Hypoxia-Inducible Factor-1α: The Master Regulator of Endothelial Cell Senescence in Vascular Aging

 ,
,  , and
, and
Abstract
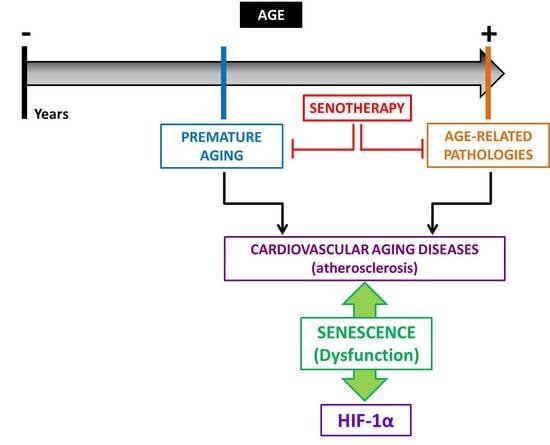
1. Aging, Vascular Senescence, and Cardiovascular Diseases
2. Hypoxia-Inducible Factors
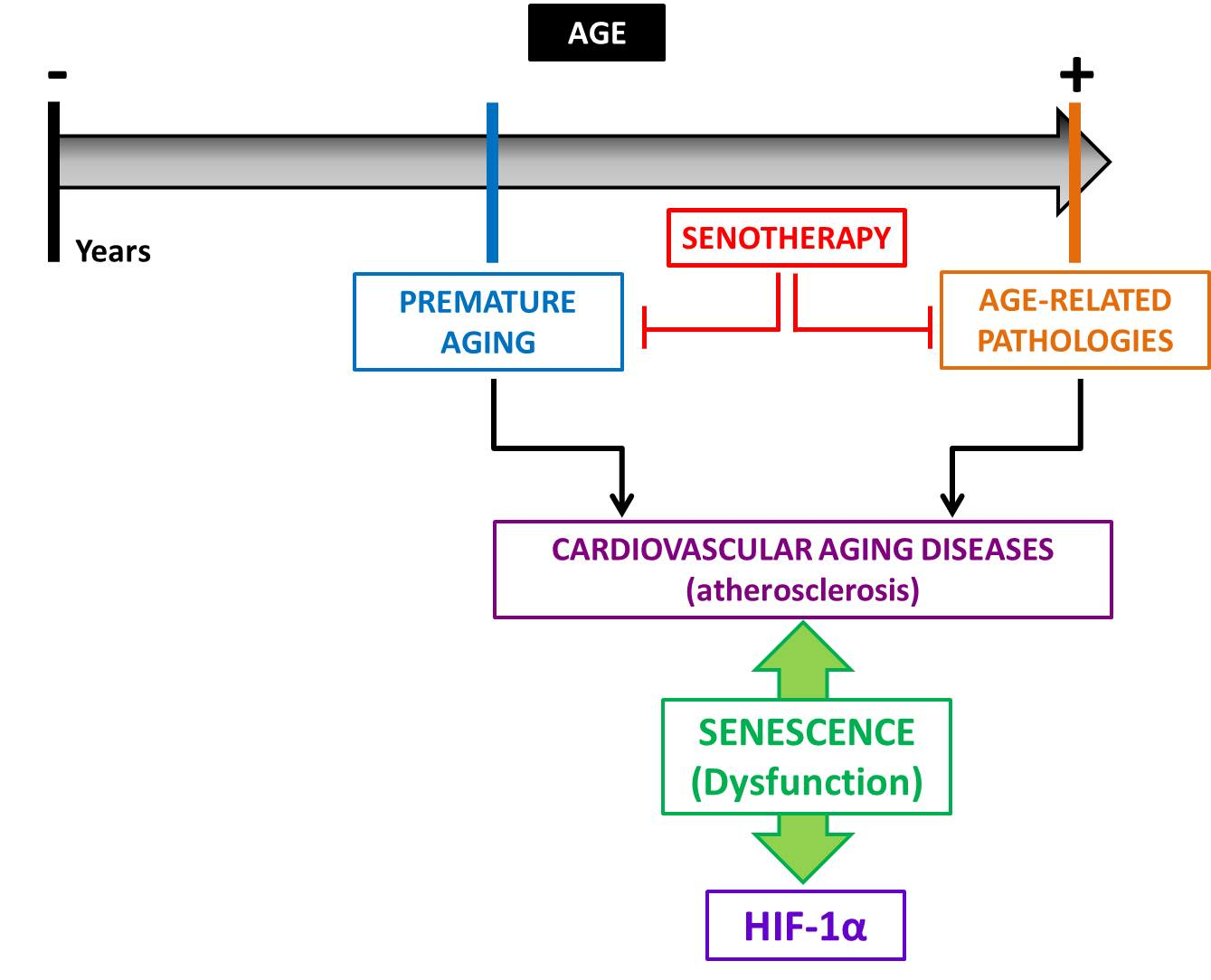
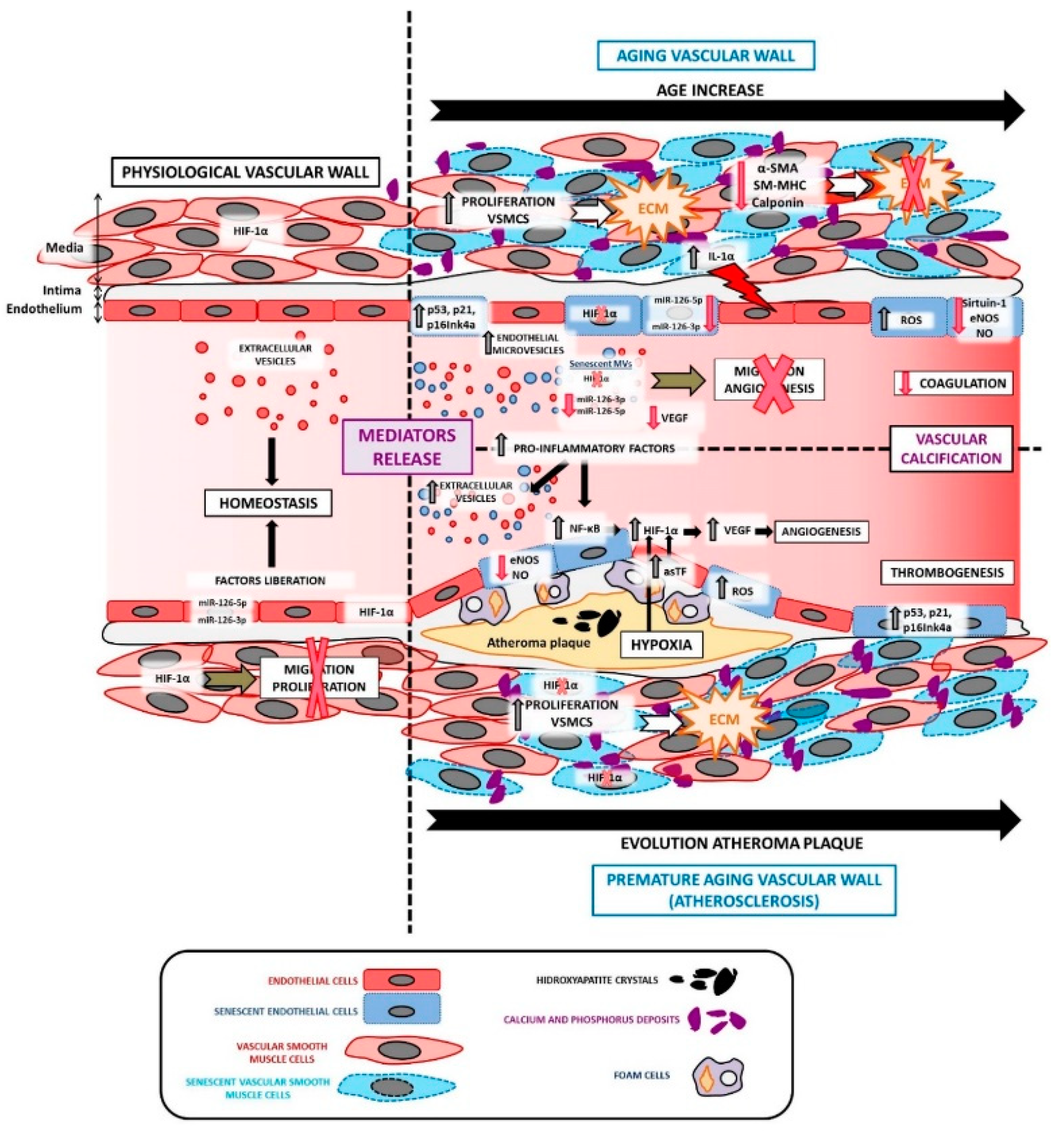
3. The Physio-Pathological Role of HIF in Aging
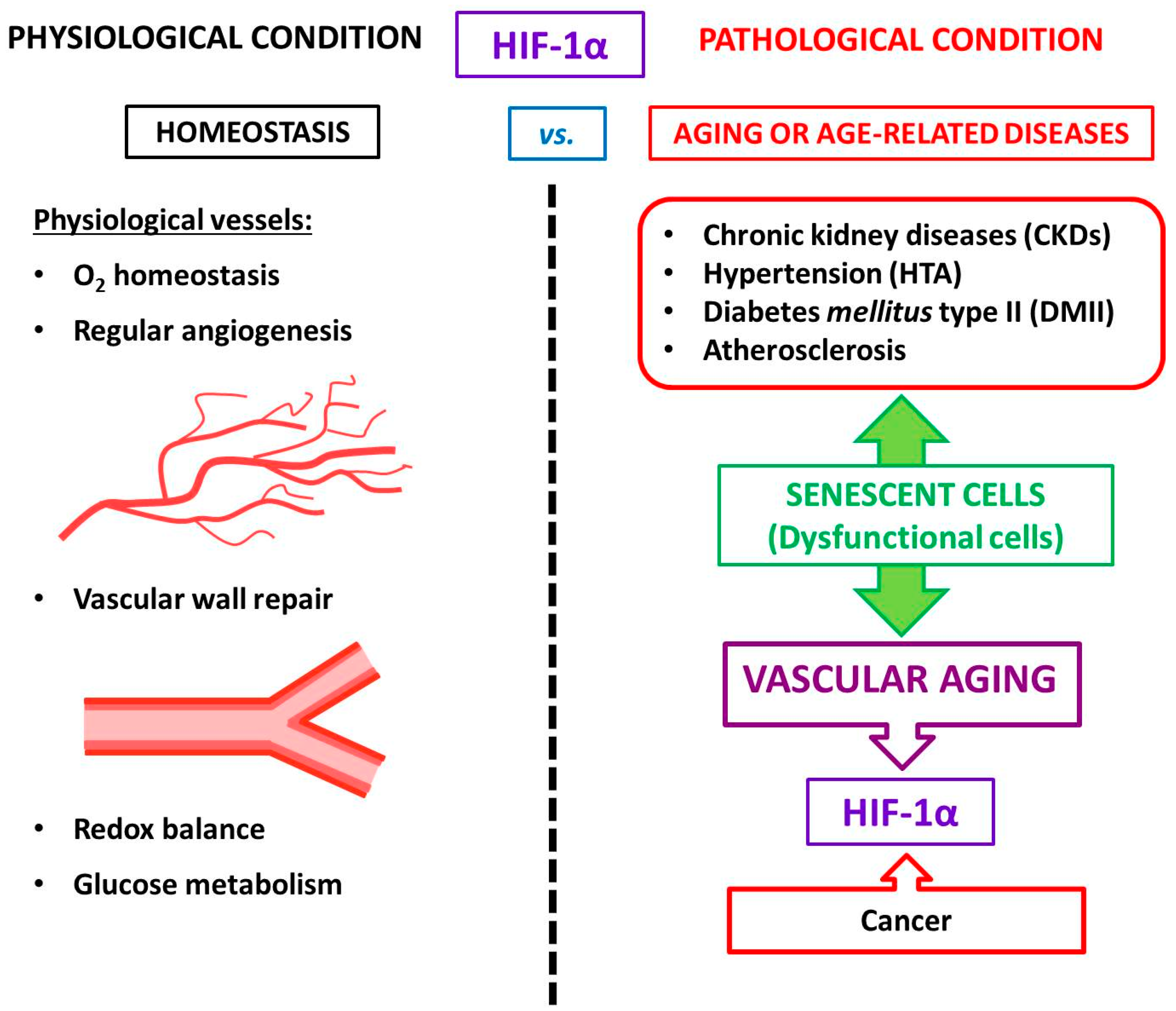
4. HIF-1α and Vascular Aging
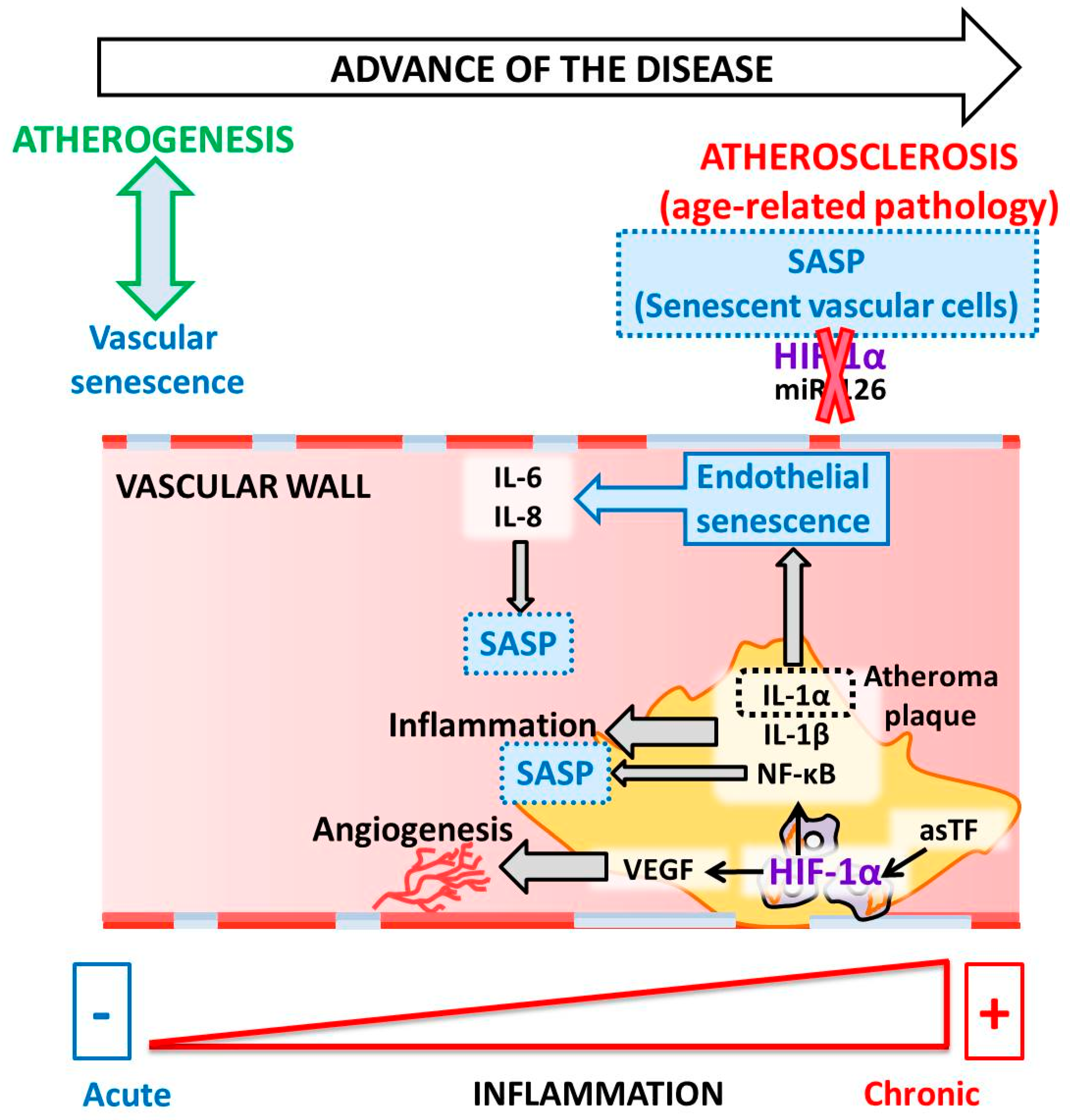
5. HIF and Atherosclerosis
6. HIF and Endothelial Cells
7. HIF and Vascular Smooth Muscle Cells
8. Conclusions
9. Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Schmitt, R. Senotherapy: Growing old and staying young? Pflugers Arch. 2017, 469, 1051–1059. [Google Scholar] [CrossRef]
- Scott, F.G. Developmental Biology, 6th ed.; Sinauer Associates: Sunderland, MA, USA, 2000. [Google Scholar]
- Kaluz, S.; Tan, C.; Van Meir, E.G. Taking a HIF pill for old age diseases? Aging 2018, 10, 290–292. [Google Scholar] [CrossRef]
- Head, T.; Daunert, S.; Goldschmidt-Clermont, P.J. The Aging Risk and Atherosclerosis: A Fresh Look at Arterial Homeostasis. Front. Genet. 2017, 8, 216. [Google Scholar] [CrossRef]
- Liu, Z.; Kuo, P.L.; Horvath, S.; Crimmins, E.; Ferrucci, L.; Levine, M. A new aging measure captures morbidity and mortality risk across diverse subpopulations from NHANES IV: A cohort study. PLoS Med. 2018, 15, e1002718. [Google Scholar] [CrossRef]
- Alique, M.; Luna, C.; Carracedo, J.; Ramirez, R. LDL biochemical modifications: A link between atherosclerosis and aging. Food Nutr. Res. 2015, 59. [Google Scholar] [CrossRef]
- Alique, M.; Ramírez-Carracedo, R.; Bodega, G.; Carracedo, J.; Ramírez, R. Senescent Microvesicles: A Novel Advance in Molecular Mechanisms of Atherosclerotic Calcification. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef]
- Katsuumi, G.; Shimizu, I.; Yoshida, Y.; Minamino, T. Vascular Senescence in Cardiovascular and Metabolic Diseases. Front. Cardiovasc. Med. 2018, 5, 18. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Eckardt, K.U. HIF Activation Against CVD in CKD: Novel Treatment Opportunities. Semin. Nephrol. 2018, 38, 267–276. [Google Scholar] [CrossRef]
- Wang, J.C.; Bennett, M. Aging and atherosclerosis: Mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ. Res. 2012, 111, 245–259. [Google Scholar] [CrossRef] [PubMed]
- Carrecedo, J.; Ramírez-Carracedo, R.; Alique, M.; Ramírez-Chamond, R. Endothelial Cell Senescence in the Pathogenesis of Endothelial Dysfunction. In Endothelial Dysfunction: Old Concepts and New Challenges; Intech Open: London, UK, 2018. [Google Scholar]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Coppe, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed]
- Ott, C.; Jung, T.; Grune, T.; Höhn, A. SIPS as a model to study age-related changes in proteolysis and aggregate formation. Mech. Ageing Dev. 2018, 170, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Yáñez-Mó, M.; Siljander, P.R.; Andreu, Z.; Zavec, A.B.; Borràs, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef]
- Dai, L.; Qureshi, A.R.; Witasp, A.; Lindholm, B.; Stenvinkel, P. Early Vascular Ageing and Cellular Senescence in Chronic Kidney Disease. Comput. Struct. Biotechnol. J. 2019, 17, 721–729. [Google Scholar] [CrossRef]
- Kirkland, J.L.; Tchkonia, T. Cellular Senescence: A Translational Perspective. EBioMedicine 2017, 21, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, I.; Minamino, T. Cellular senescence in cardiac diseases. J. Cardiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Hickson, L.J.; Langhi Prata, L.G.P.; Bobart, S.A.; Evans, T.K.; Giorgadze, N.; Hashmi, S.K.; Herrmann, S.M.; Jensen, M.D.; Jia, Q.; Jordan, K.L.; et al. Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine 2019, 47, 446–456. [Google Scholar] [CrossRef]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef]
- Justice, J.N.; Nambiar, A.M.; Tchkonia, T.; LeBrasseur, N.K.; Pascual, R.; Hashmi, S.K.; Prata, L.; Masternak, M.M.; Kritchevsky, S.B.; Musi, N.; et al. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. EBioMedicine 2019, 40, 554–563. [Google Scholar] [CrossRef]
- Palmer, A.K.; Xu, M.; Zhu, Y.; Pirtskhalava, T.; Weivoda, M.M.; Hachfeld, C.M.; Prata, L.G.; van Dijk, T.H.; Verkade, E.; Casaclang-Verzosa, G.; et al. Targeting senescent cells alleviates obesity-induced metabolic dysfunction. Aging Cell 2019, 18, e12950. [Google Scholar] [CrossRef]
