Targeting Aberrant RAS/RAF/MEK/ERK Signaling for Cancer Therapy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
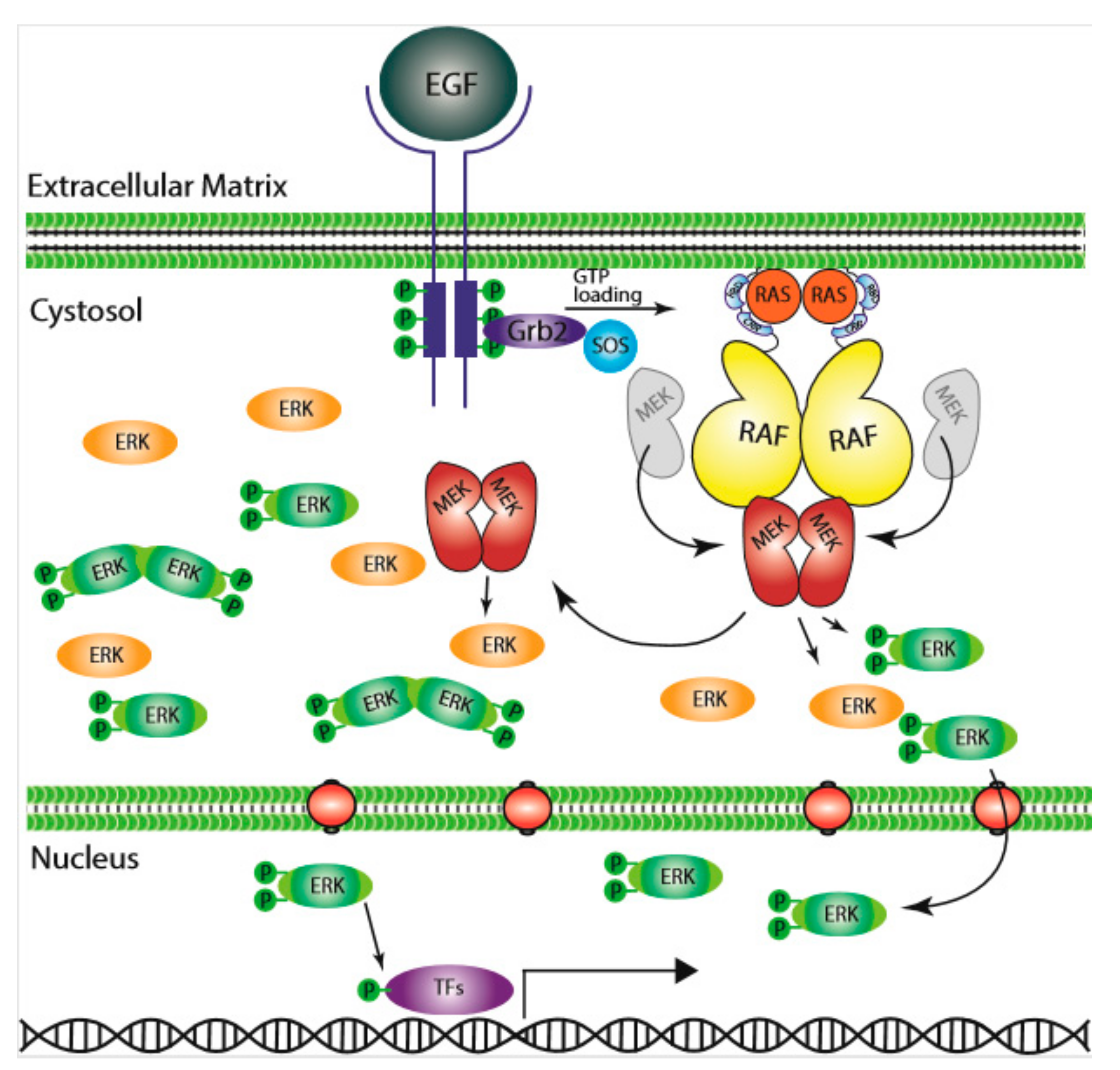
1. A Brief History of RAS/RAF/MEK/ERK Signaling Cascade
2. RAS GTPases and Their Activation
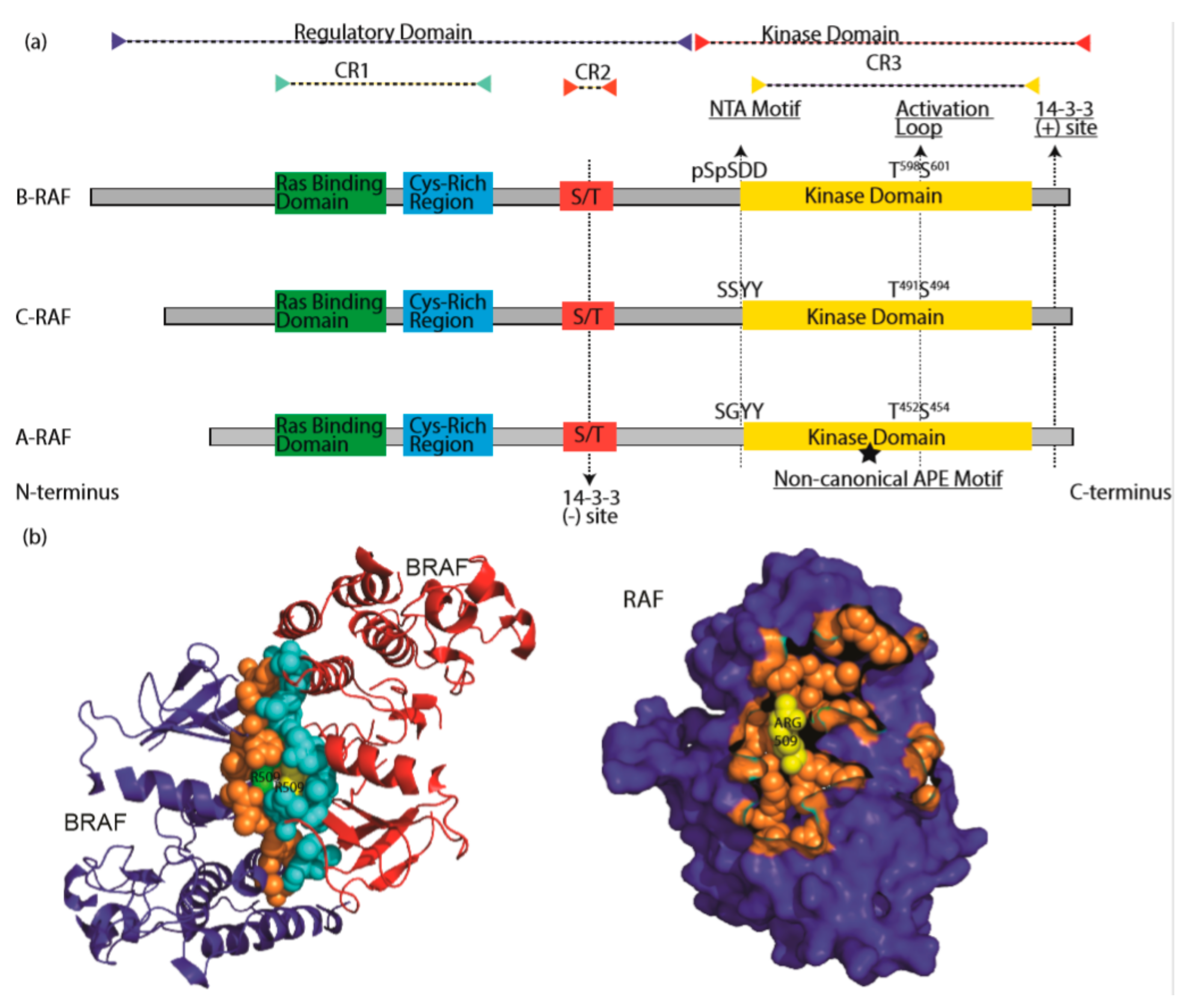
3. RAF Isoforms
4. RAF-Knockout Mouse Models
5. Activation of RAF Proteins
5.1. Auto-Inhibited State of RAF
5.2. RAF Recruitment to Plasma Membrane by Activated RAS
5.3. Dimerization Is a Key Event in RAF Activation
5.4. The Role of NTA Motif and Activation Loop Phosphorylation in RAF Activation
6. Regulation of RAF by Accessory Molecules
6.1. Hsp90/Cdc37 Chaperone Complex
6.2. KSR
6.3. Proteins 14-3-3
7. RAF Function as a Dimer
8. RAF–MEK Heterodimerization and MEK–MEK Homodimerization, Essential Events for Signaling
9. Feedback Inhibition and Return to the Inactive State
10. Mutations in the Ras/RAF/MEK/ERK Signaling Cascade
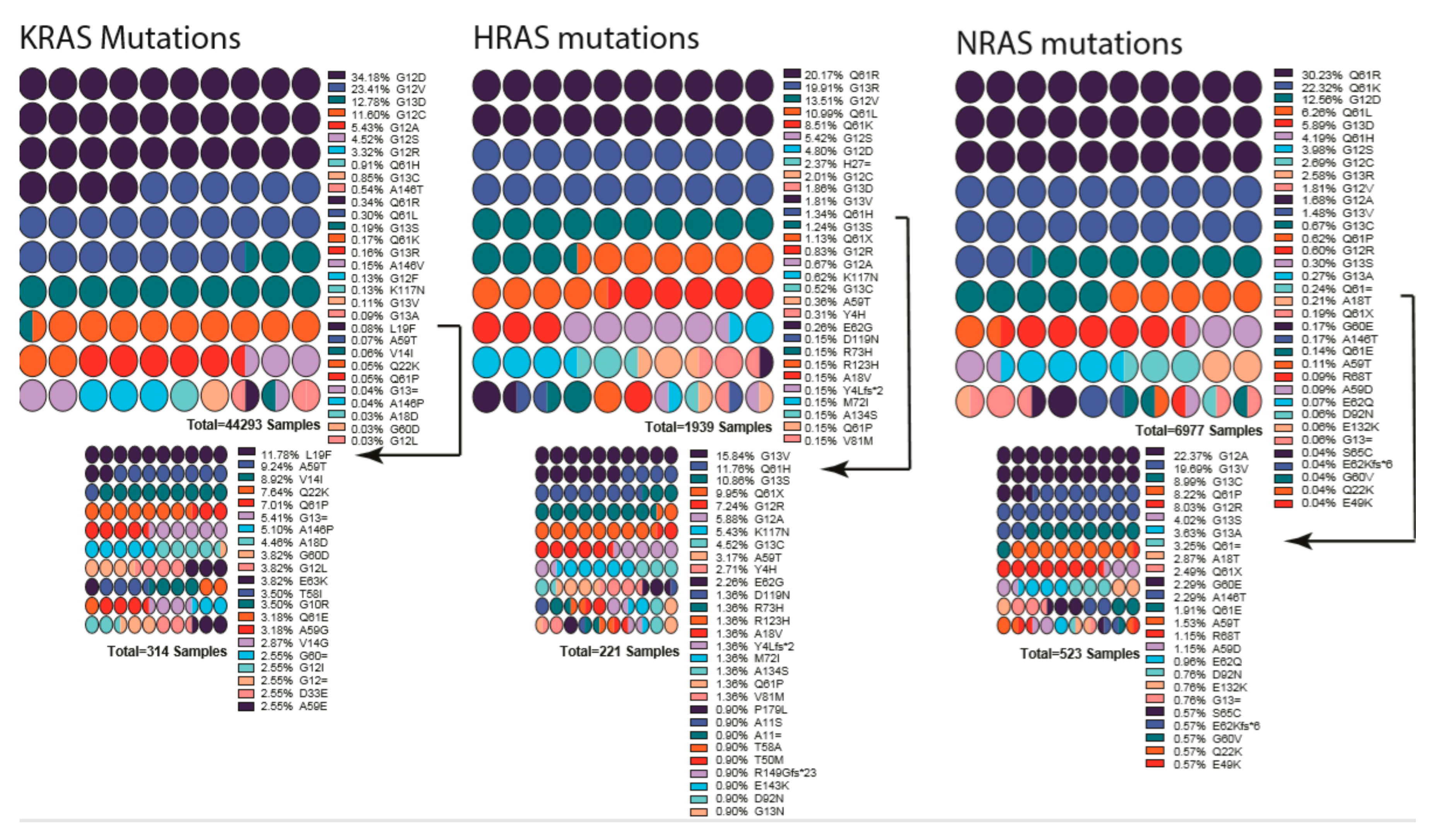
10.1. Ras Mutations
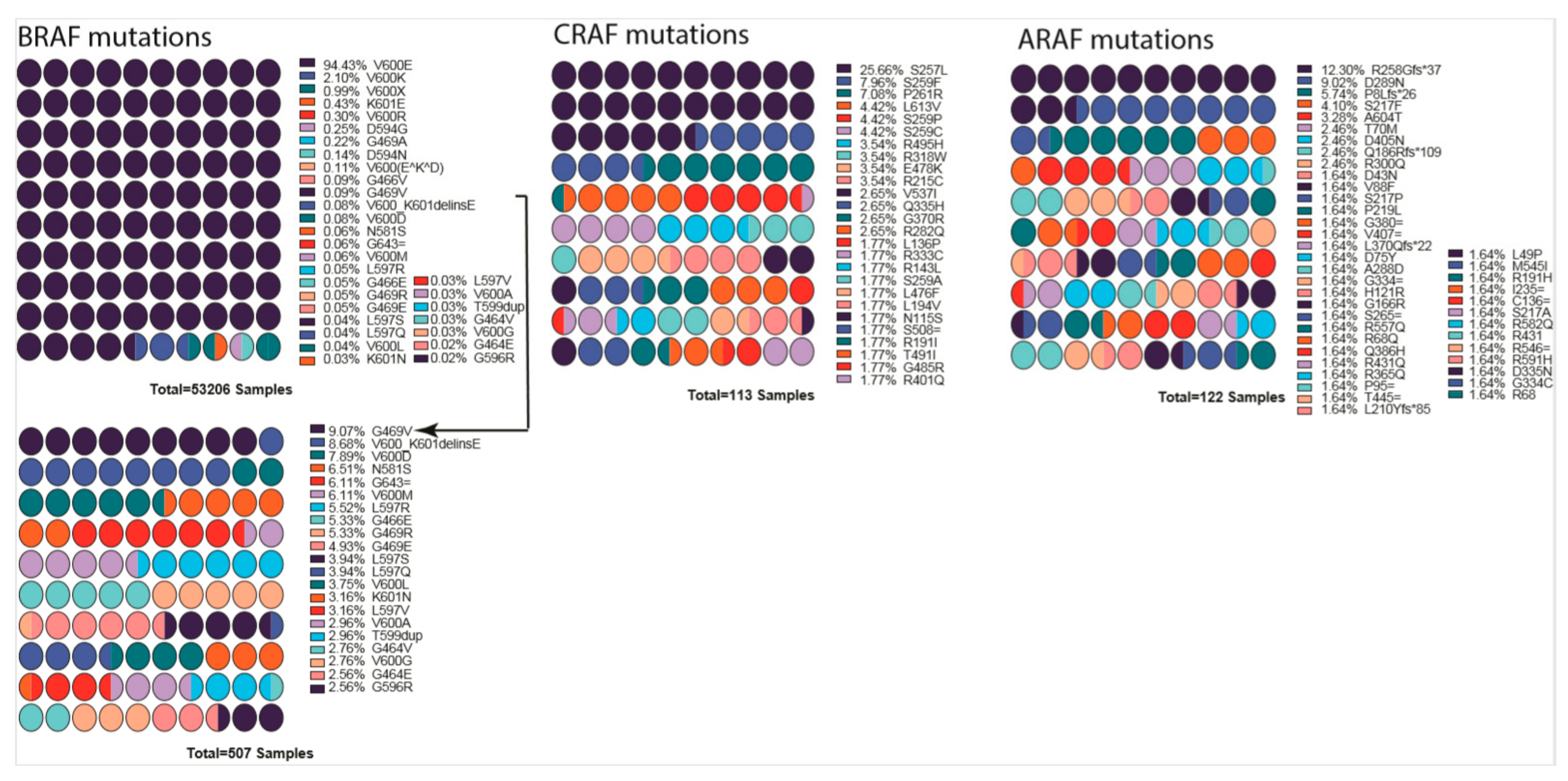
10.2. RAF Mutations
10.3. MEK and ERK Mutations
11. Targeted Therapies against Hyperactive Ras/RAF/MEK/ERK Signaling in Cancers: The Present State and Perspectives
11.1. Is RAS Druggable?
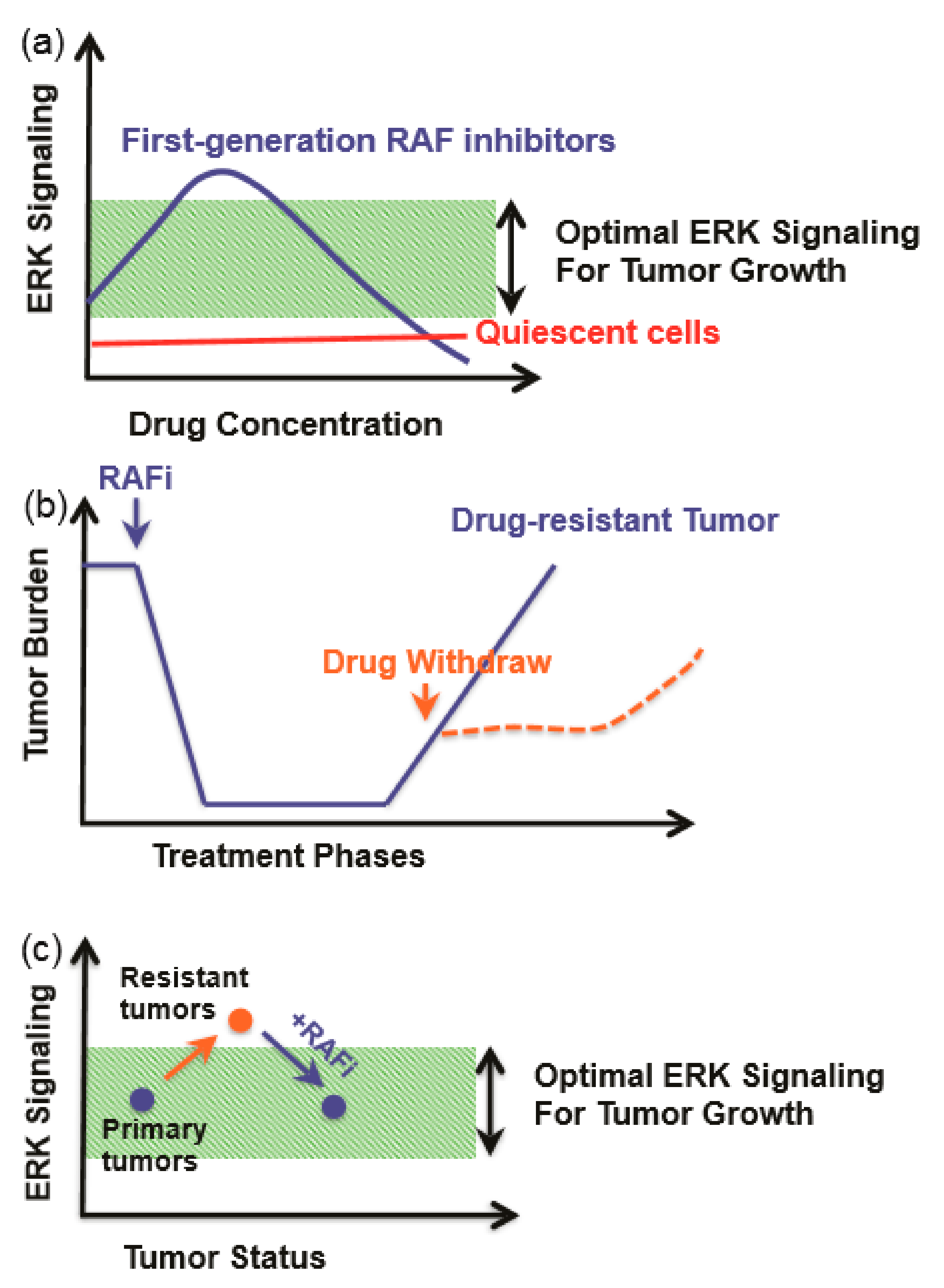
11.2. RAF/MEK/ERK Inhibitors and Resistance
12. Closing Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rapp, U.R.; Todaro, C. Generation of new mouse sarcoma viruses in cell culture. Science 1978, 201, 821–824. [Google Scholar] [CrossRef] [PubMed]
- Jansen, H.W.; Lurz, R.; Bister, K.; Bonner, T.I.; Mark, G.E.; Rapp, U.R. Homologous cell-derived oncogenes in avian carcinoma virus MH2 and murine sarcoma virus 3611. Nature 1984, 307, 281–284. [Google Scholar] [CrossRef] [PubMed]
- Jansen, H.W.; Rückert, B.; Lurz, R.; Bister, K. Two unrelated cell-derived sequences in the genome of avian leukemia and carcinoma inducing retrovirus MH2. EMBO J. 1983, 2, 1969–1975. [Google Scholar] [CrossRef] [PubMed]
- Bonner, T.; O’Brien, S.; Nash, W.; Rapp, U.; Morton, C.; Leder, P. The human homologs of the raf (mil) oncogene are located on human chromosomes 3 and 4. Science 1984, 223, 71–74. [Google Scholar] [CrossRef]
- Kozak, C.; Gunnell, M.A.; Rapp, U.R. A new oncogene, c-raf, is located on mouse chromosome 6. J. Virol. 1984, 49, 297–299. [Google Scholar] [CrossRef]
- Bonner, T.I.; Kerby, S.B.; Sutrave, P.; Gunnell, M.A.; Mark, G.; Rapp, U.R. Structure and biological activity of human homologs of the raf/mil oncogene. Mol. Cell. Biol. 1985, 5, 1400–1407. [Google Scholar] [CrossRef]
- Rapp, U.R.; Goldsborough, M.D.; Mark, G.E.; Bonner, T.I.; Groffen, J.; Reynolds, F.H.; Stephenson, J.R. Structure and biological activity of v-raf, a unique oncogene transduced by a retrovirus. Proc. Natl. Acad. Sci. USA 1983, 80, 4218–4222. [Google Scholar] [CrossRef]
- Moelling, K.; Heimann, B.; Beimling, P.; Rapp, U.R.; Sander, T. Serine- and threonine-specific protein kinase activities of purified gag–mil and gag–raf proteins. Nature 1984, 312, 558–561. [Google Scholar] [CrossRef]
- Kamata, T.; Feramisco, J.R. Epidermal growth factor stimulates guanine nucleotide binding activity and phosphorylation of ras oncogene proteins. Nature 1984, 310, 147–150. [Google Scholar] [CrossRef]
- Mulcahy, L.S.; Smith, M.R.; Stacey, D.W. Requirement for ras proto-oncogene function during serum-stimulated growth of NIH 3T3 cells. Nature 1985, 313, 241–243. [Google Scholar] [CrossRef]
- Smith, M.R.; DeGudicibus, S.J.; Stacey, D.W. Requirement for c-ras proteins during viral oncogene transformation. Nature 1986, 320, 540–543. [Google Scholar] [CrossRef] [PubMed]
- Ambrosio, L.; Mahowald, A.P.; Perrimon, N. Requirement of the Drosophila raf homologue for torso function. Nature 1989, 342, 288–291. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Golden, A.; Han, Y.; Sternberg, P.W. C. elegans lin-45 raf gene participates in let-60 ras-stimulated vulval differentiation. Nature 1993, 363, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Ahn, N.G.; Weiel, J.E.; Chan, C.P.; Krebs, E.G. Identification of multiple epidermal growth factor-stimulated protein serine/threonine kinases from Swiss 3T3 cells. J. Biol. Chem. 1990, 265, 11487–11494. [Google Scholar]
- Ray, L.B.; Sturgill, T.W. Characterization of insulin-stimulated microtubule-associated protein kinase. J. Biol. Chem. 1988, 263, 12721–12727. [Google Scholar]
- Rossomando, A.J.; Payne, D.M.; Weber, M.J.; Sturgill, T.W. Evidence that pp42, a major tyrosine kinase target protein, is a mitogen-activated serine/threonine protein kinase. Proc. Natl. Acad. Sci. USA 1989, 86, 6940–6943. [Google Scholar] [CrossRef]
- Boulton, T.G.; Nye, S.H.; Robbins, D.J.; Ip, N.Y.; Radzlejewska, E.; Morgenbesser, S.D.; Depinho, R.A.; Panayotatos, N.; Cobb, M.H.; Yancopoulos, G.D.; et al. ERKs: A family of protein-serine/threonine kinases that are activated and tyrosine phosphorylated in response to insulin and NGF. Cell 1991, 65, 663–675. [Google Scholar] [CrossRef]
- Crews, C.M.; Erikson, R.L. Purification of a murine protein-tyrosine/threonine kinase that phosphorylates and activates the Erk-1 gene product: Relationship to the fission yeast byr1 gene product. Proc. Natl. Acad. Sci. USA 1992, 89, 8205–8209. [Google Scholar] [CrossRef]
- Kyriakis, J.M.; App, H.; Zhang, X.; Banerjee, P.; Brautigan, D.L.; Rapp, U.R.; Avruch, J. Raf-1 activates MAP kinase-kinase. Nature 1992, 358, 417–421. [Google Scholar] [CrossRef]
- Alessi, D.R.; Saito, Y.; Campbell, D.G.; Cohen, P.; Sithanandam, G.; Rapp, U.; Ashworth, A.; Marshall, C.J.; Cowley, S. Identification of the sites in MAP kinase kinase-1 phosphorylated by p74raf-1. EMBO J. 1994, 13, 1610–1619. [Google Scholar] [CrossRef]
- Kolch, W.; Heidecker, G.; Lloyd, P.; Rapp, U.R. Raf-1 protein kinase is required for growth of induced NIH/3T3 cells. Nature 1991, 349, 426–428. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, X.-F.; Settleman, J.; Kyriakis, J.; Takeuchi-Suzuki, E.; Elledge, S.J.; Marshall, M.S.; Bruder, J.T.; Rapp, U.R.; Avruch, J. Normal and oncogenic p21ras proteins bind to the amino-terminal regulatory domain of c-Raf-1. Nature 1993, 364, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Van Aelst, L.; Barr, M.; Marcus, S.; Polverino, A.; Wigler, M. Complex formation between RAS and RAF and other protein kinases. Proc. Natl. Acad. Sci. USA 1993, 90, 6213–6217. [Google Scholar] [CrossRef] [PubMed]
- Vojtek, A.B.; Hollenberg, S.M.; Cooper, J.A. Mammalian Ras interacts directly with the serine/threonine kinase raf. Cell 1993, 74, 205–214. [Google Scholar] [CrossRef]
- Moodie, S.A.; Willumsen, B.M.; Weber, M.J.; Wolfman, A. Complexes of Ras.GTP with Raf-1 and mitogen-activated protein kinase kinase. Science 1993, 260, 1658–1661. [Google Scholar] [CrossRef]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Garnett, M.J.; Rana, S.; Paterson, H.; Barford, D.; Marais, R. Wild-Type and Mutant B-RAF Activate C-RAF through Distinct Mechanisms Involving Heterodimerization. Mol. Cell 2005, 20, 963–969. [Google Scholar] [CrossRef]
- Winter-Vann, A.M.; Casey, P.J. Post-prenylation-processing enzymes as new targets in oncogenesis. Nat. Rev. Cancer 2005, 5, 405–412. [Google Scholar] [CrossRef]
- Jang, H.; Abraham, S.J.; Chavan, T.S.; Hitchinson, B.; Khavrutskii, L.; Tarasova, N.I.; Nussinov, R.; Gaponenko, V. Mechanisms of Membrane Binding of Small GTPase K-Ras4B Farnesylated Hypervariable Region. J. Biol. Chem. 2015, 290, 9465–9477. [Google Scholar] [CrossRef]
- Hobbs, G.A.; Der, C.J. RAS Mutations Are Not Created Equal. Cancer Discov. 2019, 9, 696–698. [Google Scholar] [CrossRef]
- Li, S.; Balmain, A.; Counter, C.M. A model for RAS mutation patterns in cancers: Finding the sweet spot. Nat. Rev. Cancer 2018, 18, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.D.; Der, C.J. Ras history. Small GTPases 2010, 1, 2–27. [Google Scholar] [CrossRef] [PubMed]
- Castellano, E.; Santos, E. Functional specificity of ras isoforms: So similar but so different. Genes Cancer 2011, 2, 216–231. [Google Scholar] [CrossRef] [PubMed]
- Hancock, J.F. Ras proteins: Different signals from different locations. Nat. Rev. Mol. Cell Biol. 2003, 4, 373–385. [Google Scholar] [CrossRef]
- Murugan, A.K.; Grieco, M.; Tsuchida, N. RAS mutations in human cancers: Roles in precision medicine. Semin. Cancer Biol. 2019. [Google Scholar] [CrossRef]
- Yan, J.; Roy, S.; Apolloni, A.; Lane, A.; Hancock, J.F. Ras Isoforms Vary in Their Ability to Activate Raf-1 and Phosphoinositide 3-Kinase. J. Biol. Chem. 1998, 273, 24052–24056. [Google Scholar] [CrossRef]
- Millán, O.; Ballester, A.; Castrillo, A.; Oliva, J.L. de la; Través, P.G.; Rojas, J.M.; Boscá, L. H-Ras-specific activation of NF-κB protects NIH 3T3 cells against stimulus-dependent apoptosis. Oncogene 2003, 22, 477–483. [Google Scholar] [CrossRef][Green Version]
- Hobbs, G.A.; Der, C.J.; Rossman, K.L. RAS isoforms and mutations in cancer at a glance. J. Cell Sci. 2016, 129, 1287–1292. [Google Scholar] [CrossRef]
- McCormick, F. ras GTPase activating protein: Signal transmitter and signal terminator. Cell 1989, 56, 5–8. [Google Scholar] [CrossRef]
- Hennig, A.; Markwart, R.; Esparza-Franco, M.A.; Ladds, G.; Rubio, I. Ras activation revisited: Role of GEF and GAP systems. Biol. Chem. 2015, 396, 831–848. [Google Scholar] [CrossRef]
- Aoki, Y.; Niihori, T.; Inoue, S.; Matsubara, Y. Recent advances in RASopathies. J. Hum. Genet. 2015, 61, 33. [Google Scholar] [CrossRef] [PubMed]
- Quilliam, L.A.; Castro, A.F.; Rogers-Graham, K.S.; Martin, C.B.; Der, C.J.; Bi, C. M-Ras/R-Ras3, a transforming ras protein regulated by Sos1, GRF1, and p120 Ras GTPase-activating protein, interacts with the putative Ras effector AF6. J. Biol. Chem. 1999, 274, 23850–23857. [Google Scholar] [CrossRef] [PubMed]
