditioning: Differences in protection and susceptibility to blockade with single-cycle versus multicycle transient ischemia. Circulation 1997, 96, 984–995. [Google Scholar] [CrossRef]
- Altman, F.P. Tetrazolium salts and formazans. Prog. Histochem. Cytochem. 1976, 9, 1–56. [Google Scholar] [CrossRef]
- Riess, M.L.; Rhodes, S.S.; Stowe, D.F.; Aldakkak, M.; Camara, A.K. Comparison of cumulative planimetry versus manual dissection to assess experimental infarct size in isolated hearts. J. Pharmacol. Toxicol. Methods 2009, 60, 275–280. [Google Scholar] [CrossRef]
- Shidham, S.V.; Nabbi, R.; Camara, A.K.S.; Riess, M.L. Development of automated infarct size measurement in TTC stained rat isolated hearts after global ischemia/reperfusion. FASEB J. 2011, 25, 1130–1132. [Google Scholar]
- Peng, X.; Chen, R.; Wu, Y.; Huang, B.; Tang, C.; Chen, J.; Wang, Q.; Wu, Q.; Yang, J.; Qiu, H.; et al. PPARgamma-PI3K/AKT-NO signal pathway is involved in cardiomyocyte hypertrophy induced by high glucose and insulin. J. Diabetes Complicat. 2015, 29, 755–760. [Google Scholar] [CrossRef]
- Peymani, M.; Ghaedi, K.; Irani, S.; Nasr-Esfahani, M.H. Peroxisome Proliferator-Activated Receptor gamma Activity is Required for Appropriate Cardiomyocyte Differentiation. Cell J. 2016, 18, 221–228. [Google Scholar] [PubMed]
- Chance, B.; Williamson, J.R.; Jamieson, D.; Schoenner, B. Properties and kinetics of reduced pyridine nucleotide fluorescence of the isolated and in vivo rat heart. Biochem. Zeit 1965, 341, 357–377. [Google Scholar]
- Camara, A.K.; Aldakkak, M.; Heisner, J.S.; Rhodes, S.S.; Riess, M.L.; An, J.; Heinen, A.; Stowe, D.F. ROS scavenging before 27 degrees C ischemia protects hearts and reduces mitochondrial ROS, Ca2+ overload, and changes in redox state. Am. J. Physiol. Cell Physiol. 2007, 292, C2021–C2031. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Gao, H.; Li, W. Long-term risk of rosiglitazone on cardiovascular events—A systematic review and meta-analysis. Endokrynol. Pol. 2018, 69, 381–394. [Google Scholar] [PubMed]
- Varga, Z.V.; Ferdinandy, P.; Liaudet, L.; Pacher, P. Drug-induced mitochondrial dysfunction and cardiotoxicity. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1453–H1467. [Google Scholar] [CrossRef]
- Sivarajah, A.; McDonald, M.C.; Thiemermann, C. The cardioprotective effects of preconditioning with endotoxin, but not ischemia, are abolished by a peroxisome proliferator-activated receptor-gamma antagonist. J. Pharmacol. Exp. Ther. 2005, 313, 896–901. [Google Scholar] [CrossRef]
- Morrison, A.; Yan, X.; Tong, C.; Li, J. Acute rosiglitazone treatment is cardioprotective against ischemia-reperfusion injury by modulating AMPK, Akt, and JNK signaling in nondiabetic mice. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H895–H902. [Google Scholar] [CrossRef]
- Chen, T.; Jin, X.; Crawford, B.H.; Cheng, H.; Saafir, T.B.; Wagner, M.B.; Yuan, Z.; Ding, G. Cardioprotection from oxidative stress in the newborn heart by activation of PPARgamma is mediated by catalase. Free Radic. Biol. Med. 2012, 53, 208–215. [Google Scholar] [CrossRef]
- Nagashima, A.; Watanabe, R.; Ogawa, M.; Suzuki, J.; Masumura, M.; Hishikari, K.; Shimizu, T.; Takayama, K.; Hirata, Y.; Nagai, R.; et al. Different roles of PPAR-gamma activity on physiological and pathological alteration after myocardial ischemia. J. Cardiovasc. Pharmacol. 2012, 60, 158–164. [Google Scholar] [CrossRef]
- Han, J.; Wang, D.; Ye, L.; Li, P.; Hao, W.; Chen, X.; Ma, J.; Wang, B.; Shang, J.; Li, D.; et al. Rosmarinic Acid Protects against Inflammation and Cardiomyocyte Apoptosis during Myocardial Ischemia/Reperfusion Injury by Activating Peroxisome Proliferator-Activated Receptor Gamma. Front. Pharmacol. 2017, 8, 456. [Google Scholar] [CrossRef] [PubMed]