- Zhang, P.; Kishimoto, Y.; Grammatikakis, I.; Gottimukkala, K.; Cutler, R.G.; Zhang, S.; Abdelmohsen, K.; Bohr, V.A.; Misra Sen, J.; Gorospe, M.; et al. Senolytic therapy alleviates Abeta-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat. Neurosci. 2019, 22, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Kirkland, J.L.; Tchkonia, T.; Zhu, Y.; Niedernhofer, L.J.; Robbins, P.D. The Clinical Potential of Senolytic Drugs. J. Am. Geriatr. Soc. 2017, 65, 2297–2301. [Google Scholar] [CrossRef] [PubMed]
- Roos, C.M.; Zhang, B.; Palmer, A.K.; Ogrodnik, M.B.; Pirtskhalava, T.; Thalji, N.M.; Hagler, M.; Jurk, D.; Smith, L.A.; Casaclang-Verzosa, G.; et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell 2016, 15, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Wang, B.; Ren, C.; Hu, J.; Greenberg, D.A.; Chen, T.; Xie, L.; Jin, K. Recent Progress in Vascular Aging: Mechanisms and Its Role in Age-related Diseases. Aging Dis. 2017, 8, 486–505. [Google Scholar] [CrossRef] [PubMed]
- Franklin, S.S.; Gustin, W.; Wong, N.D.; Larson, M.G.; Weber, M.A.; Kannel, W.B.; Levy, D. Hemodynamic patterns of age-related changes in blood pressure. The Framingham Heart Study. Circulation 1997, 96, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Minamino, T.; Komuro, I. Vascular cell senescence: Contribution to atherosclerosis. Circ. Res. 2007, 100, 15–26. [Google Scholar] [CrossRef]
- Alique, M.; Bodega, G.; Giannarelli, C.; Carracedo, J.; Ramírez, R. MicroRNA-126 regulates Hypoxia-Inducible Factor-1α which inhibited migration, proliferation, and angiogenesis in replicative endothelial senescence. Sci. Rep. 2019, 9, 7381. [Google Scholar] [CrossRef]
- Cerutti, C.; Edwards, L.J.; de Vries, H.E.; Sharrack, B.; Male, D.K.; Romero, I.A. MiR-126 and miR-126* regulate shear-resistant firm leukocyte adhesion to human brain endothelium. Sci. Rep. 2017, 7, 45284. [Google Scholar] [CrossRef]
- Villain, G.; Poissonnier, L.; Noueihed, B.; Bonfils, G.; Rivera, J.C.; Chemtob, S.; Soncin, F.; Mattot, V. miR-126-5p promotes retinal endothelial cell survival through SetD5 regulation in neurons. Development 2018, 145. [Google Scholar] [CrossRef]
- Xie, H.; Wang, F.; Chen, X.; Ying, H. miR-126 is essential for endothelial phenotype expression during endothelial differentiation in adipose-derived stem cells. Mol. Med. Rep. 2018, 17, 442–446. [Google Scholar] [CrossRef]
- Venkat, P.; Cui, C.; Chopp, M.; Zacharek, A.; Wang, F.; Landschoot-Ward, J.; Shen, Y.; Chen, J. MiR-126 Mediates Brain Endothelial Cell Exosome Treatment-Induced Neurorestorative Effects After Stroke in Type 2 Diabetes Mellitus Mice. Stroke 2019, 50, 2865–2874. [Google Scholar] [CrossRef]
- Yang, H.H.; Chen, Y.; Gao, C.Y.; Cui, Z.T.; Yao, J.M. Protective Effects of MicroRNA-126 on Human Cardiac Microvascular Endothelial Cells Against Hypoxia/Reoxygenation-Induced Injury and Inflammatory Response by Activating PI3K/Akt/eNOS Signaling Pathway. Cell Physiol. Biochem. 2017, 42, 506–518. [Google Scholar] [CrossRef] [PubMed]
- López-Lázaro, M. HIF-1: Hypoxia-inducible factor or dysoxia-inducible factor? FASEB J. 2006, 20, 828–832. [Google Scholar] [CrossRef] [PubMed]
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell 2010, 40, 294–309. [Google Scholar] [CrossRef] [PubMed]
- Bertout, J.A.; Patel, S.A.; Simon, M.C. The impact of O2 availability on human cancer. Nat. Rev. Cancer 2008, 8, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Rey, S.; Semenza, G.L. Hypoxia-inducible factor-1-dependent mechanisms of vascularization and vascular remodelling. Cardiovasc. Res. 2010, 86, 236–242. [Google Scholar] [CrossRef]
- Krock, B.L.; Skuli, N.; Simon, M.C. Hypoxia-induced angiogenesis: Good and evil. Genes Cancer 2011, 2, 1117–1133. [Google Scholar] [CrossRef]
- Giannarelli, C.; Alique, M.; Rodriguez, D.T.; Yang, D.K.; Jeong, D.; Calcagno, C.; Hutter, R.; Millon, A.; Kovacic, J.C.; Weber, T.; et al. Alternatively Spliced Tissue Factor Promotes Plaque Angiogenesis Through the Activation of Hypoxia-Inducible Factor-1 alpha and Vascular Endothelial Growth Factor Signaling. Circulation 2014, 130, 1274–1286. [Google Scholar] [CrossRef]
- Elvidge, G.P.; Glenny, L.; Appelhoff, R.J.; Ratcliffe, P.J.; Ragoussis, J.; Gleadle, J.M. Concordant regulation of gene expression by hypoxia and 2-oxoglutarate-dependent dioxygenase inhibition: The role of HIF-1alpha, HIF-2alpha, and other pathways. J. Biol. Chem. 2006, 281, 15215–15226. [Google Scholar] [CrossRef]
- Manalo, D.J.; Rowan, A.; Lavoie, T.; Natarajan, L.; Kelly, B.D.; Ye, S.Q.; Garcia, J.G.; Semenza, G.L. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood 2005, 105, 659–669. [Google Scholar] [CrossRef]
- Lim, C.S.; Kiriakidis, S.; Sandison, A.; Paleolog, E.M.; Davies, A.H. Hypoxia-inducible factor pathway and diseases of the vascular wall. J. Vasc. Surg. 2013, 58, 219–230. [Google Scholar] [CrossRef]