- Maertens, O.; Johnson, B.; Hollstein, P.; Frederick, D.T.; Cooper, Z.A.; Messiaen, L.; Bronson, R.T.; McMahon, M.; Granter, S.; Flaherty, K.; et al. Elucidating distinct roles for NF1 in melanomagenesis. Cancer Discov. 2013, 3, 338–349. [Google Scholar] [CrossRef] [PubMed]
- Casey, P.J.; Solski, P.A.; Der, C.J.; Buss, J.E. p21ras is modified by a farnesyl isoprenoid. Proc. Natl. Acad. Sci. USA 1989, 86, 8323–8327. [Google Scholar] [CrossRef] [PubMed]
- Sebti, S.M. Protein farnesylation: Implications for normal physiology, malignant transformation, and cancer therapy. Cancer Cell 2005, 7, 297–300. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Casey, P.J. Protein prenylation: Unique fats make their mark on biology. Nat. Rev. Mol. Cell Biol. 2016, 17, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Williams, C. A new signaling paradigm to control the prenylation and trafficking of small GTPases. Cell Cycle 2013, 12, 2933–2934. [Google Scholar] [CrossRef]
- Zhang, F.L.; Casey, P.J. Protein prenylation: Molecular Mechanisms and Functional Consequences. Annu. Rev. Biochem. 1996, 65, 241–269. [Google Scholar] [CrossRef]
- Lerner, E.C.; Qian, Y.; Blaskovich, M.A.; Fossum, R.D.; Vogt, A.; Sun, J.; Cox, A.D.; Der, C.J.; Hamilton, A.D.; Sebti, S.M. Ras CAAX peptidomimetic FTI-277 selectively blocks oncogenic Ras signaling by inducing cytoplasmic accumulation of inactive Ras-Raf complexes. J. Biol. Chem. 1995, 270, 26802–26806. [Google Scholar] [CrossRef]
- Casey, P.J.; Thissen, J.A.; Moomaw, J.F. Enzymatic modification of proteins with a geranylgeranyl isoprenoid. Proc. Natl. Acad. Sci. USA 1991, 88, 8631–8635. [Google Scholar] [CrossRef]
- Hancock, J.F.; Magee, A.I.; Childs, J.E.; Marshall, C.J. All ras proteins are polyisoprenylated but only some are palmitoylated. Cell 1989, 57, 1167–1177. [Google Scholar] [CrossRef]
- Buss, J.E.; Quilliam, L.A.; Kato, K.; Casey, P.J.; Solski, P.A.; Wong, G.; Clark, R.; McCormick, F.; Bokoch, G.M.; Der, C.J. The COOH-terminal domain of the Rap1A (Krev-1) protein is isoprenylated and supports transformation by an H-Ras:Rap1A chimeric protein. Mol. Cell. Biol. 1991, 11, 1523–1530. [Google Scholar] [CrossRef] [PubMed]
- Wright, L.P.; Philips, M.R. Thematic review series: Lipid Posttranslational Modifications CAAX modification and membrane targeting of Ras. J. Lipid Res. 2006, 47, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Boyartchuk, V.L. Modulation of Ras and a-Factor Function by Carboxyl-Terminal Proteolysis. Science 1997, 275, 1796–1800. [Google Scholar] [CrossRef]
- Bergo, M.O.; Gavino, B.J.; Hong, C.; Beigneux, A.P.; McMahon, M.; Casey, P.J.; Young, S.G. Inactivation of Icmt inhibits transformation by oncogenic K-Ras and B-Raf. J. Clin. Investig. 2004, 113, 539–550. [Google Scholar] [CrossRef]
- Bergo, M.O.; Leung, G.K.; Ambroziak, P.; Otto, J.C.; Casey, P.J.; Young, S.G. Targeted Inactivation of the Isoprenylcysteine Carboxyl Methyltransferase Gene Causes Mislocalization of K-Ras in Mammalian Cells. J. Biol. Chem. 2000, 275, 17605–17610. [Google Scholar] [CrossRef]
- Winter-Vann, A.M.; Kamen, B.A.; Bergo, M.O.; Young, S.G.; Melnyk, S.; James, S.J.; Casey, P.J. Targeting Ras signaling through inhibition of carboxyl methylation: An unexpected property of methotrexate. Proc. Natl. Acad. Sci. USA 2003, 100, 6529–6534. [Google Scholar] [CrossRef]
- Inouye, K.; Mizutani, S.; Koide, H.; Kaziro, Y. Formation of the Ras Dimer Is Essential for Raf-1 Activation. J. Biol. Chem. 2000, 275, 3737–3740. [Google Scholar] [CrossRef]
- Varma, R.; Mayor, S. GPI-anchored proteins are organized in submicron domains at the cell surface. Nature 1998, 394, 798–801. [Google Scholar] [CrossRef]
- Prior, I.A.; Muncke, C.; Parton, R.G.; Hancock, J.F. Direct visualization of Ras proteins in spatially distinct cell surface microdomains. J. Cell Biol. 2003, 160, 165–170. [Google Scholar] [CrossRef]
- Plowman, S.J.; Muncke, C.; Parton, R.G.; Hancock, J.F. H-ras, K-ras, and inner plasma membrane raft proteins operate in nanoclusters with differential dependence on the actin cytoskeleton. Proc. Natl. Acad. Sci. USA 2005, 102, 15500–15505. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Hancock, J.F. Ras nanoclusters: Versatile lipid-based signaling platforms. Biochim. Biophys. Acta Mol. Cell Res. 2015, 1853, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Harding, A.; Inder, K.; Plowman, S.; Parton, R.G.; Hancock, J.F. Plasma membrane nanoswitches generate high-fidelity Ras signal transduction. Nat. Cell Biol. 2007, 9, 905–914. [Google Scholar] [CrossRef]
- Huleihel, M.; Goldsborough, M.; Cleveland, J.; Gunnell, M.; Bonner, T.; Rapp, U.R. Characterization of murine A-raf, a new oncogene related to the v-raf oncogene. Mol. Cell. Biol. 1986, 6, 2655–2662. [Google Scholar] [CrossRef] [PubMed]
- Beck, T.W.; Huleihel, M.; Gunnell, M.; Bonner, T.I.; Rapp, U.R. The complete coding sequence of the human A-raf-1 oncogene and transforming activity of a human A-raf carrying retrovirus. Nucl. Acids Res. 1987, 15, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Ikawa, S.; Fukui, M.; Ueyama, Y.; Tamaoki, N.; Yamamoto, T.; Toyoshima, K. B-raf, a new member of the raf family, is activated by DNA rearrangement. Mol. Cell. Biol. 1988, 8, 2651–2654. [Google Scholar] [CrossRef]
- Daum, G.; Eisenmann-Tappe, I.; Fries, H.-W.; Troppmair, J.; Rapp, U.R. The ins and outs of Raf kinases. Trends Biochem. Sci. 1994, 19, 474–480. [Google Scholar] [CrossRef]
- Wellbrock, C.; Karasarides, M.; Marais, R. The RAF proteins take centre stage. Nat. Rev. Mol. Cell Biol. 2004, 5, 875–885. [Google Scholar] [CrossRef]
- Nassar, N.; Horn, G.; Herrmann, C.A.; Scherer, A.; McCormick, F.; Wittinghofer, A. The 2.2 Å crystal structure of the Ras-binding domain of the serine/threonine kinase c-Raf1 in complex with RaplA and a GTP analogue. Nature 1995, 375, 554–560. [Google Scholar] [CrossRef]
- Scheffler, J.E.; Waugh, D.S.; Bekesi, E.; Kiefer, S.E.; LoSardo, J.E.; Neri, A.; Prinzo, K.M.; Tsao, K.L.; Wegrzynski, B.; Emerson, S.D.; et al. Characterization of a 78-residue fragment of c-Raf-1 that comprises a minimal binding domain for the interaction with Ras-GTP. J. Biol. Chem. 1994, 269, 22340–22346. [Google Scholar]
- Chuang, E.; Barnard, D.; Hettich, L.; Zhang, X.F.; Avruch, J.; Marshall, M.S. Critical binding and regulatory interactions between Ras and Raf occur through a small, stable N-terminal domain of Raf and specific Ras effector residues. Mol. Cell. Biol. 1994, 14, 5318–5325. [Google Scholar] [CrossRef] [PubMed]
- Emerson, S.D.; Waugh, D.S.; Scheffler, J.E.; Tsao, K.-L.; Prinzo, K.M.; Fry, D.C. Chemical Shift Assignments and Folding Topology of the RAS-Binding Domain of Human RAF-1 As Determined by Heteronuclear Three-Dimensional NMR Spectroscopy. Biochemistry 1994, 33, 7745–7752. [Google Scholar] [CrossRef] [PubMed]
- Mott, H.R.; Carpenter, J.W.; Zhong, S.; Ghosh, S.; Bell, R.M.; Campbell, S.L. The solution structure of the Raf-1 cysteine-rich domain: A novel ras and phospholipid binding site. Proc. Natl. Acad. Sci. USA 1996, 93, 8312–8317. [Google Scholar] [CrossRef] [PubMed]
- Diaz, B.; Barnard, D.; Filson, A.; MacDonald, S.; King, A.; Marshall, M. Phosphorylation of Raf-1 serine 338-serine 339 is an essential regulatory event for Ras-dependent activation and biological signaling. Mol. Cell. Biol. 1997, 17, 4509–4516. [Google Scholar] [CrossRef]
- Mason, C.S.; Springer, C.J.; Cooper, R.G.; Superti-Furga, G.; Marshall, C.J.; Marais, R. Serine and tyrosine phosphorylations cooperate in Raf-1, but not B-Raf activation. EMBO J. 1999, 18, 2137–2148. [Google Scholar] [CrossRef]
- Dhillon, A.S.; Yip, Y.Y.; Grindlay, G.J.; Pakay, J.L.; Dangers, M.; Hillmann, M.; Clark, W.; Pitt, A.; Mischak, H.; Kolch, W. The C-terminus of Raf-1 acts as a 14-3-3-dependent activation switch. Cell. Signal. 2009, 21, 1645–1651. [Google Scholar] [CrossRef]
- Kondo, Y.; Ognjenović, J.; Banerjee, S.; Karandur, D.; Merk, A.; Kulhanek, K.; Wong, K.; Roose, J.P.; Subramaniam, S.; Kuriyan, J. Cryo-EM structure of a dimeric B-Raf:14-3-3 complex reveals asymmetry in the active sites of B-Raf kinases. Science 2019, 366, 109–115. [Google Scholar] [CrossRef]
- Muslin, A.J.; Tanner, J.W.; Allen, P.M.; Shaw, A.S. Interaction of 14-3-3 with Signaling Proteins Is Mediated by the Recognition of Phosphoserine. Cell 1996, 84, 889–897. [Google Scholar] [CrossRef]
- Morrison, D.K.; Heidecker, G.; Rapp, U.R.; Copeland, T.D.; Morrisonst, D.K.; Heideckerq, G.; Rappq, U.R.; Copeland, T.D. Identification of the major phosphorylation sites of the Raf-1 kinase. J. Biol. Chem. 1993, 268, 17309–17316. [Google Scholar]
- Hu, J.; Stites, E.C.; Yu, H.; Germino, E.A.; Meharena, H.S.; Stork, P.J.S.; Kornev, A.P.; Taylor, S.S.; Shaw, A.S. Allosteric Activation of Functionally Asymmetric RAF Kinase Dimers. Cell 2013, 154, 1036–1046. [Google Scholar] [CrossRef]
- Blasco, R.B.; Francoz, S.; Santamaría, D.; Cañamero, M.; Dubus, P.; Charron, J.; Baccarini, M.; Barbacid, M. c-Raf, but Not B-Raf, Is Essential for Development of K-Ras Oncogene-Driven Non-Small Cell Lung Carcinoma. Cancer Cell 2011, 19, 652–663. [Google Scholar] [CrossRef] [PubMed]
- Karreth, F.A.; Frese, K.K.; DeNicola, G.M.; Baccarini, M.; Tuveson, D.A. C-Raf is required for the initiation of lung cancer by K-Ras(G12D). Cancer Discov. 2011, 1, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Lito, P.; Saborowski, A.; Yue, J.; Solomon, M.; Joseph, E.; Gadal, S.; Saborowski, M.; Kastenhuber, E.; Fellmann, C.; Ohara, K.; et al. Disruption of CRAF-Mediated MEK Activation Is Required for Effective MEK Inhibition in KRAS Mutant Tumors. Cancer Cell 2014, 25, 697–710. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Ng, W.H.; Lam, P.Y.P.P.; Wang, Y.; Xia, H.; Yap, J.; Guan, S.P.; Lee, A.S.G.G.; Wang, M.; Baccarini, M.; et al. The dimer-dependent catalytic activity of RAF family kinases is revealed through characterizing their oncogenic mutants. Oncogene 2018, 37, 5719–5734. [Google Scholar] [CrossRef]
