Visfatin Induces Senescence of Human Dental Pulp Cells


Abstract

1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Tissue Samples and Immunohistochemistry
2.3. Cell Culture
2.4. Gene Silencing by siRNA
2.5. RT-PCR
2.6. Western Blot Analysis
2.7. SA-β-Galactosidase Staining Assay
2.8. NADP/NADPH Assay
2.9. Immunocytochemistry
2.10. Statistical Analysis
3. Results
3.1. Visfatin Levels Increase with Increasing Age in Human Dental Pulp
3.2. Visfatin Expression is Upregulated in Premature Senescent Dental Pulp Cells
3.3. Visfatin Silencing Delays Cellular Senescence
3.4. Visfatin Treatment Accelerates Cellular Senescence
3.5. FK866 Suppresses Senescence Induced by Visfatin
3.6. Telomere Damage Mediates Visfatin-Induced Cellular Senescence
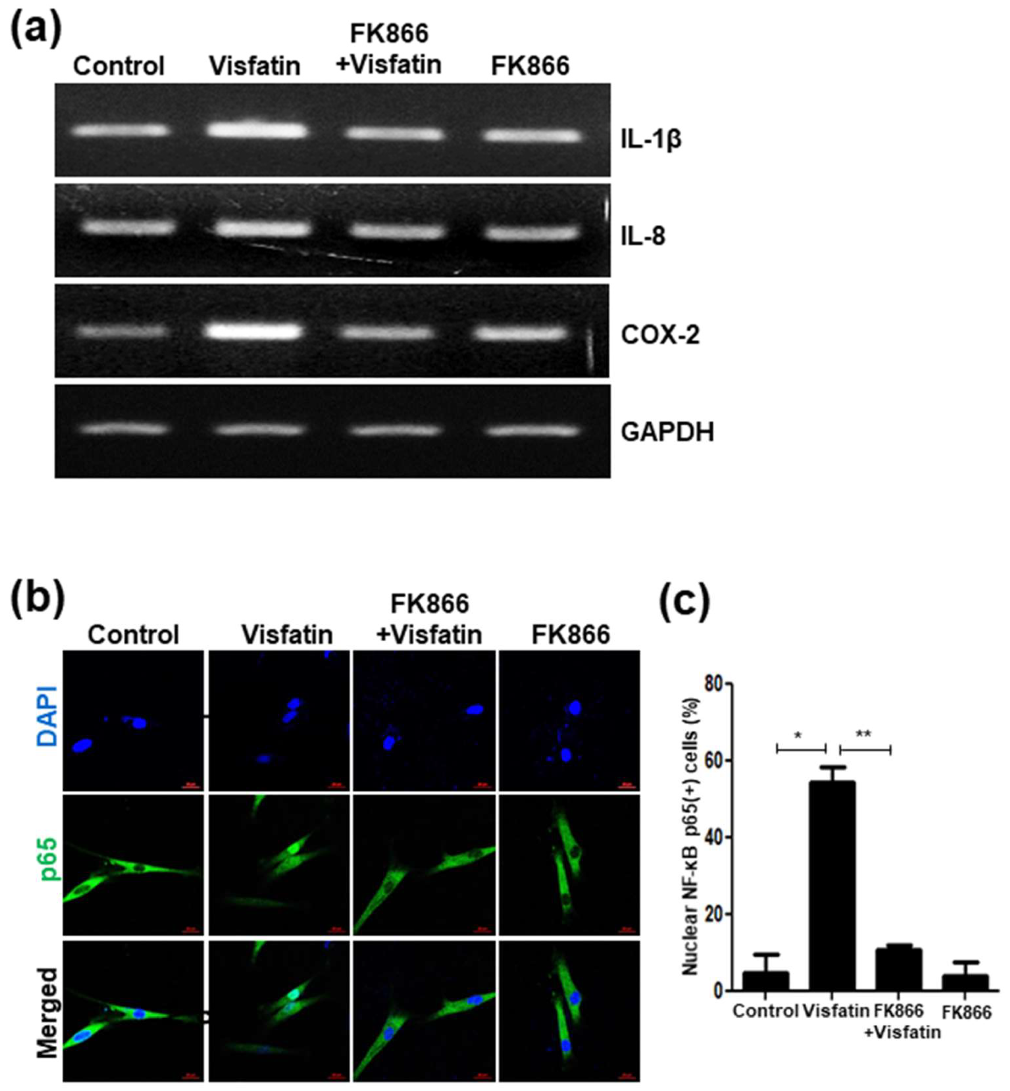
3.7. Visfatin Upregulates the Expression of SASP Factors and Activates NF-κB p65
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Goldberg, M.; Farges, J.C.; Lacerda-Pinheiro, S.; Six, N.; Jegat, N.; Decup, F.; Septier, D.; Carrouel, F.; Durand, S.; Chaussain-Miller, C.; et al. Inflammatory and Immunological Aspects of Dental Pulp Repair. Pharmacol. Res. 2008, 58, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Abbott, P.V. An Overview of the Dental Pulp: Its Functions and Responses to Injury. Aust. Dent. J. 2007, 52, S4–S16. [Google Scholar] [CrossRef] [PubMed]
- Beutler, B. Microbe Sensing, Positive Feedback Loops, and the Pathogenesis of Inflammatory Diseases. Immunol. Rev. 2009, 227, 248–263. [Google Scholar] [CrossRef] [PubMed]
- Farges, J.C. Understanding Dental Pulp Innate Immunity—A Basis for Identifying New Targets for Therapeutic Agents that Dampen Inflammation. J. Appl. Oral Sci. 2009, 17, s1678–s77572009000300001. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, T.; Mukai, K.; Hosokawa, Y.; Takegawa, D.; Matsuo, T. Catechins Inhibit Vascular Endothelial Growth Factor Production and Cyclooxygenase-2 Expression in Human Dental Pulp Cells. Int. Endod. J. 2015, 48, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.Y.; Lee, E.K.; Choi, Y.J.; Kim, J.M.; Kim, D.H.; Zou, Y.; Kim, C.H.; Lee, J.; Kim, H.S.; Kim, N.D.; et al. Molecular Inflammation as an Underlying Mechanism of the Aging Process and Age-Related Diseases. J. Dent. Res. 2011, 90, 830–840. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Kim, G.E.; Cho, H.J.; Yu, M.K.; Bhattarai, G.; Lee, N.H.; Yi, H.K. Aging of in Vitro Pulp Illustrates Change of Inflammation and Dentinogenesis. J. Endod. 2013, 39, 340–345. [Google Scholar] [CrossRef]