- Fernandez Esmerats, J.; Villa-Roel, N.; Kumar, S.; Gu, L.; Salim, M.T.; Ohh, M.; Taylor, W.R.; Nerem, R.M.; Yoganathan, A.P.; Jo, H. Disturbed Flow Increases UBE2C (Ubiquitin E2 Ligase C) via Loss of miR-483-3p, Inducing Aortic Valve Calcification by the pVHL (von Hippel-Lindau Protein) and HIF-1alpha (Hypoxia-Inducible Factor-1alpha) Pathway in Endothelial Cells. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 467–481. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Huang, R.T.; Hamanaka, R.B.; Krause, M.; Oh, M.J.; Kuo, C.H.; Nigdelioglu, R.; Meliton, A.Y.; Witt, L.; Dai, G.; et al. HIF-1alpha is required for disturbed flow-induced metabolic reprogramming in human and porcine vascular endothelium. Elife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Lei, L.; Desir, M.; Huang, Y.; Cleman, J.; Jiang, W.; Fernandez-Hernando, C.; Di Lorenzo, A.; Sessa, W.C.; Giordano, F.J. Smooth Muscle Hypoxia-Inducible Factor 1alpha Links Intravascular Pressure and Atherosclerosis--Brief Report. Arterioscle. Thromb. Vasc. Biol. 2016, 36, 442–445. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Xu, B.; Xuan, H.; Ge, Y.; Wang, Y.; Wang, L.; Huang, J.; Fu, W.; Michie, S.A.; Dalman, R.L. Hypoxia-inducible factor 1 in clinical and experimental aortic aneurysm disease. J. Vasc. Surg. 2018, 68, 1538–1550. [Google Scholar] [CrossRef] [PubMed]
- Imanishi, M.; Chiba, Y.; Tomita, N.; Matsunaga, S.; Nakagawa, T.; Ueno, M.; Yamamoto, K.; Tamaki, T.; Tomita, S. Hypoxia-Inducible Factor-1alpha in Smooth Muscle Cells Protects Against Aortic Aneurysms-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2158–2162. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. HIF-1: Mediator of physiological and pathophysiological responses to hypoxia. J. Appl. Physiol. 2000, 88, 1474–1480. [Google Scholar] [CrossRef]
- Abe, H.; Semba, H.; Takeda, N. The Roles of Hypoxia Signaling in the Pathogenesis of Cardiovascular Diseases. J. Atheroscler. Thromb. 2017, 24, 884–894. [Google Scholar] [CrossRef]
- Hölscher, M.; Schäfer, K.; Krull, S.; Farhat, K.; Hesse, A.; Silter, M.; Lin, Y.; Pichler, B.J.; Thistlethwaite, P.; El-Armouche, A.; et al. Unfavourable consequences of chronic cardiac HIF-1α stabilization. Cardiovasc. Res. 2012, 94, 77–86. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factor 1 and cardiovascular disease. Annu. Rev. Physiol. 2014, 76, 39–56. [Google Scholar] [CrossRef] [PubMed]
- Bosch-Marce, M.; Okuyama, H.; Wesley, J.B.; Sarkar, K.; Kimura, H.; Liu, Y.V.; Zhang, H.; Strazza, M.; Rey, S.; Savino, L.; et al. Effects of aging and hypoxia-inducible factor-1 activity on angiogenic cell mobilization and recovery of perfusion after limb ischemia. Circ. Res. 2007, 101, 1310–1318. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, G.; Di Giulio, C.; Rapino, C.; Rapino, M.; Antonucci, A.; Cataldi, A. p53 and p66 proteins compete for hypoxia-inducible factor 1 alpha stabilization in young and old rat hearts exposed to intermittent hypoxia. Gerontology 2006, 52, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Rapino, C.; Bianchi, G.; Di Giulio, C.; Centurione, L.; Cacchio, M.; Antonucci, A.; Cataldi, A. HIF-1alpha cytoplasmic accumulation is associated with cell death in old rat cerebral cortex exposed to intermittent hypoxia. Aging Cell 2005, 4, 177–185. [Google Scholar] [CrossRef]
- Mehta, R.; Steinkraus, K.A.; Sutphin, G.L.; Ramos, F.J.; Shamieh, L.S.; Huh, A.; Davis, C.; Chandler-Brown, D.; Kaeberlein, M. Proteasomal regulation of the hypoxic response modulates aging in C. elegans. Science 2009, 324, 1196–1198. [Google Scholar] [CrossRef]
- Leiser, S.F.; Begun, A.; Kaeberlein, M. HIF-1 modulates longevity and healthspan in a temperature-dependent manner. Aging Cell 2011, 10, 318–326. [Google Scholar] [CrossRef]
- Leiser, S.F.; Fletcher, M.; Begun, A.; Kaeberlein, M. Life-span extension from hypoxia in Caenorhabditis elegans requires both HIF-1 and DAF-16 and is antagonized by SKN-1. J. Gerontol. A Biol. Sci. Med. Sci. 2013, 68, 1135–1144. [Google Scholar] [CrossRef]
- Leiser, S.F.; Kaeberlein, M. The hypoxia-inducible factor HIF-1 functions as both a positive and negative modulator of aging. Biol. Chem. 2010, 391, 1131–1137. [Google Scholar] [CrossRef]
- Regina, C.; Panatta, E.; Candi, E.; Melino, G.; Amelio, I.; Balistreri, C.R.; Annicchiarico-Petruzzelli, M.; Di Daniele, N.; Ruvolo, G. Vascular ageing and endothelial cell senescence: Molecular mechanisms of physiology and diseases. Mech. Ageing Dev. 2016, 159, 14–21. [Google Scholar] [CrossRef]