- Yap, J.; Yuan, J.; Tee, Z.H.; Huang, X.; Ng, W.H.; Hu, J. Characterize Disease-related Mutants of RAF Family Kinases by Using a Set of Practical and Feasible Methods. JoVE 2019, 149, e59795. [Google Scholar] [CrossRef]
- Haling, J.R.; Sudhamsu, J.; Yen, I.; Sideris, S.; Sandoval, W.; Phung, W.; Bravo, B.J.; Giannetti, A.M.; Peck, A.; Masselot, A.; et al. Structure of the BRAF-MEK Complex Reveals a Kinase Activity Independent Role for BRAF in MAPK Signaling. Cancer Cell 2014, 26, 402–413. [Google Scholar] [CrossRef]
- Kornev, A.P.; Haste, N.M.; Taylor, S.S.; Ten Eyck, L.F. Surface comparison of active and inactive protein kinases identifies a conserved activation mechanism. Proc. Natl. Acad. Sci. USA 2006, 103, 17783–17788. [Google Scholar] [CrossRef]
- Taylor, S.S.; Kornev, A.P. Protein kinases: Evolution of dynamic regulatory proteins. Trends Biochem. Sci. 2011, 36, 65–77. [Google Scholar] [CrossRef]
- Taylor, S.S.; Shaw, A.; Hu, J.; Meharena, H.S.; Kornev, A. Pseudokinases from a structural perspective. Biochem. Soc. Trans. 2013, 41, 981–986. [Google Scholar] [CrossRef]
- Shaw, A.S.; Kornev, A.P.; Hu, J.; Ahuja, L.G.; Taylor, S.S. Kinases and Pseudokinases: Lessons from RAF. Mol. Cell. Biol. 2014, 34, 1538–1546. [Google Scholar] [CrossRef]
- Hu, J.; Ahuja, L.G.; Meharena, H.S.; Kannan, N.; Kornev, A.P.; Taylor, S.S.; Shaw, A.S. Kinase Regulation by Hydrophobic Spine Assembly in Cancer. Mol. Cell. Biol. 2015, 35, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Ahuja, L.G.; Chao, F.; Xia, Y.; Mcclendon, C.L.; Kornev, A.P.; Taylor, S.S.; Veglia, G. A dynamic hydrophobic core orchestrates allostery in protein kinases. Sci. Adv. 2017, 3, e1600663. [Google Scholar] [CrossRef] [PubMed]
- Vijayan, R.S.K.; He, P.; Modi, V.; Duong-Ly, K.C.; Ma, H.; Peterson, J.R.; Dunbrack, R.L., Jr.; Levy, R.M. Conformational analysis of the DFG-out kinase motif and biochemical profiling of structurally validated type II inhibitors. J. Med. Chem. 2015, 58, 466–479. [Google Scholar] [CrossRef] [PubMed]
- Treiber, D.K.; Shah, N.P. Ins and Outs of Kinase DFG Motifs. Chem. Biol. 2013, 20, 745–746. [Google Scholar] [CrossRef] [PubMed]
- Thevakumaran, N.; Lavoie, H.; Critton, D.A.; Tebben, A.; Marinier, A.; Sicheri, F.; Therrien, M. Crystal structure of a BRAF kinase domain monomer explains basis for allosteric regulation. Nat. Struct. Mol. Biol. 2015, 22, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Terrell, E.M.; Morrison, D.K. Ras-Mediated Activation of the Raf Family Kinases. Cold Spring Harb. Perspect. Med. 2019, 9, a033746. [Google Scholar] [CrossRef] [PubMed]
- Wojnowski, L.; Stancato, L.F.; Zimmer, A.M.; Hahn, H.; Beck, T.W.; Larner, A.C.; Rapp, U.R.; Zimmer, A. Craf-1 protein kinase is essential for mouse development. Mech. Dev. 1998, 76, 141–149. [Google Scholar] [CrossRef]
- Mikula, M.; Schreiber, M.; Husak, Z.; Kucerova, L.; Rüth, J.; Wieser, R.; Zatloukal, K.; Beug, H.; Wagner, E.F.; Baccarini, M. Embryonic lethality and fetal liver apoptosis in mice lacking the c-raf-1 gene. EMBO J. 2001, 20, 1952–1962. [Google Scholar] [CrossRef]
- Wojnowski, L.; Zimmer, A.M.; Beck, T.W.; Hahn, H.; Bernal, R.; Rapp, U.R.; Zimmer, A. Endothelial apoptosis in Braf-deficient mice. Nat. Genet. 1997, 16, 293–297. [Google Scholar] [CrossRef]
- Wiese, S.; Pei, G.; Karch, C.; Troppmair, J.; Holtmann, B.; Rapp, U.R.; Sendtner, M. Specific function of B-Raf in mediating survival of embryonic motoneurons and sensory neurons. Nat. Neurosci. 2001, 4, 137–142. [Google Scholar] [CrossRef]
- Pritchard, C.A.; Bolin, L.; Slattery, R.; Murray, R.; McMahon, M. Post-natal lethality and neurological and gastrointestinal defects in mice with targeted disruption of the A-Raf protein kinase gene. Curr. Biol. 1996, 6, 614–617. [Google Scholar] [CrossRef]
- Hüser, M.; Luckett, J.; Chiloeches, A.; Mercer, K.; Iwobi, M.; Giblett, S.; Sun, X.M.; Brown, J.; Marais, R.; Pritchard, C.; et al. MEK kinase activity is not necessary for Raf-1 function. EMBO J. 2001, 20, 1940–1951. [Google Scholar] [CrossRef] [PubMed]
- Piazzolla, D.; Meissl, K.; Kucerova, L.; Rubiolo, C.; Baccarini, M. Raf-1 sets the threshold of Fas sensitivity by modulating Rok-alpha signaling. J. Cell Biol. 2005, 171, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Ehrenreiter, K.; Piazzolla, D.; Velamoor, V.; Sobczak, I.; Small, J.V.; Takeda, J.; Leung, T.; Baccarini, M. Raf-1 regulates Rho signaling and cell migration. J. Cell Biol. 2005, 168, 955–964. [Google Scholar] [CrossRef]
- Niault, T.; Sobczak, I.; Meissl, K.; Weitsman, G.; Piazzolla, D.; Maurer, G.; Kern, F.; Ehrenreiter, K.; Hamerl, M.; Moarefi, I.; et al. From autoinhibition to inhibition in trans: The Raf-1 regulatory domain inhibits Rok-alpha kinase activity. J. Cell Biol. 2009, 187, 335–342. [Google Scholar] [CrossRef]
- Ehrenreiter, K.; Kern, F.; Velamoor, V.; Meissl, K.; Galabova-Kovacs, G.; Sibilia, M.; Baccarini, M. Raf-1 Addiction in Ras-Induced Skin Carcinogenesis. Cancer Cell 2009, 16, 149–160. [Google Scholar] [CrossRef]
- Sanclemente, M.; Francoz, S.; Esteban-Burgos, L.; Bousquet-Mur, E.; Djurec, M.; Lopez-Casas, P.P.; Hidalgo, M.; Guerra, C.; Drosten, M.; Musteanu, M.; et al. c-RAF Ablation Induces Regression of Advanced Kras/Trp53 Mutant Lung Adenocarcinomas by a Mechanism Independent of MAPK Signaling. Cancer Cell 2018, 33, 217–228.e4. [Google Scholar] [CrossRef]
- Rubiolo, C.; Piazzolla, D.; Meissl, K.; Beug, H.; Huber, J.C.; Kolbus, A.; Baccarini, M. A balance between Raf-1 and Fas expression sets the pace of erythroid differentiation. Blood 2006, 108, 152–159. [Google Scholar] [CrossRef][Green Version]
- Kolbus, A.; Pilat, S.; Husak, Z.; Deiner, E.M.; Stengl, G.; Beug, H.; Baccarini, M. Raf-1 Antagonizes Erythroid Differentiation by Restraining Caspase Activation. J. Exp. Med. 2002, 196, 1347–1353. [Google Scholar] [CrossRef]
- Wang, H.-G.; Rapp, U.R.; Reed, J.C. Bcl-2 Targets the Protein Kinase Raf-1 to Mitochondria. Cell 1996, 87, 629–638. [Google Scholar] [CrossRef]
- Panka, D.J.; Atkins, M.B.; Mier, J.W. Targeting the Mitogen-Activated Protein Kinase Pathway in the Treatment of Malignant Melanoma. Clin. Cancer Res. 2006, 12, 2371s–2375s. [Google Scholar] [CrossRef] [PubMed]
- Baumann, B.; Weber, C.K.; Troppmair, J.; Whiteside, S.; Israel, A.; Rapp, U.R.; Wirth, T. Raf induces NF-kappaB by membrane shuttle kinase MEKK1, a signaling pathway critical for transformation. Proc. Natl. Acad. Sci. USA 2000, 97, 4615–4620. [Google Scholar] [CrossRef] [PubMed]
- Troppmair, J.; Hartkamp, J.; Rapp, U.R. Activation of NF-κB by oncogenic Raf in HEK 293 cells occurs through autocrine recruitment of the stress kinase cascade. Oncogene 1998, 17, 685–690. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Norris, J.L.; Baldwin, A.S. Oncogenic Ras Enhances NF-κB Transcriptional Activity through Raf-dependent and Raf-independent Mitogen-activated Protein Kinase Signaling Pathways. J. Biol. Chem. 1999, 274, 13841–13846. [Google Scholar] [CrossRef]
- Majewski, M.; Nieborowska-Skorska, M.; Salomoni, P.; Slupianek, A.; Reiss, K.; Trotta, R.; Calabretta, B.; Skorski, T. Activation of Mitochondrial Raf-1 Is Involved in the Antiapoptotic Effects of Akt. Cancer Res. 1999, 59, 2815–2819. [Google Scholar]
- Wu, X.; Carr, H.S.; Dan, I.; Ruvolo, P.P.; Frost, J.A. p21 activated kinase 5 activates Raf-1 and targets it to mitochondria. J. Cell. Biochem. 2008, 105, 167–175. [Google Scholar] [CrossRef]
- Jin, S.; Zhuo, Y.; Guo, W.; Field, J. p21-activated Kinase 1 (Pak1)-dependent Phosphorylation of Raf-1 Regulates Its Mitochondrial Localization, Phosphorylation of BAD, and Bcl-2 Association. J. Biol. Chem. 2005, 280, 24698–24705. [Google Scholar] [CrossRef]
- Hindley, A.; Kolch, W. Raf-1 and B-Raf promote protein kinase C theta interaction with BAD. Cell. Signal. 2007, 19, 547–555. [Google Scholar] [CrossRef]
- Le Mellay, V.; Troppmair, J.; Benz, R.; Rapp, U.R. Negative regulation of mitochondrial VDAC channels by C-Raf kinase. BMC Cell Biol. 2002, 3, 14. [Google Scholar] [CrossRef]
- Chen, J.; Fujii, K.; Zhang, L.; Roberts, T.; Fu, H. Raf-1 promotes cell survival by antagonizing apoptosis signal-regulating kinase 1 through a MEK-ERK independent mechanism. Proc. Natl. Acad. Sci. USA 2001, 98, 7783–7788. [Google Scholar] [CrossRef]
- Alavi, A.S.; Acevedo, L.; Min, W.; Cheresh, D.A. Chemoresistance of Endothelial Cells Induced by Basic Fibroblast Growth Factor Depends on Raf-1–Mediated Inhibition of the Proapoptotic Kinase, ASK1. Cancer Res. 2007, 67, 2766–2772. [Google Scholar] [CrossRef] [PubMed]
- Tobiume, K.; Matsuzawa, A.; Takahashi, T.; Nishitoh, H.; Morita, K.; Takeda, K.; Minowa, O.; Miyazono, K.; Noda, T.; Ichijo, H. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep. 2001, 2, 222–228. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, E.; Rushworth, L.; Baccarini, M.; Kolch, W. Role of the kinase MST2 in suppression of apoptosis by the proto-oncogene product Raf-1. Science 2004, 306, 2267–2270. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, O.; Watanabe, T.; Nishida, K.; Kashiwase, K.; Higuchi, Y.; Takeda, T.; Hikoso, S.; Hirotani, S.; Asahi, M.; Taniike, M.; et al. Cardiac-specific disruption of the c-raf-1 gene induces cardiac dysfunction and apoptosis. J. Clin. Investig. 2004, 114, 937–943. [Google Scholar] [CrossRef] [PubMed]
- Rauch, J.; Kolch, W. Spatial regulation of ARAF controls the MST2-Hippo pathway. Small GTPases 2019, 10, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Rauch, J.; O’Neill, E.; Mack, B.; Matthias, C.; Munz, M.; Kolch, W.; Gires, O. Heterogeneous nuclear ribonucleoprotein H blocks MST2-mediated apoptosis in cancer cells by regulating A-Raf transcription. Cancer Res. 2010, 70, 1679–1688. [Google Scholar] [CrossRef] [PubMed]