- Murray, P.E.; Stanley, H.R.; Matthews, J.B.; Sloan, A.J.; Smith, A.J. Age-Related Odontometric Changes of Human Teeth. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 2002, 93, 474–482. [Google Scholar] [CrossRef]
- Kitani, T.; Okuno, S.; Fujisawa, H. Growth Phase-Dependent Changes in the Subcellular Localization of Pre-B-Cell Colony-Enhancing Factor. FEBS Lett. 2003, 544, 74–78. [Google Scholar] [CrossRef]
- Sonoli, S.S.; Shivprasad, S.; Prasad, C.V.; Patil, A.B.; Desai, P.B.; Somannavar, M.S. Visfatin—A Review. Eur. Rev. Med. Pharmacol. Sci. 2011, 15, 9–14. [Google Scholar]
- Bae, Y.H.; Park, H.J.; Kim, S.R.; Kim, J.Y.; Kang, Y.; Kim, J.A.; Wee, H.J.; Kageyama, R.; Jung, J.S.; Bae, M.K.; et al. Notch1 Mediates Visfatin-Induced FGF-2 Up-Regulation and Endothelial Angiogenesis. Cardiovasc. Res. 2011, 89, 436–445. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.S.; Bae, M.K.; Kim, J.Y.; Jeong, J.W.; Yun, I.; Jang, H.O.; Bae, S.K. Visfatin Induces Neurite Outgrowth in PC12 Cells Via ERK1/2 Signaling Pathway. Neurosci. Lett. 2011, 504, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.S.; Kang, Y.G.; Park, H.J.; Wee, H.J.; Jang, H.O.; Bae, M.K.; Bae, S.K. Melatonin Inhibits Visfatin-Induced Inducible Nitric Oxide Synthase Expression and Nitric Oxide Production in Macrophages. J. Pineal Res. 2013, 55, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.R.; Bae, S.K.; Choi, K.S.; Park, S.Y.; Jun, H.O.; Lee, J.Y.; Jang, H.O.; Yun, I.; Yoon, K.H.; Kim, Y.J.; et al. Visfatin Promotes Angiogenesis by Activation of Extracellular Signal-Regulated Kinase 1/2. Biochem. Biophys. Res. Commun. 2007, 357, 150–156. [Google Scholar] [CrossRef]
- Laiguillon, M.C.; Houard, X.; Bougault, C.; Gosset, M.; Nourissat, G.; Sautet, A.; Jacques, C.; Berenbaum, F.; Sellam, J. Expression and Function of Visfatin (Nampt), an Adipokine-Enzyme Involved in Inflammatory Pathways of Osteoarthritis. Arthritis Res. Ther. 2014, 16, R38. [Google Scholar] [CrossRef]