- Rezvani, H.R.; Ali, N.; Serrano-Sanchez, M.; Dubus, P.; Varon, C.; Ged, C.; Pain, C.; Cario-André, M.; Seneschal, J.; Taïeb, A.; et al. Loss of epidermal hypoxia-inducible factor-1α accelerates epidermal aging and affects re-epithelialization in human and mouse. J. Cell Sci. 2011, 124, 4172–4183. [Google Scholar] [CrossRef]
- Ebersole, J.L.; Novak, M.J.; Orraca, L.; Martinez-Gonzalez, J.; Kirakodu, S.; Chen, K.C.; Stromberg, A.; Gonzalez, O.A. Hypoxia-inducible transcription factors, HIF1A and HIF2A, increase in aging mucosal tissues. Immunology 2018, 154, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Gardner, S.E.; Humphry, M.; Bennett, M.R.; Clarke, M.C. Senescent Vascular Smooth Muscle Cells Drive Inflammation Through an Interleukin-1α-Dependent Senescence-Associated Secretory Phenotype. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1963–1974. [Google Scholar] [CrossRef] [PubMed]
- Matthews, C.; Gorenne, I.; Scott, S.; Figg, N.; Kirkpatrick, P.; Ritchie, A.; Goddard, M.; Bennett, M. Vascular smooth muscle cells undergo telomere-based senescence in human atherosclerosis: Effects of telomerase and oxidative stress. Circ. Res. 2006, 99, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.G.; Ives, S.J.; Lesniewski, L.A.; Cawthon, R.M.; Andtbacka, R.H.; Noyes, R.D.; Richardson, R.S.; Donato, A.J. Age-related telomere uncapping is associated with cellular senescence and inflammation independent of telomere shortening in human arteries. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H251–H258. [Google Scholar] [CrossRef]
- Ortiz-Montero, P.; Londoño-Vallejo, A.; Vernot, J.P. Senescence-associated IL-6 and IL-8 cytokines induce a self- and cross-reinforced senescence/inflammatory milieu strengthening tumorigenic capabilities in the MCF-7 breast cancer cell line. Cell Commun. Signal 2017, 15, 17. [Google Scholar] [CrossRef]
- Young, A.P.; Schlisio, S.; Minamishima, Y.A.; Zhang, Q.; Li, L.; Grisanzio, C.; Signoretti, S.; Kaelin, W.G. VHL loss actuates a HIF-independent senescence programme mediated by Rb and p400. Nat. Cell Biol. 2008, 10, 361–369. [Google Scholar] [CrossRef]
- Rossman, M.J.; Kaplon, R.E.; Hill, S.D.; McNamara, M.N.; Santos-Parker, J.R.; Pierce, G.L.; Seals, D.R.; Donato, A.J. Endothelial cell senescence with aging in healthy humans: Prevention by habitual exercise and relation to vascular endothelial function. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H890–H895. [Google Scholar] [CrossRef]
- Montgomery, R.L.; van Rooij, E. MicroRNA regulation as a therapeutic strategy for cardiovascular disease. Curr. Drug Targets 2010, 11, 936–942. [Google Scholar] [CrossRef]
- Small, E.M.; Olson, E.N. Pervasive roles of microRNAs in cardiovascular biology. Nature 2011, 469, 336–342. [Google Scholar] [CrossRef]
- Hergenreider, E.; Heydt, S.; Tréguer, K.; Boettger, T.; Horrevoets, A.J.; Zeiher, A.M.; Scheffer, M.P.; Frangakis, A.S.; Yin, X.; Mayr, M.; et al. Atheroprotective communication between endothelial cells and smooth muscle cells through miRNAs. Nat. Cell Biol. 2012, 14, 249–256. [Google Scholar] [CrossRef]
- Zernecke, A.; Bidzhekov, K.; Noels, H.; Shagdarsuren, E.; Gan, L.; Denecke, B.; Hristov, M.; Köppel, T.; Jahantigh, M.N.; Lutgens, E.; et al. Delivery of microRNA-126 by apoptotic bodies induces CXCL12-dependent vascular protection. Sci. Signal 2009, 2, ra81. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Hernando, C.; Suárez, Y. MicroRNAs in endothelial cell homeostasis and vascular disease. Curr. Opin. Hematol. 2018, 25, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Van Solingen, C.; de Boer, H.C.; Bijkerk, R.; Monge, M.; van Oeveren-Rietdijk, A.M.; Seghers, L.; de Vries, M.R.; van der Veer, E.P.; Quax, P.H.; Rabelink, T.J.; et al. MicroRNA-126 modulates endothelial SDF-1 expression and mobilization of Sca-1+/Lin− progenitor cells in ischaemia. Cardiovasc. Res. 2011, 92, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Stellos, K.; Bigalke, B.; Langer, H.; Geisler, T.; Schad, A.; Kögel, A.; Pfaff, F.; Stakos, D.; Seizer, P.; Müller, I.; et al. Expression of stromal-cell-derived factor-1 on circulating platelets is increased in patients with acute coronary syndrome and correlates with the number of CD34+ progenitor cells. Eur. Heart J. 2009, 30, 584–593. [Google Scholar] [CrossRef]
- Qu, M.; Pan, J.; Shi, X.; Zhang, Z.; Tang, Y.; Yang, G. MicroRNA-126 is a prospective target for vascular disease. Neuroimmunol. Neuroinflam. 2018, 5, 10. [Google Scholar] [CrossRef]
- Zhou, J.; Li, Y.S.; Nguyen, P.; Wang, K.C.; Weiss, A.; Kuo, Y.C.; Chiu, J.J.; Shyy, J.Y.; Chien, S. Regulation of vascular smooth muscle cell turnover by endothelial cell-secreted microRNA-126: Role of shear stress. Circ. Res. 2013, 113, 40–51. [Google Scholar] [CrossRef]