- Christofk, H.R.; Vander Heiden, M.G.; Harris, M.H.; Ramanathan, A.; Gerszten, R.E.; Wei, R.; Fleming, M.D.; Schreiber, S.L.; Cantley, L.C. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008, 452, 230–233. [Google Scholar] [CrossRef]
- Le Mellay, V.; Houben, R.; Troppmair, J.; Hagemann, C.; Mazurek, S.; Frey, U.; Beigel, J.; Weber, C.; Benz, R.; Eigenbrodt, E.; et al. Regulation of glycolysis by Raf protein serine/threonine kinases. Adv. Enzyme Regul. 2002, 42, 317–332. [Google Scholar] [CrossRef]
- Mercer, K.; Giblett, S.; Oakden, A.; Brown, J.; Marais, R.; Pritchard, C. A-Raf and Raf-1 work together to influence transient ERK phosphorylation and Gl/S cell cycle progression. Oncogene 2005, 24, 5207–5217. [Google Scholar] [CrossRef]
- Wojnowski, L.; Stancato, L.F.; Larner, A.C.; Rapp, U.R.; Zimmer, A. Overlapping and specific functions of Braf and Craf-1 proto-oncogenes during mouse embryogenesis. Mech. Dev. 2000, 91, 97–104. [Google Scholar] [CrossRef]
- Mercer, K.; Giblett, S.; Green, S.; Lloyd, D.; DaRocha Dias, S.; Plumb, M.; Marais, R.; Pritchard, C. Expression of endogenous oncogenic V600EB-raf induces proliferation and developmental defects in mice and transformation of primary fibroblasts. Cancer Res. 2005, 65, 11493–11500. [Google Scholar] [CrossRef] [PubMed]
- Cutler, R.E.; Stephens, R.M.; Saracino, M.R.; Morrison, D.K. Autoregulation of the Raf-1 serine/threonine kinase. Proc. Natl. Acad. Sci. USA 1998, 95, 9214–9219. [Google Scholar] [CrossRef] [PubMed]
- Morrison, D.K.; Cutler, R.E. The complexity of Raf-1 regulation. Curr. Opin. Cell Biol. 1997, 9, 174–179. [Google Scholar] [CrossRef]
- Jin, T.; Lavoie, H.; Sahmi, M.; David, M.; Hilt, C.; Hammell, A.; Therrien, M. RAF inhibitors promote RAS-RAF interaction by allosterically disrupting RAF autoinhibition. Nat. Commun. 2017, 8, 1211. [Google Scholar] [CrossRef] [PubMed]
- Hey, F.; Pritchard, C. A New Mode of RAF Autoregulation: A Further Complication in the Inhibitor Paradox. Cancer Cell 2013, 23, 561–563. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337.e10. [Google Scholar] [CrossRef]
- Ahearn, I.M.; Haigis, K.; Bar-Sagi, D.; Philips, M.R. Regulating the regulator: Post-translational modification of RAS. Nat. Rev. Mol. Cell Biol. 2011, 13, 39. [Google Scholar] [CrossRef]
- Clark, G.J.; Drugan, J.K.; Terrell, R.S.; Bradham, C.; Der, C.J.; Bell, R.M.; Campbell, S. Peptides containing a consensus Ras binding sequence from Raf-1 and theGTPase activating protein NF1 inhibit Ras function. Proc. Natl. Acad. Sci. USA 1996, 93, 1577–1581. [Google Scholar] [CrossRef]
- Pacold, M.E.; Suire, S.; Perisic, O.; Lara-Gonzalez, S.; Davis, C.T.; Walker, E.H.; Hawkins, P.T.; Stephens, L.; Eccleston, J.F.; Williams, R.L. Crystal Structure and Functional Analysis of Ras Binding to Its Effector Phosphoinositide 3-Kinase γ. Cell 2000, 103, 931–944. [Google Scholar] [CrossRef]
- Fabian, J.R.; Vojtek, A.B.; Cooper, J.A.; Morrison, D.K. A single amino acid change in Raf-1 inhibits Ras binding and alters Raf-1 function. Proc. Natl. Acad. Sci. USA 1994, 91, 5982–5986. [Google Scholar] [CrossRef]
- Im, E.; von Lintig, F.C.; Chen, J.; Zhuang, S.; Qui, W.; Chowdhury, S.; Worley, P.F.; Boss, G.R.; Pilz, R.B. Rheb is in a high activation state and inhibits B-Raf kinase in mammalian cells. Oncogene 2002, 21, 6356–6365. [Google Scholar] [CrossRef] [PubMed]
- Vossler, M.R.; Yao, H.; York, R.D.; Pan, M.-G.; Rim, C.S.; Stork, P.J.S. cAMP Activates MAP Kinase and Elk-1 through a B-Raf- and Rap1-Dependent Pathway. Cell 1997, 89, 73–82. [Google Scholar] [CrossRef]
- Fischer, A.; Hekman, M.; Kuhlmann, J.; Rubio, I.; Wiese, S.; Rapp, U.R. B- and C-RAF Display Essential Differences in Their Binding to Ras: The isotype-specific n terminus of b-RAF facilitates ras binding. J. Biol. Chem. 2007, 282, 26503–26516. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Diaz, B.; Marshall, M.S.; Avruch, J. An intact Raf zinc finger is required for optimal binding to processed Ras and for ras-dependent Raf activation in situ. Mol. Cell. Biol. 1997, 17, 46–53. [Google Scholar] [CrossRef]
- Williams, J.G.; Drugan, J.K.; Yi, G.S.; Clark, G.J.; Der, C.J.; Campbell, S.L. Elucidation of binding determinants and functional consequences of Ras/Raf-cysteine-rich domain interactions. J. Biol. Chem. 2000, 275, 22172–22179. [Google Scholar] [CrossRef]
- Winkler, D.G.; Cutler, R.E.; Drugan, J.K.; Campbell, S.; Morrison, D.K.; Cooper, J.A. Identification of Residues in the Cysteine-rich Domain of Raf-1 That Control Ras Binding and Raf-1 Activity. J. Biol. Chem. 1998, 273, 21578–21584. [Google Scholar] [CrossRef]
- Thapar, R.; Williams, J.G.; Campbell, S.L. NMR Characterization of Full-length Farnesylated and Non-farnesylated H-Ras and its Implications for Raf Activation. J. Mol. Biol. 2004, 343, 1391–1408. [Google Scholar] [CrossRef]
- Ding, J.; Tchaicheeyan, O.; Ambrosio, L. Drosophila Raf’s N Terminus Contains a Novel Conserved Region and Can Contribute to Torso RTK Signaling. Genetics 2010, 184, 717–729. [Google Scholar] [CrossRef][Green Version]
- Kubicek, M.; Pacher, M.; Abraham, D.; Podar, K.; Eulitz, M.; Baccarini, M. Dephosphorylation of Ser-259 regulates Raf-1 membrane association. J. Biol. Chem. 2002, 277, 7913–7919. [Google Scholar] [CrossRef]
- Tran, N.H.; Wu, X.; Frost, J.A. B-Raf and Raf-1 Are Regulated by Distinct Autoregulatory Mechanisms. J. Biol. Chem. 2005, 280, 16244–16253. [Google Scholar] [CrossRef]
- Abraham, D.; Podar, K.; Pacher, M.; Kubicek, M.; Welzel, N.; Hemmings, B.A.; Dilworth, S.M.; Mischak, H.; Kolch, W.; Baccarini, M. Raf-1-associated protein phosphatase 2A as a positive regulator of kinase activation. J. Biol. Chem. 2000, 275, 22300–22304. [Google Scholar] [CrossRef] [PubMed]
- Jaumot, M.; Hancock, J.F. Protein phosphatases 1 and 2A promote Raf-1 activation by regulating 14-3-3 interactions. Oncogene 2001, 20, 3949–3958. [Google Scholar] [CrossRef] [PubMed]
- Ory, S.; Zhou, M.; Conrads, T.P.; Veenstra, T.D.; Morrison, D.K. Protein Phosphatase 2A Positively Regulates Ras Signaling by Dephosphorylating KSR1 and Raf-1 on Critical 14-3-3 Binding Sites. Curr. Biol. 2003, 13, 1356–1364. [Google Scholar] [CrossRef]
- Eisenhardt, A.E.; Sprenger, A.; Röring, M.; Herr, R.; Weinberg, F.; Köhler, M.; Braun, S.; Orth, J.; Diedrich, B.; Lanner, U.; et al. Phospho-proteomic analyses of B-Raf protein complexes reveal new regulatory principles. Oncotarget 2016, 7, 26628–26652. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Tzivion, G.; Belshaw, P.J.; Vavvas, D.; Marshall, M.; Avruch, J. Oligomerization activates c-Raf-1 through a Ras-dependent mechanism. Nature 1996, 383, 181–185. [Google Scholar] [CrossRef]
- Farrar, M.A.; Alberola-lla, J.; Perlmutter, R.M. Activation of the Raf-1 kinase cascade by coumermycin-induced dimerization. Nature 1996, 383, 178–181. [Google Scholar] [CrossRef]
- Weber, C.K.; Slupsky, J.R.; Kalmes, H.A.; Rapp, U.R. Active Ras Induces Heterodimerization of cRaf and BRaf 1. Cancer Res. 2001, 61, 3595–3598. [Google Scholar]
- Rajakulendran, T.; Sahmi, M.; Lefrançois, M.; Sicheri, F.; Therrien, M. A dimerization-dependent mechanism drives RAF catalytic activation. Nature 2009, 461, 542–545. [Google Scholar] [CrossRef]
- Rushworth, L.K.; Hindley, A.D.; O’Neill, E.; Kolch, W. Regulation and role of Raf-1/B-Raf heterodimerization. Mol. Cell. Biol. 2006, 26, 2262–2272. [Google Scholar] [CrossRef]
- Heidorn, S.J.; Milagre, C.; Whittaker, S.; Nourry, A.; Niculescu-Duvas, I.; Dhomen, N.; Hussain, J.; Reis-Filho, J.S.; Springer, C.J.; Pritchard, C.; et al. Kinase-Dead BRAF and Oncogenic RAS Cooperate to Drive Tumor Progression through CRAF. Cell 2010, 140, 209–221. [Google Scholar] [CrossRef]
- Kwong, L.N.; Chin, L. The Brothers RAF. Cell 2010, 2008–2010. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Yu, H.; Kornev, A.P.; Zhao, J.; Filbert, E.L.; Taylor, S.S.; Shaw, A.S. Mutation that blocks ATP binding creates a pseudokinase stabilizing the scaffolding function of kinase suppressor of Ras, CRAF and BRAF. Proc. Natl. Acad. Sci. USA 2011, 108, 6067–6072. [Google Scholar] [CrossRef] [PubMed]
- Baljuls, A.; Mahr, R.; Schwarzenau, I.; Müller, T.; Polzien, L.; Hekman, M.; Rapp, U.R. Single Substitution within the RKTR Motif Impairs Kinase Activity but Promotes Dimerization of RAF Kinase. J. Biol. Chem. 2011, 286, 16491–16503. [Google Scholar] [CrossRef] [PubMed]
- Jambrina, P.G.; Rauch, N.; Pilkington, R.; Rybakova, K.; Nguyen, L.K.; Kholodenko, B.N.; Buchete, N.-V.; Kolch, W.; Rosta, E. Phosphorylation of RAF Kinase Dimers Drives Conformational Changes that Facilitate Transactivation. Angew. Chem. Int. Ed. 2016, 55, 983–986. [Google Scholar] [CrossRef] [PubMed]
- Poulikakos, P.I.; Persaud, Y.; Janakiraman, M.; Kong, X.; Ng, C.; Moriceau, G.; Shi, H.; Atefi, M.; Titz, B.; Gabay, M.T.; et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature 2011, 480, 387–390. [Google Scholar] [CrossRef]
- Dunham, I.; Kundaje, A.; Aldred, S.F.; Collins, P.J.; Davis, C.A.; Doyle, F.; Epstein, C.B.; Frietze, S.; Harrow, J.; Kaul, R.; et al. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef]
- Palanisamy, N.; Ateeq, B.; Kalyana-Sundaram, S.; Pflueger, D.; Ramnarayanan, K.; Shankar, S.; Han, B.; Cao, Q.; Cao, X.; Suleman, K.; et al. Rearrangements of the RAF kinase pathway in prostate cancer, gastric cancer and melanoma. Nat. Med. 2010, 16, 793–798. [Google Scholar] [CrossRef]
- Jain, P.; Fierst, T.M.; Han, H.J.; Smith, T.E.; Vakil, A.; Storm, P.B.; Resnick, A.C.; Waanders, A.J. CRAF gene fusions in pediatric low-grade gliomas define a distinct drug response based on dimerization profiles. Oncogene 2017, 36, 6348–6358. [Google Scholar] [CrossRef]
- Stransky, N.; Cerami, E.; Schalm, S.; Kim, J.L.; Lengauer, C. The landscape of kinase fusions in cancer. Nat. Commun. 2014, 5, 4846. [Google Scholar] [CrossRef]