- Ravingerova, T.; Adameova, A.; Carnicka, S.; Nemcekova, M.; Kelly, T.; Matejikova, J.; Galatou, E.; Barlaka, E.; Lazou, A. The role of PPAR in myocardial response to ischemia in normal and diseased heart. Gen. Physiol. Biophys. 2011, 30, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, B.R.; El-Mansy, M.F.; Sem, D.S.; Greene, A.S. Chemical proteomics-based analysis of off-target binding profiles for rosiglitazone and pioglitazone: Clues for assessing potential for cardiotoxicity. J. Med. Chem. 2012, 55, 8260–8271. [Google Scholar] [CrossRef] [PubMed]
- Feinstein, D.L.; Spagnolo, A.; Akar, C.; Weinberg, G.; Murphy, P.; Gavrilyuk, V.; Dello Russo, C. Receptor-independent actions of PPAR thiazolidinedione agonists: Is mitochondrial function the key? Biochem. Pharmacol. 2005, 70, 177–188. [Google Scholar] [CrossRef]
- Gardner, O.S.; Shiau, C.W.; Chen, C.S.; Graves, L.M. Peroxisome proliferator-activated receptor gamma-independent activation of p38 MAPK by thiazolidinediones involves calcium/calmodulin-dependent protein kinase II and protein kinase R: Correlation with endoplasmic reticulum stress. J. Biol. Chem. 2005, 280, 10109–10118. [Google Scholar] [CrossRef] [PubMed]
- Mughal, R.S.; Warburton, P.; O’Regan, D.J.; Ball, S.G.; Turner, N.A.; Porter, K.E. Peroxisome proliferator-activated receptor gamma-independent effects of thiazolidinediones on human cardiac myofibroblast function. Clin. Exp. Pharmacol. Physiol. 2009, 36, 478–486. [Google Scholar] [CrossRef]
- He, H.; Tao, H.; Xiong, H.; Duan, S.Z.; McGowan, F.X., Jr.; Mortensen, R.M.; Balschi, J.A. Rosiglitazone causes cardiotoxicity via peroxisome proliferator-activated receptor gamma-independent mitochondrial oxidative stress in mouse hearts. Toxicol. Sci. 2014, 138, 468–481. [Google Scholar] [CrossRef]
- Chen, Q.; Vazquez, E.J.; Moghaddas, S.; Hoppel, C.L.; Lesnefsky, E.J. Production of reactive oxygen species by mitochondria: Central role of complex III. J. Biol. Chem. 2003, 278, 36027–36031. [Google Scholar] [CrossRef]
- Krenz, M.; Oldenburg, O.; Wimpee, H.; Cohen, M.V.; Garlid, K.D.; Critz, S.D.; Downey, J.M.; Benoit, J.N. Opening of ATP-sensitive potassium channels causes generation of free radicals in vascular smooth muscle cells. Basic Res. Cardiol. 2002, 97, 365–373. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| During Application | 120 min Reperfusion | |||
|---|---|---|---|---|
| Con | Rosi | Con | Rosi | |
| sysLVP (%bl) | 95.5 ± 2.0 | 101.9 ± 2.8 | 70.5 ± 4.9 | 60.3 ± 3.9 |
| diaLVP (mmHg) | 10.5 ± 2.1 | 9.9 ± 0.8 | 32.3 ± 4.3 | 31.8 ± 2.7 |
| devLVP (%bl) | 92.6 ± 3.8 | 102.4 ± 3.6 | 33.8 ± 3.7 | 29.7 ± 1.0 |
| RPP (%bl) | 96.4 ± 5.5 | 101.3 ± 4.4 | 33.5 ± 3.7 | 30.9 ± 0.7 |
| dLVP/dtmax (%bl) | 94.4 ± 3.5 | 106.6 ± 4.7 | 35.9 ± 2.6 | 32.8 ± 1.3 |
| dLVP/dtmin (%bl) | 97.2 ± 2.4 | 100.6 ± 4.5 | 38.9 ± 5.0 | 30.4 ± 1.1 |
| HR (%bl) | 105.3 ± 2.8 | 98.8 ± 2.2 | 98.4 ± 2.7 | 104.3 ± 1.6 |
| CF (%bl) | 100.0 ± 1.0 | * 124.0 ± 6.1 | 64.6 ± 5.3 | 64.5 ± 3.0 |
| IS (%) | 36.2 ± 3.4 | * 45.3 ± 0.7 | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Riess, M.L.; Elorbany, R.; Weihrauch, D.; Stowe, D.F.; Camara, A.K.S. PPARγ-Independent Side Effects of Thiazolidinediones on Mitochondrial Redox State in Rat Isolated Hearts. Cells 2020, 9, 252. https://doi.org/10.3390/cells9010252
Riess ML, Elorbany R, Weihrauch D, Stowe DF, Camara AKS. PPARγ-Independent Side Effects of Thiazolidinediones on Mitochondrial Redox State in Rat Isolated Hearts. Cells. 2020; 9(1):252. https://doi.org/10.3390/cells9010252
Chicago/Turabian StyleRiess, Matthias L., Reem Elorbany, Dorothee Weihrauch, David F. Stowe, and Amadou K.S. Camara. 2020. "PPARγ-Independent Side Effects of Thiazolidinediones on Mitochondrial Redox State in Rat Isolated Hearts" Cells 9, no. 1: 252. https://doi.org/10.3390/cells9010252
APA StyleRiess, M. L., Elorbany, R., Weihrauch, D., Stowe, D. F., & Camara, A. K. S. (2020). PPARγ-Independent Side Effects of Thiazolidinediones on Mitochondrial Redox State in Rat Isolated Hearts. Cells, 9(1), 252. https://doi.org/10.3390/cells9010252