- Hao, X.Z.; Fan, H.M. Identification of miRNAs as atherosclerosis biomarkers and functional role of miR-126 in atherosclerosis progression through MAPK signalling pathway. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 2725–2733. [Google Scholar]
- Semenza, G.L. Pharmacologic Targeting of Hypoxia-Inducible Factors. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 379–403. [Google Scholar] [CrossRef]
- Zhang, X.; Sarkar, K.; Rey, S.; Sebastian, R.; Andrikopoulou, E.; Marti, G.P.; Fox-Talbot, K.; Semenza, G.L.; Harmon, J.W. Aging impairs the mobilization and homing of bone marrow-derived angiogenic cells to burn wounds. J. Mol. Med. 2011, 89, 985–995. [Google Scholar] [CrossRef]
- Nilsson, P.M.; Lurbe, E.; Laurent, S. The early life origins of vascular ageing and cardiovascular risk: The EVA syndrome. J. Hypertens. 2008, 26, 1049–1057. [Google Scholar] [CrossRef]
- Shanahan, C.M. Mechanisms of vascular calcification in CKD-evidence for premature ageing? Nat. Rev. Nephrol. 2013, 9, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Shroff, R.; Long, D.A.; Shanahan, C. Mechanistic insights into vascular calcification in CKD. J. Am. Soc. Nephrol. 2013, 24, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Bedja, D.; Koitabashi, N.; Xing, D.; Chen, J.; Fox-Talbot, K.; Rouf, R.; Chen, S.; Steenbergen, C.; Harmon, J.W.; et al. Endothelial expression of hypoxia-inducible factor 1 protects the murine heart and aorta from pressure overload by suppression of TGF-β signaling. Proc. Natl. Acad. Sci. USA 2012, 109, E841–E850. [Google Scholar] [CrossRef] [PubMed]
- Yeo, E.J. Hypoxia and aging. Exp. Mol. Med. 2019, 51, 67. [Google Scholar] [CrossRef]
- Satoh, A.; Brace, C.S.; Rensing, N.; Cliften, P.; Wozniak, D.F.; Herzog, E.D.; Yamada, K.A.; Imai, S. Sirt1 extends life span and delays aging in mice through the regulation of Nk2 homeobox 1 in the DMH and LH. Cell Metab. 2013, 18, 416–430. [Google Scholar] [CrossRef]
- Kume, S.; Uzu, T.; Horiike, K.; Chin-Kanasaki, M.; Isshiki, K.; Araki, S.; Sugimoto, T.; Haneda, M.; Kashiwagi, A.; Koya, D. Calorie restriction enhances cell adaptation to hypoxia through Sirt1-dependent mitochondrial autophagy in mouse aged kidney. J. Clin. Investig. 2010, 120, 1043–1055. [Google Scholar] [CrossRef]
- Ryu, D.R.; Yu, M.R.; Kong, K.H.; Kim, H.; Kwon, S.H.; Jeon, J.S.; Han, D.C.; Noh, H. Sirt1-hypoxia-inducible factor-1α interaction is a key mediator of tubulointerstitial damage in the aged kidney. Aging Cell 2019, 18, e12904. [Google Scholar] [CrossRef]
- Laemmle, A.; Lechleiter, A.; Roh, V.; Schwarz, C.; Portmann, S.; Furer, C.; Keogh, A.; Tschan, M.P.; Candinas, D.; Vorburger, S.A.; et al. Inhibition of SIRT1 impairs the accumulation and transcriptional activity of HIF-1α protein under hypoxic conditions. PLoS ONE 2012, 7, e33433. [Google Scholar] [CrossRef]
- Yuan, Y.; Cruzat, V.F.; Newsholme, P.; Cheng, J.; Chen, Y.; Lu, Y. Regulation of SIRT1 in aging: Roles in mitochondrial function and biogenesis. Mech. Ageing Dev. 2016, 155, 10–21. [Google Scholar] [CrossRef]
- Kitada, M.; Ogura, Y.; Koya, D. The protective role of Sirt1 in vascular tissue: Its relationship to vascular aging and atherosclerosis. Aging 2016, 8, 2290–2307. [Google Scholar] [CrossRef]
- Orimo, M.; Minamino, T.; Miyauchi, H.; Tateno, K.; Okada, S.; Moriya, J.; Komuro, I. Protective role of SIRT1 in diabetic vascular dysfunction. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 889–894. [Google Scholar] [CrossRef] [PubMed]
- Ota, H.; Eto, M.; Ogawa, S.; Iijima, K.; Akishita, M.; Ouchi, Y. SIRT1/eNOS axis as a potential target against vascular senescence, dysfunction and atherosclerosis. J. Atheroscler. Thromb. 2010, 17, 431–435. [Google Scholar] [CrossRef]
- Zu, Y.; Liu, L.; Lee, M.Y.; Xu, C.; Liang, Y.; Man, R.Y.; Vanhoutte, P.M.; Wang, Y. SIRT1 promotes proliferation and prevents senescence through targeting LKB1 in primary porcine aortic endothelial cells. Circ. Res. 2010, 106, 1384–1393. [Google Scholar] [CrossRef] [PubMed]
- Rivard, A.; Berthou-Soulie, L.; Principe, N.; Kearney, M.; Curry, C.; Branellec, D.; Semenza, G.L.; Isner, J.M. Age-dependent defect in vascular endothelial growth factor expression is associated with reduced hypoxia-inducible factor 1 activity. J. Biol. Chem. 2000, 275, 29643–29647. [Google Scholar] [CrossRef] [PubMed]
- Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group. Lancet 1998, 352, 837–853. [Google Scholar]
- Macciò, A.; Madeddu, C. Management of anemia of inflammation in the elderly. Anemia 2012, 2012, 563251. [Google Scholar] [CrossRef]