- Tran, N.H.; Frost, J.A. Phosphorylation of Raf-1 by p21-activated Kinase 1 and Src Regulates Raf-1 Autoinhibition. J. Biol. Chem. 2003, 278, 11221–11226. [Google Scholar] [CrossRef]
- Williams, N.G.; Roberts, T.M.; Li, P. Both p21ras and pp60v-src are required, but neither alone is sufficient, to activate the Raf-1 kinase. Proc. Natl. Acad. Sci. USA 1992, 89, 2922–2926. [Google Scholar] [CrossRef] [PubMed]
- Thompson, P.A.; Ledbetter, J.A.; Rapp, U.R.; Bolen, J.B. The Raf-1 serine-threonine kinase is a substrate for the p56lck protein tyrosine kinase in human T-cells. Cell Growth Differ. 1991, 2, 609–617. [Google Scholar] [PubMed]
- Fabian, J.R.; Daar, I.R.A.; Morrison, D.K. Critical Tyrosine Residues Regulate the Enzymatic and Biological Activity of Raf-1 Kinase. Mol. Cell. Biol. 1993, 13, 7170–7179. [Google Scholar] [CrossRef] [PubMed]
- Cleghon, V.; Morrison, D.K. Raf-1 Interacts with Fyn and Src in a Non-phosphotyrosine-dependent Manner. J. Biol. Chem. 1994, 269, 17749–17755. [Google Scholar] [PubMed]
- Marais, R.; Light, Y.; Paterson, H.F.; Marshall, C.J. Ras recruits Raf-1 to the plasma membrane for activation by tyrosine phosphorylation. EMBO J. 1995, 14, 3136–3145. [Google Scholar] [CrossRef]
- Chiloeches, A.; Mason, C.S.; Marais, R. S338 phosphorylation of Raf-1 is independent of phosphatidylinositol 3-kinase and Pak3. Mol. Cell. Biol. 2001, 21, 2423–2434. [Google Scholar] [CrossRef]
- Chaudhary, A.; King, W.G.; Mattaliano, M.D.; Frost, J.A.; Diaz, B.; Morrison, D.K.; Cobb, M.H.; Marshall, M.S.; Brugge, J.S. Phosphatidylinositol 3-kinase regulates Raf1 through Pak phosphorylation of serine 338. Curr. Biol. 2000, 10, 551–554. [Google Scholar] [CrossRef]
- King, A.J.; Sun, H.; Diaz, B.; Barnard, D.; Miao, W.; Bagrodia, S.; Marshall, M.S. The protein kinase Pak3 positively regulates Raf-1 activity through phosphorylation of serine 338. Nature 1998, 396, 180–183. [Google Scholar] [CrossRef]
- Xia, K.; Mukhopadhyay, N.K.; Inhorn, R.C.; Barber, D.L.; Rose, P.E.; Lee, R.S.; Narsimhan, R.P.; D’Andrea, A.D.; Griffin, J.D.; Roberts, T.M. The cytokine-activated tyrosine kinase JAK2 activates Raf-1 in a p21ras-dependent manner. Proc. Natl. Acad. Sci. USA 1996, 93, 11681–11686. [Google Scholar] [CrossRef]
- Ritt, D.A.; Zhou, M.; Conrads, T.P.; Veenstra, T.D.; Copeland, T.D.; Morrison, D.K. CK2 Is a Component of the KSR1 Scaffold Complex that Contributes to Raf Kinase Activation. Curr. Biol. 2007, 17, 179–184. [Google Scholar] [CrossRef]
- Salzano, M.; Rusciano, M.R.; Russo, E.; Bifulco, M.; Postiglione, L.; Vitale, M. Calcium/calmodulin-dependent protein kinase II (CaMKII) phosphorylates Raf-1 at serine 338 and mediates Ras-stimulated Raf-1 activation. Cell Cycle 2012, 11, 2100–2106. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Baljuls, A.; Schmitz, W.; Mueller, T.; Zahedi, R.P.; Sickmann, A.; Hekman, M.; Rapp, U.R. Positive Regulation of A-RAF by Phosphorylation of Isoform-specific Hinge Segment and Identification of Novel Phosphorylation Sites. J. Biol. Chem. 2008, 283, 27239–27254. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.-H.; Guan, K.-L. Activation of B-Raf kinase requires phosphorylation of the conserved residues Thr598 and Ser601. EMBO J. 2000, 19, 5429–5439. [Google Scholar] [CrossRef] [PubMed]
- Chong, H.; Lee, J.; Guan, K. Positive and negative regulation of Raf kinase activity and function by phosphorylation. EMBO J. 2001, 20, 3716–3727. [Google Scholar] [CrossRef]
- Köhler, M.; Röring, M.; Schorch, B.; Heilmann, K.; Stickel, N.; Fiala, G.J.; Schmitt, L.C.; Braun, S.; Ehrenfeld, S.; Uhl, F.M.; et al. Activation loop phosphorylation regulates B-Raf in vivo and transformation by B-Raf mutants. EMBO J. 2016, 35, 143–161. [Google Scholar] [CrossRef]
- Köhler, M.; Brummer, T. B-Raf activation loop phosphorylation revisited. Cell Cycle 2016, 15, 1171–1173. [Google Scholar] [CrossRef]
- Varga, A.; Baccarini, M. Knock-in(g) RAF for a loop. EMBO J. 2016, 35, 118–120. [Google Scholar] [CrossRef]
- Grammatikakis, N.; Lin, J.; Grammatikakis, A.; Tsichlis, P.N.; Cochran, B.H. p50 cdc37 Acting in Concert with Hsp90 Is Required for Raf-1 Function. Mol. Cell. Biol. 1999, 19, 1661–1672. [Google Scholar] [CrossRef]
- Da Rocha Dias, S.; Friedlos, F.; Light, Y.; Springer, C.; Workman, P.; Marais, R. Activated B-RAF is an Hsp90 client protein that is targeted by the anticancer drug 17-allylamino-17-demethoxygeldanamycin. Cancer Res. 2005, 65, 10686–10691. [Google Scholar] [CrossRef]
- Dent, P.; Reardon, D.B.; Morrison, D.K.; Sturgill, T.W. Regulation of Raf-1 and Raf-1 mutants by Ras-dependent and Ras-independent mechanisms in vitro. Mol. Cell. Biol. 1995, 15, 4125–4135. [Google Scholar] [CrossRef]
- Fantl, W.J.; Muslin, A.J.; Kikuchi, A.; Martin, J.A.; MacNicol, A.M.; Grosst, R.W.; Williams, L.T. Activation of Raf-1 by 14-3-3 proteins. Nature 1994, 371, 612–614. [Google Scholar] [CrossRef] [PubMed]
- Freed, E.; Symons, M.; Macdonald, S.G.; McCormick, F.; Ruggieri, R. Binding of 14-3-3 proteins to the protein kinase Raf and effects on its activation. Science 1994, 265, 1713–1716. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Xia, K.; Pallas, D.C.; Cui, C.; Conroy, K.; Narsimhan, R.P.; Mamon, H.; Collier, R.J.; Roberts, T.M. Interaction of the protein kinase Raf-1 with 14-3-3 proteins. Science 1994, 266, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Mandal, A.K.; Lee, P.; Chen, J.A.; Nillegoda, N.; Heller, A.; DiStasio, S.; Oen, H.; Victor, J.; Nair, D.M.; Brodsky, J.L.; et al. Cdc37 has distinct roles in protein kinase quality control that protect nascent chains from degradation and promote posttranslational maturation. J. Cell Biol. 2007, 176, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Diedrich, B.; Rigbolt, K.T.; Röring, M.; Herr, R.; Kaeser-Pebernard, S.; Gretzmeier, C.; Murphy, R.F.; Brummer, T.; Dengjel, J. Discrete cytosolic macromolecular BRAF complexes exhibit distinct activities and composition. EMBO J. 2017, 36, 646–663. [Google Scholar] [CrossRef] [PubMed]
- Hunter, T.; Poon, R.Y.C. Cdc37: A protein kinase chaperone? Trends Cell Biol. 1997, 7, 157–161. [Google Scholar] [CrossRef]
- Grbovic, O.M.; Basso, A.D.; Sawai, A.; Ye, Q.; Friedlander, P.; Solit, D.; Rosen, N. V600E B-Raf requires the Hsp90 chaperone for stability and is degraded in response to Hsp90 inhibitors. Proc. Natl. Acad. Sci. USA 2006, 103, 57–62. [Google Scholar] [CrossRef]
- Smyth, T.; Paraiso, K.H.T.; Hearn, K.; Rodriguez-Lopez, A.M.; Munck, J.M.; Haarberg, H.E.; Sondak, V.K.; Thompson, N.T.; Azab, M.; Lyons, J.F.; et al. Inhibition of HSP90 by AT13387 Delays the Emergence of Resistance to BRAF Inhibitors and Overcomes Resistance to Dual BRAF and MEK Inhibition in Melanoma Models. Mol. Cancer Ther. 2014, 13, 2793–2804. [Google Scholar] [CrossRef]
- Paraiso, K.H.T.; Haarberg, H.E.; Wood, E.; Rebecca, V.W.; Chen, Y.A.; Xiang, Y.; Ribas, A.; Lo, R.S.; Weber, J.S.; Sondak, V.K.; et al. The HSP90 Inhibitor XL888 Overcomes BRAF Inhibitor Resistance Mediated through Diverse Mechanisms. Clin. Cancer Res. 2012, 18, 2502–2514. [Google Scholar] [CrossRef]
- Sullivan, R.J. Forestalling BRAF-Inhibitor Resistance in a Shocking Way. Clin. Cancer Res. 2018, 24, 5496–5498. [Google Scholar] [CrossRef]
- Kornfeld, K.; Hom, D.B.; Horvitz, H.R. The ksr-1 gene encodes a novel protein kinase involved in Ras-mediated signaling in C. elegans. Cell 1995, 83, 903–913. [Google Scholar] [CrossRef]
- Boudeau, J.; Miranda-Saavedra, D.; Barton, G.J.; Alessi, D.R. Emerging roles of pseudokinases. Trends Cell Biol. 2006, 16, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Michaud, N.R.; Therrien, M.; Cacace, A.; Edsall, L.C.; Spiegel, S.; Rubin, G.M.; Morrison, D.K. KSR stimulates Raf-1 activity in a kinase-independent manner. Proc. Natl. Acad. Sci. USA 1997, 94, 12792–12796. [Google Scholar] [CrossRef] [PubMed]
- Douziech, M.; Sahmi, M.; Laberge, G.; Therrien, M. A KSR/CNK complex mediated by HYP, a novel SAM domain-containing protein, regulates RAS-dependent RAF activation in Drosophila. Genes Dev. 2006, 20, 807–819. [Google Scholar] [CrossRef]
- Sundaram, M.; Han, M.; The, C. elegans ksr-1 gene encodes a novel raf-related kinase involved in Ras-mediated signal transduction. Cell 1995, 83, 889–901. [Google Scholar] [CrossRef]
- Therrien, M.; Chang, H.C.; Solomon, N.M.; Karim, F.D.; Wassarman, D.A.; Rubin, G.M. KSR, a novel protein kinase required for RAS signal transduction. Cell 1995, 83, 879–888. [Google Scholar] [CrossRef]
- Lavoie, H.; Sahmi, M.; Maisonneuve, P.; Marullo, S.A.; Thevakumaran, N.; Jin, T.; Kurinov, I.; Sicheri, F.; Therrien, M. MEK drives BRAF activation through allosteric control of KSR proteins. Nature 2018, 554, 549–553. [Google Scholar] [CrossRef]
- Brennan, D.F.; Dar, A.C.; Hertz, N.T.; Chao, W.C.H.; Burlingame, A.L.; Shokat, K.M.; Barford, D. A Raf-induced allosteric transition of KSR stimulates phosphorylation of MEK. Nature 2011, 472, 366–369. [Google Scholar] [CrossRef]
- Pennington, K.L.; Chan, T.Y.; Torres, M.P.; Andersen, J.L. The dynamic and stress-adaptive signaling hub of 14-3-3: Emerging mechanisms of regulation and context-dependent protein–protein interactions. Oncogene 2018, 37, 5587–5604. [Google Scholar] [CrossRef]
- Yaffe, M.B. How do 14-3-3 proteins work?—Gatekeeper phosphorylation and the molecular anvil hypothesis. FEBS Lett. 2002, 513, 53–57. [Google Scholar] [CrossRef]
- Freeman, A.K.; Morrison, D.K. 14-3-3 Proteins: Diverse functions in cell proliferation and cancer progression. Semin. Cell Dev. Biol. 2011, 22, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Mischak, H.; Seitz, T.; Janosch, P.; Eulitz, M.; Steen, H.; Schellerer, M.; Philipp, A.; Kolch, W. Negative regulation of Raf-1 by phosphorylation of serine 621. Mol. Cell. Biol. 1996, 16, 5409–5418. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Rawson, S.; Li, K.; Kim, B.-W.; Ficarro, S.B.; Pino, G.G.-D.; Sharif, H.; Marto, J.A.; Jeon, H.; Eck, M.J. Architecture of autoinhibited and active BRAF–MEK1–14-3-3 complexes. Nature 2019, 575, 545–550. [Google Scholar] [CrossRef] [PubMed]
- Cook, S.J.; McCormick, F. Inhibition by cAMP of Ras-dependent activation of Raf. Science 1993, 262, 1069–1072. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, A.S.; Pollock, C.; Steen, H.; Shaw, P.E.; Mischak, H.; Kolch, W. Cyclic AMP-dependent kinase regulates Raf-1 kinase mainly by phosphorylation of serine 259. Mol. Cell. Biol. 2002, 22, 3237–3246. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Dent, P.; Jelinek, T.; Wolfman, A.; Weber, M.J.; Sturgill, T.W. Inhibition of the EGF-activated MAP kinase signaling pathway by adenosine 3’,5’-monophosphate. Science 1993, 262, 1065–1069. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, S.; Moelling, K. Phosphorylation and Regulation of Raf by Akt (Protein Kinase B). Science 1999, 286, 1741–1744. [Google Scholar] [CrossRef]
- Rommel, C.; Clarke, B.A.; Zimmermann, S.; Nuñez, L.; Rossman, R.; Reid, K.; Moelling, K.; Yancopoulos, G.D.; Glass, D.J. Differentiation Stage-Specific Inhibition of the Raf-MEK-ERK Pathway by Akt. Science 1999, 286, 1738–1741. [Google Scholar] [CrossRef]
- Yuan, J.; Ng, W.H.; Yap, J.; Chia, B.; Huang, X.; Wang, M.; Hu, J. The AMPK inhibitor overcomes the paradoxical effect of RAF inhibitors through blocking phospho–Ser-621 in the C terminus of CRAF. J. Biol. Chem. 2018, 293, 14276–14284. [Google Scholar] [CrossRef]
- Sprenkle, A.B.; Davies, S.P.; Carling, D.; Hardie, D.G.; Sturgill, T.W. Identification of Raf-1 Ser621 kinase activity from NIH 3T3 cells as AMP-activated protein kinase. FEBS Lett. 1997, 403, 254–258. [Google Scholar] [CrossRef]
- Romano, D.; Nguyen, L.K.; Matallanas, D.; Halasz, M.; Doherty, C.; Kholodenko, B.N.; Kolch, W. Protein interaction switches coordinate Raf-1 and MST2/Hippo signalling. Nat. Cell Biol. 2014, 16, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Ng, W.H.; Tian, Z.; Yap, J.; Baccarini, M.; Chen, Z.; Hu, J. Activating mutations in MEK1 enhance homodimerization and promote tumorigenesis. Sci. Signal. 2018, 11, eaar6795. [Google Scholar] [CrossRef] [PubMed]
- Röring, M.; Herr, R.; Fiala, G.J.; Heilmann, K.; Braun, S.; Eisenhardt, A.E.; Halbach, S.; Capper, D.; von Deimling, A.; Schamel, W.W.; et al. Distinct requirement for an intact dimer interface in wild-type, V600E and kinase-dead B-Raf signalling. EMBO J. 2012, 31, 2629–2647. [Google Scholar] [CrossRef] [PubMed]
- Dent, P.; Haser, W.; Haystead, T.A.; Vincent, L.A.; Roberts, T.M.; Sturgill, T.W. Activation of mitogen-activated protein kinase kinase by v-Raf in NIH 3T3 cells and in vitro. Science 1992, 257, 1404–1407. [Google Scholar] [CrossRef] [PubMed]
- Haystead, C.M.M.; Wu, J.; Gregory, P.; Sturgill, T.W.; Haystead, T.A.J. Functional expression of a MAP kinase kinase in COS cells and recognition by an anti-STE7/byrl antibody. FEBS Lett. 1993, 317, 12–16. [Google Scholar] [CrossRef]
- Papin, C.; Denouel, A.; Calothy, G.; Eychène, A. Identification of signalling proteins interacting with B-Raf in the yeast two-hybrid system. Oncogene 1996, 12, 2213–2221. [Google Scholar]
- Papin, C.; Eychne, A.; Brunet, A.; Pages, G.; Pouyssegur, J.; Calothy, G.; Barnier, J.V. B-Raf protein isoforms interact with and phosphorylate Mek-1 on serine. Oncogene 1995, 10, 1647–1651. [Google Scholar]
- Corbalan-Garcia, S.; Yang, S.S.; Degenhardt, K.R.; Bar-Sagi, D. Identification of the mitogen-activated protein kinase phosphorylation sites on human Sos1 that regulate interaction with Grb2. Mol. Cell. Biol. 1996, 16, 5674–5682. [Google Scholar] [CrossRef]
- Saha, M.; Carriere, A.; Cheerathodi, M.; Zhang, X.; Lavoie, G.; Rush, J.; Roux, P.P.; Ballif, B.A. RSK phosphorylates SOS1 creating 14-3-3-docking sites and negatively regulating MAPK activation. Biochem. J. 2012, 447, 159–166. [Google Scholar] [CrossRef]
- Brummer, T.; Naegele, H.; Reth, M.; Misawa, Y. Identification of novel ERK-mediated feedback phosphorylation sites at the C-terminus of B-Raf. Oncogene 2003, 22, 8823–8834. [Google Scholar] [CrossRef]
- Dougherty, M.K.; Müller, J.; Ritt, D.A.; Zhou, M.; Zhou, X.Z.; Copeland, T.D.; Conrads, T.P.; Veenstra, T.D.; Lu, K.P.; Morrison, D.K. Regulation of Raf-1 by Direct Feedback Phosphorylation. Mol. Cell 2005, 17, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Ritt, D.A.; Monson, D.M.; Specht, S.I.; Morrison, D.K. Impact of Feedback Phosphorylation and Raf Heterodimerization on Normal and Mutant B-Raf Signaling. Mol. Cell. Biol. 2010, 30, 806–819. [Google Scholar] [CrossRef] [PubMed]
- Ritt, D.A.; Abreu-Blanco, M.T.; Bindu, L.; Durrant, D.E.; Zhou, M.; Specht, S.I.; Stephen, A.G.; Holderfield, M.; Morrison, D.K.; Abreu-blanco, T.; et al. Inhibition of Ras/Raf/MEK/ERK Pathway Signaling by a Stress-Induced Phospho-Regulatory Circuit. Mol. Cell 2016, 64, 875–887. [Google Scholar] [CrossRef] [PubMed]
- Holderfield, M.; Merritt, H.; Chan, J.; Wallroth, M.; Tandeske, L.; Zhai, H.; Tellew, J.; Hardy, S.; Hekmat-Nejad, M.; Stuart, D.D.; et al. RAF Inhibitors Activate the MAPK Pathway by Relieving Inhibitory Autophosphorylation. Cancer Cell 2013, 23, 594–602. [Google Scholar] [CrossRef]
- Tassin, T.C.; Benavides, D.R.; Plattner, F.; Nishi, A.; Bibb, J.A. Regulation of ERK kinase by MEK1 kinase inhibition in the brain. J. Biol. Chem. 2015, 290, 16319–16329. [Google Scholar] [CrossRef]
- Von Kriegsheim, A.; Pitt, A.; Grindlay, G.J.; Kolch, W.; Dhillon, A.S. Regulation of the Raf-MEK-ERK pathway by protein phosphatase 5. Nat. Cell Biol. 2006, 8, 1011–1016. [Google Scholar] [CrossRef]
- Huang, C.Y.; Tan, T.H. DUSPs, to MAP kinases and beyond. Cell Biosci. 2012, 2, 1. [Google Scholar] [CrossRef]
- Zhang, Z.; Kobayashi, S.; Borczuk, A.C.; Leidner, R.S.; LaFramboise, T.; Levine, A.D.; Halmos, B. Dual specificity phosphatase 6 (DUSP6) is an ETS-regulated negative feedback mediator of oncogenic ERK signaling in lung cancer cells. Carcinogenesis 2010, 31, 577–586. [Google Scholar] [CrossRef]
- Ekerot, M.; Stavridis, M.P.; Delavaine, L.; Mitchell, M.P.; Staples, C.; Owens, D.M.; Keenan, I.D.; Dickinson, R.J.; Storey, K.G.; Keyse, S.M. Negative-feedback regulation of FGF signalling by DUSP6/MKP-3 is driven by ERK1/2 and mediated by Ets factor binding to a conserved site within the DUSP6/MKP-3 gene promoter. Biochem. J. 2008, 412, 287–298. [Google Scholar] [CrossRef]
- Hacohen, N.; Kramer, S.; Sutherland, D.; Hiromi, Y.; Krasnow, M.A. sprouty Encodes a Novel Antagonist of FGF Signaling that Patterns Apical Branching of the Drosophila Airways. Cell 1998, 92, 253–263. [Google Scholar] [CrossRef]
- Yusoff, P.; Lao, D.-H.; Ong, S.H.; Wong, E.S.M.; Lim, J.; Lo, T.L.; Leong, H.F.; Fong, C.W.; Guy, G.R. Sprouty2 Inhibits the Ras/MAP Kinase Pathway by Inhibiting the Activation of Raf. J. Biol. Chem. 2002, 277, 3195–3201. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, A.; Taketomi, T.; Kato, R.; Saeki, K.; Nonami, A.; Sasaki, M.; Kuriyama, M.; Saito, N.; Shibuya, M.; Yoshimura, A. Mammalian Sprouty4 suppresses Ras-independent ERK activation by binding to Raf1. Nat. Cell Biol. 2003, 5, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Haigis, K.M.; Kendall, K.R.; Wang, Y.; Cheung, A.; Haigis, M.C.; Glickman, J.N.; Niwa-Kawakita, M.; Sweet-Cordero, A.; Sebolt-Leopold, J.; Shannon, K.M.; et al. Differential effects of oncogenic K-Ras and N-Ras on proliferation, differentiation and tumor progression in the colon. Nat. Genet. 2008, 40, 600–608. [Google Scholar] [CrossRef]
- Drosten, M.; Simón-Carrasco, L.; Hernández-Porras, I.; Lechuga, C.G.; Blasco, M.T.; Jacob, H.K.C.; Fabbiano, S.; Potenza, N.; Bustelo, X.R.; Guerra, C.; et al. H-Ras and K-Ras Oncoproteins Induce Different Tumor Spectra When Driven by the Same Regulatory Sequences. Cancer Res. 2017, 77, 707–718. [Google Scholar] [CrossRef]
- Scheffzek, K.; Ahmadian, M.R.; Kabsch, W.; Wiesmüller, L.; Lautwein, A.; Schmitz, F.; Wittinghofer, A. The Ras-RasGAP complex: Structural basis for GTPase activation and its loss in oncogenic ras mutants. Science 1997, 277, 333–338. [Google Scholar] [CrossRef]
- Rabara, D.; Tran, T.H.; Dharmaiah, S.; Stephens, R.M.; McCormick, F.; Simanshu, D.K.; Holderfield, M. KRAS G13D sensitivity to neurofibromin-mediated GTP hydrolysis. Proc. Natl. Acad. Sci. USA 2019, 116, 22122–22131. [Google Scholar] [CrossRef]
- Gremer, L.; Gilsbach, B.; Reza Ahmadian, M.; Wittinghofer, A. Fluoride complexes of oncogenic Ras mutants to study the Ras-RasGAP interaction. Biol. Chem. 2008, 389, 1163. [Google Scholar] [CrossRef]
- Fukushima, T.; Suzuki, S.; Mashiko, M.; Ohtake, T.; Endo, Y.; Takebayashi, Y.; Sekikawa, K.; Hagiwara, K.; Takenoshita, S. BRAF mutations in papillary carcinomas of the thyroid. Oncogene 2003, 22, 6455–6457. [Google Scholar] [CrossRef]
- Forbes, S.A.; Bindal, N.; Bamford, S.; Cole, C.; Kok, C.Y.; Beare, D.; Jia, M.; Shepherd, R.; Leung, K.; Menzies, A.; et al. COSMIC: Mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucl. Acids Res. 2010, 39, D945–D950. [Google Scholar] [CrossRef]
- Flaherty, K.T.; McArthur, G. BRAF, a target in melanoma. Cancer 2010, 116, 4902–4913. [Google Scholar] [CrossRef] [PubMed]
- Wan, P.T.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Project, C.G.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; et al. Mechanism of Activation of the RAF-ERK Signaling Pathway by Oncogenic Mutations of B-RAF. Cell 2004, 116, 855–867. [Google Scholar] [CrossRef]
- Lavoie, H.; Therrien, M. Regulation of RAF protein kinases in ERK signalling. Nat. Rev. Mol. Cell Biol. 2015, 16, 281–298. [Google Scholar] [CrossRef] [PubMed]