- Nakayama, T.; Kurobe, H.; Sugasawa, N.; Kinoshita, H.; Higashida, M.; Matsuoka, Y.; Yoshida, Y.; Hirata, Y.; Sakata, M.; Maxfield, M.W.; et al. Role of macrophage-derived hypoxia-inducible factor (HIF)-1α as a mediator of vascular remodelling. Cardiovasc. Res. 2013, 99, 705–715. [Google Scholar] [CrossRef]
- Hansson, G.K. Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 2005, 352, 1685–1695. [Google Scholar] [CrossRef]
- Childs, B.G.; Baker, D.J.; Wijshake, T.; Conover, C.A.; Campisi, J.; van Deursen, J.M. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 2016, 354, 472–477. [Google Scholar] [CrossRef]
- Minamino, T.; Miyauchi, H.; Yoshida, T.; Ishida, Y.; Yoshida, H.; Komuro, I. Endothelial cell senescence in human atherosclerosis: Role of telomere in endothelial dysfunction. Circulation 2002, 105, 1541–1544. [Google Scholar] [CrossRef]
- Minamino, T.; Yoshida, T.; Tateno, K.; Miyauchi, H.; Zou, Y.; Toko, H.; Komuro, I. Ras induces vascular smooth muscle cell senescence and inflammation in human atherosclerosis. Circulation 2003, 108, 2264–2269. [Google Scholar] [CrossRef] [PubMed]
- Minamino, T.; Miyauchi, H.; Yoshida, T.; Tateno, K.; Kunieda, T.; Komuro, I. Vascular cell senescence and vascular aging. J. Mol. Cell Cardiol. 2004, 36, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Ferns, G.A.A.; Heikal, L. Hypoxia in Atherogenesis. Angiology 2017, 68, 472–493. [Google Scholar] [CrossRef] [PubMed]
- Vink, A.; Schoneveld, A.H.; Lamers, D.; Houben, A.J.; van der Groep, P.; van Diest, P.J.; Pasterkamp, G. HIF-1 alpha expression is associated with an atheromatous inflammatory plaque phenotype and upregulated in activated macrophages. Atherosclerosis 2007, 195, e69–e75. [Google Scholar] [CrossRef] [PubMed]
- Camaré, C.; Pucelle, M.; Nègre-Salvayre, A.; Salvayre, R. Angiogenesis in the atherosclerotic plaque. Redox Biol. 2017, 12, 18–34. [Google Scholar] [CrossRef]
- Parma, L.; Baganha, F.; Quax, P.H.A.; de Vries, M.R. Plaque angiogenesis and intraplaque hemorrhage in atherosclerosis. Eur. J. Pharmacol. 2017, 816, 107–115. [Google Scholar] [CrossRef]
- Ramkhelawon, B.; Yang, Y.; van Gils, J.M.; Hewing, B.; Rayner, K.J.; Parathath, S.; Guo, L.; Oldebeken, S.; Feig, J.L.; Fisher, E.A.; et al. Hypoxia induces netrin-1 and Unc5b in atherosclerotic plaques: Mechanism for macrophage retention and survival. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1180–1188. [Google Scholar] [CrossRef]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef]
- Mohr, T.; Haudek-Prinz, V.; Slany, A.; Grillari, J.; Micksche, M.; Gerner, C. Proteome profiling in IL-1beta and VEGF-activated human umbilical vein endothelial cells delineates the interlink between inflammation and angiogenesis. PLoS ONE 2017, 12, e0179065. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Mariotti, M.; Castiglioni, S.; Bernardini, D.; Maier, J.A. Interleukin 1 alpha is a marker of endothelial cellular senescent. Immun. Ageing 2006, 3, 4. [Google Scholar] [CrossRef] [PubMed]
- Apte, R.N.; Dotan, S.; Elkabets, M.; White, M.R.; Reich, E.; Carmi, Y.; Song, X.; Dvozkin, T.; Krelin, Y.; Voronov, E. The involvement of IL-1 in tumorigenesis, tumor invasiveness, metastasis and tumor-host interactions. Cancer Metastasis. Rev. 2006, 25, 387–408. [Google Scholar] [CrossRef] [PubMed]
- Orjalo, A.V.; Bhaumik, D.; Gengler, B.K.; Scott, G.K.; Campisi, J. Cell surface-bound IL-1alpha is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc. Natl. Acad. Sci. USA 2009, 106, 17031–17036. [Google Scholar] [CrossRef] [PubMed]
- Zouein, F.A.; Booz, G.W.; Altara, R. STAT3 and Endothelial Cell-Cardiomyocyte Dialog in Cardiac Remodeling. Front. Cardiovasc. Med. 2019, 6, 50. [Google Scholar] [CrossRef]
- Chen, Q.; Lv, J.; Yang, W.; Xu, B.; Wang, Z.; Yu, Z.; Wu, J.; Yang, Y.; Han, Y. Targeted inhibition of STAT3 as a potential treatment strategy for atherosclerosis. Theranostics 2019, 9, 6424–6442. [Google Scholar] [CrossRef]
- Ribeiro, S.; Belo, L.; Reis, F.; Santos-Silva, A. The HIF System Response to ESA Therapy in CKD-Anemia. In Hypoxia and Human Diseases; InTech Open: London, UK, 2017. [Google Scholar]
- Cao, L.; Li, W.; Kim, S.; Brodie, S.G.; Deng, C.X. Senescence, aging, and malignant transformation mediated by p53 in mice lacking the Brca1 full-length isoform. Genes Dev. 2003, 17, 201–213. [Google Scholar] [CrossRef]
- Lacolley, P.; Regnault, V.; Avolio, A.P. Smooth muscle cell and arterial aging: Basic and clinical aspects. Cardiovasc. Res. 2018, 114, 513–528. [Google Scholar] [CrossRef]
- Akhtar, S.; Hartmann, P.; Karshovska, E.; Rinderknecht, F.A.; Subramanian, P.; Gremse, F.; Grommes, J.; Jacobs, M.; Kiessling, F.; Weber, C.; et al. Endothelial Hypoxia-Inducible Factor-1α Promotes Atherosclerosis and Monocyte Recruitment by Upregulating MicroRNA-19a. Hypertension 2015, 66, 1220–1226. [Google Scholar] [CrossRef]