- Ikenoue, T.; Hikiba, Y.; Kanai, F.; Tanaka, Y.; Imamura, J.; Imamura, T.; Ohta, M.; Ijichi, H.; Tateishi, K.; Kawakami, T.; et al. Functional Analysis of Mutations within the Kinase Activation Segment of B-Raf in Human Colorectal Tumors. Cancer Res. 2003, 63, 8132–8137. [Google Scholar]
- Andreadi, C.; Cheung, L.-K.; Giblett, S.; Patel, B.; Jin, H.; Mercer, K.; Kamata, T.; Lee, P.; Williams, A.; McMahon, M.; et al. The intermediate-activity (L597V)BRAF mutant acts as an epistatic modifier of oncogenic RAS by enhancing signaling through the RAF/MEK/ERK pathway. Genes Dev. 2012, 26, 1945–1958. [Google Scholar] [CrossRef]
- Dahlman, K.B.; Xia, J.; Hutchinson, K.; Ng, C.; Hucks, D.; Jia, P.; Atefi, M.; Su, Z.; Branch, S.; Lyle, P.L.; et al. BRAF L597 Mutations in Melanoma Are Associated with Sensitivity to MEK Inhibitors. Cancer Discov. 2012, 2, 791–797. [Google Scholar] [CrossRef]
- Houben, R.; Becker, J.; Kappel, A.; Terheyden, P.; Brocker, E.-B.; Goetz, R.; Rapp, U. Constitutive activation of the Ras-Raf signaling pathway in metastatic melanoma is associated with poor prognosis. J. Carcinog. 2004, 3, 6. [Google Scholar] [CrossRef][Green Version]
- Ikenoue, T.; Hikiba, Y.; Kanai, F.; Aragaki, J.; Tanaka, Y.; Imamura, J.; Imamura, T.; Ohta, M.; Ijichi, H.; Tateishi, K.; et al. Different Effects of Point Mutations within the B-Raf Glycine-Rich Loop in Colorectal Tumors on Mitogen-Activated Protein/Extracellular Signal-Regulated Kinase Kinase/Extracellular Signal-Regulated Kinase and Nuclear Factor κB Pathway and Cellular Transfo. Cancer Res. 2004, 64, 3428–3435. [Google Scholar] [CrossRef]
- Noeparast, A.; Teugels, E.; Giron, P.; Verschelden, G.; Brakeleer, S. De; Decoster, L.; Grève, J. De Non-V600 BRAF mutations recurrently found in lung cancer predict sensitivity to the combination of Trametinib and Dabrafenib. Oncotarget 2016, 8, 60094–60108. [Google Scholar]
- Swofford, B.P.; Homsi, J. Uncommon BRAF Mutations Associated with Durable Response to Immunotherapy in Patients with Metastatic Melanoma. Case Rep. Oncol. Med. 2017, 2017, 8241624. [Google Scholar] [CrossRef]
- Smalley, K.S.M.; Xiao, M.; Villanueva, J.; Nguyen, T.K.; Flaherty, K.T.; Letrero, R.; Van Belle, P.; Elder, D.E.; Wang, Y.; Nathanson, K.L.; et al. CRAF inhibition induces apoptosis in melanoma cells with non-V600E BRAF mutations. Oncogene 2009, 28, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Torres, N.M.; Tao, A.; Gao, Y.; Luo, L.; Li, Q.; de Stanchina, E.; Abdel-Wahab, O.; Solit, D.B.; Poulikakos, P.I.; et al. BRAF Mutants Evade ERK-Dependent Feedback by Different Mechanisms that Determine Their Sensitivity to Pharmacologic Inhibition. Cancer Cell 2015, 28, 370–383. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T.W.; Kocialkowski, S.; Liu, L.; Pearson, D.M.; Bäcklund, L.M.; Ichimura, K.; Collins, V.P. Tandem Duplication Producing a Novel Oncogenic BRAF Fusion Gene Defines the Majority of Pilocytic Astrocytomas. Cancer Res. 2008, 68, 8673–8677. [Google Scholar] [CrossRef] [PubMed]
- Cin, H.; Meyer, C.; Herr, R.; Janzarik, W.G.; Lambert, S.; Jones, D.T.W.; Jacob, K.; Benner, A.; Witt, H.; Remke, M.; et al. Oncogenic FAM131B–BRAF fusion resulting from 7q34 deletion comprises an alternative mechanism of MAPK pathway activation in pilocytic astrocytoma. Acta Neuropathol. 2011, 121, 763–774. [Google Scholar] [CrossRef] [PubMed]
- Ciampi, R.; Knauf, J.A.; Kerler, R.; Gandhi, M.; Zhu, Z.; Nikiforova, M.N.; Rabes, H.M.; Fagin, J.A.; Nikiforov, Y.E. Oncogenic AKAP9-BRAF fusion is a novel mechanism of MAPK pathway activation in thyroid cancer. J. Clin. Investig. 2005, 115, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703. [Google Scholar] [CrossRef] [PubMed]
- Sarkozy, A.; Carta, C.; Moretti, S.; Zampino, G.; Digilio, M.C.; Pantaleoni, F.; Scioletti, A.P.; Esposito, G.; Cordeddu, V.; Lepri, F.; et al. Germline BRAF mutations in Noonan, LEOPARD, and cardiofaciocutaneous syndromes: Molecular diversity and associated phenotypic spectrum. Hum. Mutat. 2009, 30, 695–702. [Google Scholar] [CrossRef]
- Travers, T.; López, C.A.; Van, Q.N.; Neale, C.; Tonelli, M.; Stephen, A.G.; Gnanakaran, S. Molecular recognition of RAS/RAF complex at the membrane: Role of RAF cysteine-rich domain. Sci. Rep. 2018, 8, 8461. [Google Scholar] [CrossRef]
- Fetics, S.K.; Guterres, H.; Kearney, B.M.; Buhrman, G.; Ma, B.; Nussinov, R.; Mattos, C. Allosteric effects of the oncogenic rasq61l mutant on raf-RBD. Structure 2015, 23, 505–516. [Google Scholar] [CrossRef]
- Leicht, D.T.; Balan, V.; Kaplun, A.; Singh-Gupta, V.; Kaplun, L.; Dobson, M.; Tzivion, G. Raf kinases: Function, regulation and role in human cancer. Biochim. Biophys. Acta Mol. Cell Res. 2007, 1773, 1196–1212. [Google Scholar] [CrossRef]
- Molzan, M.; Schumacher, B.; Ottmann, C.; Baljuls, A.; Polzien, L.; Weyand, M.; Thiel, P.; Rose, R.; Rose, M.; Kuhenne, P.; et al. Impaired Binding of 14-3-3 to C-RAF in Noonan Syndrome Suggests New Approaches in Diseases with Increased Ras Signaling. Mol. Cell. Biol. 2010, 30, 4698–4711. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.A.; Furtado, L.V.; Betz, B.L.; Kiel, M.J.; Weigelin, H.C.; Lim, M.S.; Elenitoba-Johnson, K.S.J. High prevalence of somatic MAK1 mutations in BRAF V600E–negative Langerhans cell histiocytosis. Blood 2014, 124, 1655–1658. [Google Scholar] [CrossRef] [PubMed]
- Brenan, L.; Andreev, A.; Cohen, O.; Pantel, S.; Kamburov, A.; Cacchiarelli, D.; Persky, N.S.; Zhu, C.; Bagul, M.; Goetz, E.M.; et al. Phenotypic Characterization of a Comprehensive Set of MAPK1/ERK2 Missense Mutants. Cell Rep. 2016, 17, 1171–1183. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the undruggable RAS: Mission Possible? Nat. Rev. Drug Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548. [Google Scholar] [CrossRef]
- Fakih, M.; O’Neil, B.; Price, T.J.; Falchook, G.S.; Desai, J.; Kuo, J.; Govindan, R.; Rasmussen, E.; Morrow, P.K.H.; Ngang, J.; et al. Phase 1 study evaluating the safety, tolerability, pharmacokinetics (PK), and efficacy of AMG 510, a novel small molecule KRASG12C inhibitor, in advanced solid tumors. J. Clin. Oncol. 2019, 37, 3003. [Google Scholar] [CrossRef]
- Christensen, J.G.; Fell, J.B.; Marx, M.A.; Fischer, J.; Hallin, J.; Calinisan, A.; Baer, B.; Burkhard, M.; Blake, J.; Vigers, G.; et al. Abstract LB-271: Insight towards therapeutic susceptibility of KRAS mutant cancers from MRTX1257, a novel KRAS G12C mutant selective small molecule inhibitor. Cancer Res. 2019, 79. [Google Scholar] [CrossRef]
- Zeitouni, D.; Pylayeva-Gupta, Y.; Der, C.J.; Bryant, K.L. KRAS Mutant Pancreatic Cancer: No Lone Path to an Effective Treatment. Cancers 2016, 8, 45. [Google Scholar] [CrossRef]
- Gibbs, J.B.; Oliff, A.; Kohl, N.E. Farnesyltransferase inhibitors: Ras research yields a potential cancer therapeutic. Cell 1994, 77, 175–178. [Google Scholar] [CrossRef]
- Furfine, E.S.; Leban, J.J.; Landavazo, A.; Moomaw, J.F.; Casey, P.J. Protein farnesyltransferase: Kinetics of farnesyl pyrophosphate binding and product release. Biochemistry 1995, 34, 6857–6862. [Google Scholar] [CrossRef]
- Long, S.B.; Casey, P.J.; Beese, L.S. Cocrystal Structure of Protein Farnesyltransferase Complexed with a Farnesyl Diphosphate Substrate. Biochemistry 1998, 37, 9612–9618. [Google Scholar] [CrossRef] [PubMed]
- Long, S.B.; Casey, P.J.; Beese, L.S. Reaction path of protein farnesyltransferase at atomic resolution. Nature 2002, 419, 645–650. [Google Scholar] [CrossRef] [PubMed]
- Mijimolle, N.; Velasco, J.; Dubus, P.; Guerra, C.; Weinbaum, C.A.; Casey, P.J.; Campuzano, V.; Barbacid, M. Protein farnesyltransferase in embryogenesis, adult homeostasis, and tumor development. Cancer Cell 2005, 7, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Whyte, D.B.; Kirschmeier, P.; Hockenberry, T.N.; Nunez-Oliva, I.; James, L.; Catino, J.J.; Bishop, W.R.; Pai, J.-K. K- and N-Ras Are Geranylgeranylated in Cells Treated with Farnesyl Protein Transferase Inhibitors. J. Biol. Chem. 1997, 272, 14459–14464. [Google Scholar] [CrossRef] [PubMed]
- Fiordalisi, J.J.; Johnson, R.L., 2nd; Weinbaum, C.A.; Sakabe, K.; Chen, Z.; Casey, P.J.; Cox, A.D. High affinity for farnesyltransferase and alternative prenylation contribute individually to K-Ras4B resistance to farnesyltransferase inhibitors. J. Biol. Chem. 2003, 278, 41718–41727. [Google Scholar] [CrossRef] [PubMed]
- Berndt, N.; Hamilton, A.D.; Sebti, S.M. Targeting protein prenylation for cancer therapy. Nat. Rev. Cancer 2011, 11, 775–791. [Google Scholar] [CrossRef]
- Peterson, Y.K.; Kelly, P.; Weinbaum, C.A.; Casey, P.J. A Novel Protein Geranylgeranyltransferase-I Inhibitor with High Potency, Selectivity, and Cellular Activity. J. Biol. Chem. 2006, 281, 12445–12450. [Google Scholar] [CrossRef]
- Sjogren, A.-K.M.; Andersson, K.M.E.; Liu, M.; Cutts, B.A.; Karlsson, C.; Wahlstrom, A.M.; Dalin, M.; Weinbaum, C.; Casey, P.J.; Tarkowski, A.; et al. GGTase-I deficiency reduces tumor formation and improves survival in mice with K-RAS–induced lung cancer. J. Clin. Investig. 2007, 117, 1294–1304. [Google Scholar] [CrossRef]
- Lobell, R.B.; Liu, D.; Buser, C.A.; Davide, J.P.; DePuy, E.; Hamilton, K.; Koblan, K.S.; Lee, Y.; Mosser, S.; Motzel, S.L.; et al. Preclinical and Clinical Pharmacodynamic Assessment of L-778,123, a Dual Inhibitor of Farnesyl:Protein Transferase and Geranylgeranyl: Protein Transferase Type-I. Mol. Cancer Ther. 2002, 1, 747–758. [Google Scholar]
- Wahlstrom, A.M.; Cutts, B.A.; Liu, M.; Lindskog, A.; Karlsson, C.; Sjogren, A.-K.M.; Andersson, K.M.E.; Young, S.G.; Bergo, M.O. Inactivating Icmt ameliorates K-RAS–induced myeloproliferative disease. Blood 2008, 112, 1357–1365. [Google Scholar] [CrossRef]