- Aarup, A.; Pedersen, T.X.; Junker, N.; Christoffersen, C.; Bartels, E.D.; Madsen, M.; Nielsen, C.H.; Nielsen, L.B. Hypoxia-Inducible Factor-1α Expression in Macrophages Promotes Development of Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1782–1790. [Google Scholar] [CrossRef]
- Sage, A.P.; Tintut, Y.; Demer, L.L. Regulatory mechanisms in vascular calcification. Nat. Rev. Cardiol. 2010, 7, 528–536. [Google Scholar] [CrossRef]
- Durham, A.L.; Speer, M.Y.; Scatena, M.; Giachelli, C.M.; Shanahan, C.M. Role of smooth muscle cells in vascular calcification: Implications in atherosclerosis and arterial stiffness. Cardiovasc. Res. 2018, 114, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Pescatore, L.A.; Gamarra, L.F.; Liberman, M. Multifaceted Mechanisms of Vascular Calcification in Aging. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1307–1316. [Google Scholar] [CrossRef] [PubMed]
- Bodega, G.; Alique, M.; Bohórquez, L.; Ciordia, S.; Mena, M.C.; Ramírez, M.R. The Antioxidant Machinery of Young and Senescent Human Umbilical Vein Endothelial Cells and Their Microvesicles. Oxid. Med. Cell Longev. 2017, 2017, 7094781. [Google Scholar] [CrossRef] [PubMed]
- Bodega, G.; Alique, M.; Bohórquez, L.; Morán, M.; Magro, L.; Puebla, L.; Ciordia, S.; Mena, M.C.; Arza, E.; Ramirez, M.R. Young and Especially Senescent Endothelial Microvesicles Produce NADPH: The Fuel for Their Antioxidant Machinery. Oxid. Med. Cell. Longev. 2018, 2018, 12. [Google Scholar] [CrossRef]
- Bodega, G.; Alique, M.; Puebla, L.; Carracedo, J.; Ramírez, R.M. Microvesicles: ROS scavengers and ROS producers. J. Extracell. Vesicles 2019, 8, 1626654. [Google Scholar] [CrossRef]
- Van der Vorst, E.P.C.; de Jong, R.J.; Donners, M.M.P.C. Message in a Microbottle: Modulation of Vascular Inflammation and Atherosclerosis by Extracellular Vesicles. Front. Cardiovasc. Med. 2018, 5, 2. [Google Scholar] [CrossRef]
- Boulanger, C.M.; Loyer, X.; Rautou, P.E.; Amabile, N. Extracellular vesicles in coronary artery disease. Nat. Rev. Cardiol. 2017, 14, 259–272. [Google Scholar] [CrossRef]
- Nomura, S. Extracellular vesicles and blood diseases. Int. J. Hematol. 2017, 105, 392–405. [Google Scholar] [CrossRef]
- New, S.E.; Aikawa, E. Role of extracellular vesicles in de novo mineralization: An additional novel mechanism of cardiovascular calcification. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1753–1758. [Google Scholar] [CrossRef]
- Hwang, H.V.; Lin, Y.; Rebuffatti, M.N.; Tran, D.T.; Lee, L.; Gomes, A.V.; Li, C.S.; Knowlton, A.A. Impaired proteostasis in senescent vascular endothelial cells: A perspective on estrogen and oxidative stress in the aging vasculature. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H421–H429. [Google Scholar] [CrossRef]
- Alique, M.; Ruíz-Torres, M.P.; Bodega, G.; Noci, M.V.; Troyano, N.; Bohórquez, L.; Luna, C.; Luque, R.; Carmona, A.; Carracedo, J.; et al. Microvesicles from the plasma of elderly subjects and from senescent endothelial cells promote vascular calcification. Aging 2017, 9, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Shimizu, I.; Katsuumi, G.; Jiao, S.; Suda, M.; Hayashi, Y.; Minamino, T. p53-Induced inflammation exacerbates cardiac dysfunction during pressure overload. J. Mol. Cell Cardiol. 2015, 85, 183–198. [Google Scholar] [CrossRef] [PubMed]
- Déry, M.A.; Michaud, M.D.; Richard, D.E. Hypoxia-inducible factor 1: Regulation by hypoxic and non-hypoxic activators. Int. J. Biochem. Cell Biol. 2005, 37, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Hewitson, K.S.; Schofield, C.J. The HIF pathway as a therapeutic target. Drug Discov. Today 2004, 9, 704–711. [Google Scholar] [CrossRef]
- Carbajo-Pescador, S.; Ordoñez, R.; Benet, M.; Jover, R.; García-Palomo, A.; Mauriz, J.L.; González-Gallego, J. Inhibition of VEGF expression through blockade of Hif1α and STAT3 signalling mediates the anti-angiogenic effect of melatonin in HepG2 liver cancer cells. Br. J. Cancer 2013, 109, 83–91. [Google Scholar] [CrossRef]
- Cesselli, D.; Aleksova, A.; Sponga, S.; Cervellin, C.; Di Loreto, C.; Tell, G.; Beltrami, A.P. Cardiac Cell Senescence and Redox Signaling. Front. Cardiovasc. Med. 2017, 4, 38. [Google Scholar] [CrossRef]
- Jansen, F.; Yang, X.; Hoelscher, M.; Cattelan, A.; Schmitz, T.; Proebsting, S.; Wenzel, D.; Vosen, S.; Franklin, B.S.; Fleischmann, B.K.; et al. Endothelial microparticle-mediated transfer of MicroRNA-126 promotes vascular endothelial cell repair via SPRED1 and is abrogated in glucose-damaged endothelial microparticles. Circulation 2013, 128, 2026–2038. [Google Scholar] [CrossRef]