- Bergman, J.A.; Hahne, K.; Song, J.; Hrycyna, C.A.; Gibbs, R.A. S-Farnesyl-Thiopropionic Acid Triazoles as Potent Inhibitors of Isoprenylcysteine Carboxyl Methyltransferase. ACS Med. Chem. Lett. 2012, 3, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Lau, H.Y.; Ramanujulu, P.M.; Guo, D.; Yang, T.; Wirawan, M.; Casey, P.J.; Go, M.-L.; Wang, M. An improved isoprenylcysteine carboxylmethyltransferase inhibitor induces cancer cell death and attenuates tumor growth in vivo. Cancer Biol. Ther. 2014, 15, 1280–1291. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Hossain, M.S.; Tan, W.; Coolman, B.; Zhou, J.; Liu, S.; Casey, P.J. Inhibition of isoprenylcysteine carboxylmethyltransferase induces autophagic-dependent apoptosis and impairs tumor growth. Oncogene 2010, 29, 4959–4970. [Google Scholar] [CrossRef] [PubMed]
- Athuluri-Divakar, S.K.; Vasquez-Del Carpio, R.; Dutta, K.; Baker, S.J.; Cosenza, S.C.; Basu, I.; Gupta, Y.K.; Reddy, M.V.R.; Ueno, L.; Hart, J.R.; et al. A Small Molecule RAS-Mimetic Disrupts RAS Association with Effector Proteins to Block Signaling. Cell 2016, 165, 643–655. [Google Scholar] [CrossRef] [PubMed]
- Keeton, A.B.; Salter, E.A.; Piazza, G.A. The RAS–Effector Interaction as a Drug Target. Cancer Res. 2017, 77, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Kato-Stankiewicz, J.; Hakimi, I.; Zhi, G.; Zhang, J.; Serebriiskii, I.; Guo, L.; Edamatsu, H.; Koide, H.; Menon, S.; Eckl, R.; et al. Inhibitors of Ras/Raf-1 interaction identified by two-hybrid screening revert Ras-dependent transformation phenotypes in human cancer cells. Proc. Natl. Acad. Sci. USA 2002, 99, 14398–14403. [Google Scholar] [CrossRef]
- Wang, W.; Fang, G.; Rudolph, J. Ras inhibition via direct Ras binding—Is there a path forward? Bioorg. Med. Chem. Lett. 2012, 22, 5766–5776. [Google Scholar] [CrossRef]
- Bryant, K.L.; Stalnecker, C.A.; Zeitouni, D.; Klomp, J.E.; Peng, S.; Tikunov, A.P.; Gunda, V.; Pierobon, M.; Waters, A.M.; George, S.D.; et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat. Med. 2019, 25, 628–640. [Google Scholar] [CrossRef]
- Kinsey, C.G.; Camolotto, S.A.; Boespflug, A.M.; Guillen, K.P.; Foth, M.; Truong, A.; Schuman, S.S.; Shea, J.E.; Seipp, M.T.; Yap, J.T.; et al. Protective autophagy elicited by RAF→MEK→ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat. Med. 2019, 25, 620–627. [Google Scholar] [CrossRef]
- Wang, M.; Tan, W.; Zhou, J.; Leow, J.; Go, M.; Lee, H.S.; Casey, P.J. A Small Molecule Inhibitor of Isoprenylcysteine Carboxymethyltransferase Induces Autophagic Cell Death in PC3 Prostate Cancer Cells. J. Biol. Chem. 2008, 283, 18678–18684. [Google Scholar] [CrossRef]
- Teh, J.T.; Zhu, W.L.; Ilkayeva, O.R.; Li, Y.; Gooding, J.; Casey, P.J.; Summers, S.A.; Newgard, C.B.; Wang, M. Isoprenylcysteine carboxylmethyltransferase regulates mitochondrial respiration and cancer cell metabolism. Oncogene 2015, 34, 3296–3304. [Google Scholar] [CrossRef] [PubMed]
- Manu, K.A.; Chai, T.F.; Teh, J.T.; Zhu, W.L.; Casey, P.J.; Wang, M. Inhibition of Isoprenylcysteine Carboxylmethyltransferase Induces Cell-Cycle Arrest and Apoptosis through p21 and p21-Regulated BNIP3 Induction in Pancreatic Cancer. Mol. Cancer Ther. 2017, 16, 914–923. [Google Scholar] [CrossRef] [PubMed]
- Thompson, N.; Adams, D.J.; Ranzani, M. Synthetic lethality: Emerging targets and opportunities in melanoma. Pigment Cell Melanoma Res. 2017, 30, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.P.; Zhao, D.; Sasik, R.; Luebeck, J.; Birmingham, A.; Bojorquez-Gomez, A.; Licon, K.; Klepper, K.; Pekin, D.; Beckett, A.N.; et al. Combinatorial CRISPR-Cas9 screens for de novo mapping of genetic interactions. Nat. Methods 2017, 14, 573–576. [Google Scholar] [CrossRef]
- Flaherty, K.; Puzanov, I.; Sosman, J.; Kim, K.; Ribas, A.; McArthur, G.; Lee, R.J.; Grippo, J.F.; Nolop, K.; Chapman, P. Phase I study of PLX4032: Proof of concept for V600E BRAF mutation as a therapeutic target in human cancer. J. Clin. Oncol. 2009, 27, 9000. [Google Scholar] [CrossRef]
- Tsai, J.; Lee, J.T.; Wang, W.; Zhang, J.; Cho, H.; Mamo, S.; Bremer, R.; Gillette, S.; Kong, J.; Haass, N.K.; et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc. Natl. Acad. Sci. USA 2008, 105, 3041–3046. [Google Scholar] [CrossRef]
- Hauschild, A.; Grob, J.-J.; Demidov, L.V.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Miller, W.H., Jr.; Kaempgen, E.; et al. Dabrafenib in BRAF mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012, 380, 358–365. [Google Scholar] [CrossRef]
- Li, Z.; Jiang, K.; Zhu, X.; Lin, G.; Song, F.; Zhao, Y.; Piao, Y.; Liu, J.; Cheng, W.; Bi, X.; et al. Encorafenib (LGX818), a potent BRAF inhibitor, induces senescence accompanied by autophagy in BRAFV600E melanoma cells. Cancer Lett. 2016, 370, 332–344. [Google Scholar] [CrossRef]
- Wang, L.; Leite de Oliveira, R.; Huijberts, S.; Bosdriesz, E.; Pencheva, N.; Brunen, D.; Bosma, A.; Song, J.Y.; Zevenhoven, J.; Los-de Vries, G.T.; et al. An Acquired Vulnerability of Drug-Resistant Melanoma with Therapeutic Potential. Cell 2018, 173, 1413–1425.e14. [Google Scholar] [CrossRef]
- Seghers, A.C.; Wilgenhof, S.; Lebbé, C.; Neyns, B. Successful rechallenge in two patients with BRAF-V600-mutant melanoma who experienced previous progression during treatment with a selective BRAF inhibitor. Melanoma Res. 2012, 22, 466–472. [Google Scholar] [CrossRef]
- Peng, S.-B.; Henry, J.R.; Kaufman, M.D.; Lu, W.-P.; Smith, B.D.; Vogeti, S.; Rutkoski, T.J.; Wise, S.; Chun, L.; Zhang, Y.; et al. Inhibition of RAF Isoforms and Active Dimers by LY3009120 Leads to Anti-tumor Activities in RAS or BRAF Mutant Cancers. Cancer Cell 2015, 28, 384–398. [Google Scholar] [CrossRef] [PubMed]
- Okaniwa, M.; Hirose, M.; Arita, T.; Yabuki, M.; Nakamura, A.; Takagi, T.; Kawamoto, T.; Uchiyama, N.; Sumita, A.; Tsutsumi, S.; et al. Discovery of a Selective Kinase Inhibitor (TAK-632) Targeting Pan-RAF Inhibition: Design, Synthesis, and Biological Evaluation of C-7-Substituted 1,3-Benzothiazole Derivatives. J. Med. Chem. 2013, 56, 6478–6494. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Alberta, J.A.; Pilarz, C.; Calligaris, D.; Chadwick, E.J.; Ramkissoon, S.H.; Ramkissoon, L.A.; Garcia, V.M.; Mazzola, E.; Goumnerova, L.; et al. A brain-penetrant RAF dimer antagonist for the noncanonical BRAF oncoprotein of pediatric low-grade astrocytomas. Neuro. Oncol. 2017, 19, 774–785. [Google Scholar] [CrossRef] [PubMed]
- Dean, E.J.; Banerji, U.; Girotti, R.; Niculescu-Duvaz, I.; Lopes, F.; Davies, L.; Niculescu-Duvaz, D.; Dhomen, N.; Ellis, S.; Ali, Z.; et al. A Phase 1 first-in-human trial to evaluate the safety and tolerability of CCT3833, an oral panRAF inhibitor, in patients with advanced solid tumours, including metastatic melanoma. J. Clin. Oncol. 2016, 34, TPS9597. [Google Scholar] [CrossRef]
- Saturno, G.; Lopes, F.; Girotti, M.R.; Niculescu-Duvaz, I.; Niculescu-Duvaz, D.; Zambon, A.; Davies, L.; Johnson, L.; Preece, N.; Viros, A.; et al. Therapeutic efficacy of the paradox-breaking panRAF and SRC drug CCT3833/BAL3833 in KRAS-driven cancer models. Eur. J. Cancer 2016, 61, S199. [Google Scholar] [CrossRef]
- Ramurthy, S.; Taft, B.R.; Aversa, R.J.; Barsanti, P.A.; Burger, M.T.; Lou, Y.; Nishiguchi, G.A.; Rico, A.; Setti, L.; Smith, A.; et al. Design and Discovery of N-(3-(2-(2-Hydroxyethoxy)-6-morpholinopyridin-4-yl)-4-methylphenyl)-2-(trifluoromethyl)isonicotinamide, a Selective, Efficacious, and Well-Tolerated RAF Inhibitor Targeting RAS Mutant Cancers: The Path to the Clinic. J. Med. Chem. 2019. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.E.; Subramanian, S.; Verhagen, J.; McBride, C.M.; Costales, A.; Sung, L.; Antonios-McCrea, W.; McKenna, M.; Louie, A.K.; Ramurthy, S.; et al. Discovery of RAF265: A Potent mut-B-RAF Inhibitor for the Treatment of Metastatic Melanoma. ACS Med. Chem. Lett. 2015, 6, 961–965. [Google Scholar] [CrossRef]
- Yao, Z.; Gao, Y.; Su, W.; Yaeger, R.; Tao, J.; Na, N.; Zhang, Y.; Zhang, C.; Rymar, A.; Tao, A.; et al. RAF inhibitor PLX8394 selectively disrupts BRAF dimers and RAS-independent BRAF-mutant-driven signaling. Nat. Med. 2019, 25, 284–291. [Google Scholar] [CrossRef]
- Tutuka, C.S.A.; Andrews, M.C.; Mariadason, J.M.; Ioannidis, P.; Hudson, C.; Cebon, J.; Behren, A. PLX8394, a new generation BRAF inhibitor, selectively inhibits BRAF in colonic adenocarcinoma cells and prevents paradoxical MAPK pathway activation. Mol. Cancer 2017, 16, 112. [Google Scholar] [CrossRef]
- Zhang, C.; Spevak, W.; Zhang, Y.; Burton, E.A.; Ma, Y.; Habets, G.; Zhang, J.; Lin, J.; Ewing, T.; Matusow, B.; et al. RAF inhibitors that evade paradoxical MAPK pathway activation. Nature 2015, 526. [Google Scholar] [CrossRef]
- Cheng, Y.; Tian, H. Current development status of MEK inhibitors. Molecules 2017, 22, 1551. [Google Scholar] [CrossRef] [PubMed]
- Welsh, S.J.; Corrie, P.G. Management of BRAF and MEK inhibitor toxicities in patients with metastatic melanoma. Ther. Adv. Med. Oncol. 2015, 7, 122–136. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.-F.; Qiu, H.-Y.; Zhu, H.-L. A patent review of BRAF inhibitors: 2013–2018. Expert Opin. Ther. Pat. 2019, 29, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Molina-Arcas, M.; Downward, J. How to fool a wonder drug: Truncate and dimerize. Cancer Cell 2012, 21, 7–9. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Degirmenci, U.; Wang, M.; Hu, J. Targeting Aberrant RAS/RAF/MEK/ERK Signaling for Cancer Therapy. Cells 2020, 9, 198. https://doi.org/10.3390/cells9010198
Degirmenci U, Wang M, Hu J. Targeting Aberrant RAS/RAF/MEK/ERK Signaling for Cancer Therapy. Cells. 2020; 9(1):198. https://doi.org/10.3390/cells9010198
Chicago/Turabian StyleDegirmenci, Ufuk, Mei Wang, and Jiancheng Hu. 2020. "Targeting Aberrant RAS/RAF/MEK/ERK Signaling for Cancer Therapy" Cells 9, no. 1: 198. https://doi.org/10.3390/cells9010198
APA StyleDegirmenci, U., Wang, M., & Hu, J. (2020). Targeting Aberrant RAS/RAF/MEK/ERK Signaling for Cancer Therapy. Cells, 9(1), 198. https://doi.org/10.3390/cells9010198