- Schurgers, L.J.; Akbulut, A.C.; Kaczor, D.M.; Halder, M.; Koenen, R.R.; Kramann, R. Initiation and Propagation of Vascular Calcification Is Regulated by a Concert of Platelet- and Smooth Muscle Cell-Derived Extracellular Vesicles. Front. Cardiovasc. Med. 2018, 5, 36. [Google Scholar] [CrossRef]
- Sinha, S.; Iyer, D.; Granata, A. Embryonic origins of human vascular smooth muscle cells: Implications for in vitro modeling and clinical application. Cell Mol. Life Sci. 2014, 71, 2271–2288. [Google Scholar] [CrossRef]
- Pfaltzgraff, E.R.; Bader, D.M. Heterogeneity in vascular smooth muscle cell embryonic origin in relation to adult structure, physiology, and disease. Dev. Dyn. 2015, 244, 410–416. [Google Scholar] [CrossRef]
- Bennett, M.R.; Sinha, S.; Owens, G.K. Vascular Smooth Muscle Cells in Atherosclerosis. Circ. Res. 2016, 118, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Alexander, M.R.; Owens, G.K. Epigenetic control of smooth muscle cell differentiation and phenotypic switching in vascular development and disease. Annu. Rev. Physiol. 2012, 74, 13–40. [Google Scholar] [CrossRef] [PubMed]
- Lacolley, P.; Regnault, V.; Nicoletti, A.; Li, Z.; Michel, J.B. The vascular smooth muscle cell in arterial pathology: A cell that can take on multiple roles. Cardiovasc. Res. 2012, 95, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Shroff, R.C.; Shanahan, C.M. The vascular biology of calcification. Semin. Dial. 2007, 20, 103–109. [Google Scholar] [CrossRef]
- Wang, J.; Uryga, A.K.; Reinhold, J.; Figg, N.; Baker, L.; Finigan, A.; Gray, K.; Kumar, S.; Clarke, M.; Bennett, M. Vascular Smooth Muscle Cell Senescence Promotes Atherosclerosis and Features of Plaque Vulnerability. Circulation 2015, 132, 1909–1919. [Google Scholar] [CrossRef]
- Uryga, A.K.; Bennett, M.R. Ageing induced vascular smooth muscle cell senescence in atherosclerosis. J. Physiol. 2016, 594, 2115–2124. [Google Scholar] [CrossRef]
- Liu, K.; Fang, C.; Shen, Y.; Liu, Z.; Zhang, M.; Ma, B.; Pang, X. Hypoxia-inducible factor 1a induces phenotype switch of human aortic vascular smooth muscle cell through PI3K/AKT/AEG-1 signaling. Oncotarget 2017, 8, 33343–33352. [Google Scholar] [CrossRef]
- Afzal, T.A.; Luong, L.A.; Chen, D.; Zhang, C.; Yang, F.; Chen, Q.; An, W.; Wilkes, E.; Yashiro, K.; Cutillas, P.R.; et al. NCK Associated Protein 1 Modulated by miRNA-214 Determines Vascular Smooth Muscle Cell Migration, Proliferation, and Neointima Hyperplasia. J. Am. Heart Assoc. 2016, 5. [Google Scholar] [CrossRef]
- Kleefeldt, F.; Bömmel, H.; Broede, B.; Thomsen, M.; Pfeiffer, V.; Wörsdörfer, P.; Karnati, S.; Wagner, N.; Rueckschloss, U.; Ergün, S. Aging-related carcinoembryonic antigen-related cell adhesion molecule 1 signaling promotes vascular dysfunction. Aging Cell 2019, 18, e13025. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HIF Levels | Observations | Reference | |
|---|---|---|---|
| Pathology/Effect | Tissue/Cell Type | ||
| ↑ | Ischemic cardiovascular disease | Ischemic limb (ischemic calf muscle) (young WT mice) | [54] |
| ↓ | Ischemic limb ischemic calf muscle (aging WT mice) | ||
| ↓ | Vascular remodeling | Femoral artery ligation in aging mice | [53] |
| ↓ | Femoral artery ligation in Hif1a+/− mice | ||
| ↑ (short-term) | Heart homeostasis | Heart | [52] |
| ↑ (long-term) | Cardiomyopathy | ||
| ↓ | Atherosclerosis (Premature aging disease) | Pathogenesis of plaques (Promote angiogenesis, production of factor pro-atherosclerosis and recruitment of inflammatory cells) | [3] |
| ↓ | Replicative senescence in vitro | Endothelial cells | [29] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alique, M.; Sánchez-López, E.; Bodega, G.; Giannarelli, C.; Carracedo, J.; Ramírez, R. Hypoxia-Inducible Factor-1α: The Master Regulator of Endothelial Cell Senescence in Vascular Aging. Cells 2020, 9, 195. https://doi.org/10.3390/cells9010195
Alique M, Sánchez-López E, Bodega G, Giannarelli C, Carracedo J, Ramírez R. Hypoxia-Inducible Factor-1α: The Master Regulator of Endothelial Cell Senescence in Vascular Aging. Cells. 2020; 9(1):195. https://doi.org/10.3390/cells9010195
Chicago/Turabian StyleAlique, Matilde, Elsa Sánchez-López, Guillermo Bodega, Chiara Giannarelli, Julia Carracedo, and Rafael Ramírez. 2020. "Hypoxia-Inducible Factor-1α: The Master Regulator of Endothelial Cell Senescence in Vascular Aging" Cells 9, no. 1: 195. https://doi.org/10.3390/cells9010195
APA StyleAlique, M., Sánchez-López, E., Bodega, G., Giannarelli, C., Carracedo, J., & Ramírez, R. (2020). Hypoxia-Inducible Factor-1α: The Master Regulator of Endothelial Cell Senescence in Vascular Aging. Cells, 9(1), 195. https://doi.org/10.3390/cells9010195