- Moschen, A.R.; Gerner, R.R.; Tilg, H. Pre-B Cell Colony Enhancing factor/NAMPT/visfatin in Inflammation and Obesity-Related Disorders. Curr. Pharm. Des. 2010, 16, 1913–1920. [Google Scholar] [CrossRef]
- Park, H.J.; Kim, S.R.; Kim, S.S.; Wee, H.J.; Bae, M.K.; Ryu, M.H.; Bae, S.K. Visfatin Promotes Cell and Tumor Growth by Upregulating Notch1 in Breast Cancer. Oncotarget 2014, 5, 5087–5099. [Google Scholar] [CrossRef]
- Abolfazli, N.; Jabali, S.; Saleh Saber, F.; Babaloo, Z.; Shirmohammadi, A. Effect of Non-Surgical Periodontal Therapy on Serum and Salivary Concentrations of Visfatin in Patients with Chronic Periodontitis. J. Dent. Res. Dent. Clin. Dent. Prospects 2015, 9, 11–17. [Google Scholar] [CrossRef]
- Damanaki, A.; Memmert, S.; Nokhbehsaim, M.; Sanyal, A.; Gnad, T.; Pfeifer, A.; Deschner, J. Impact of Obesity and Aging on Crestal Alveolar Bone Height in Mice. Ann. Anat. 2018, 218, 227–235. [Google Scholar] [CrossRef]
- Bayani, M.; Pourali, M.; Keivan, M. Possible Interaction between Visfatin, Periodontal Infection, and Other Systemic Diseases: A Brief Review of Literature. Eur. J. Dent. 2017, 11, 407–410. [Google Scholar] [CrossRef]
- Mishra, V.; Shettar, L.; Bajaj, M.; Math, A.S.; Thakur, S.L. Interlinking Periodontitis and Type 2 Diabetes Mellitus by Assessment of Crevicular Visfatin Levels in Health and in Disease before and After Initial Periodontal Therapy. J. Clin. Diagn. Res. 2016, 10, ZC67–ZC71. [Google Scholar] [CrossRef] [PubMed]
- Nokhbehsaim, M.; Keser, S.; Jager, A.; Jepsen, S.; Deschner, J. Regulation of Regenerative Periodontal Healing by NAMPT. Mediators Inflamm. 2013, 2013, 202530. [Google Scholar] [CrossRef] [PubMed]
- Park, K.H.; Kim, D.K.; Huh, Y.H.; Lee, G.; Lee, S.H.; Hong, Y.; Kim, S.H.; Kook, M.S.; Koh, J.T.; Chun, J.S.; et al. NAMPT Enzyme Activity Regulates Catabolic Gene Expression in Gingival Fibroblasts during Periodontitis. Exp. Mol. Med. 2017, 49, e368. [Google Scholar] [CrossRef] [PubMed]
- Tabari, Z.A.; Keshani, F.; Sharbatdaran, M.; Banishahabadi, A.; Nejatifard, M.; Ghorbani, H. Visfatin Expression in Gingival Tissues of Chronic Periodontitis and Aggressive Periodontitis Patients: An Immunohistochemical Analysis. Dent. Res. J. 2018, 15, 104–110. [Google Scholar]
- Romacho, T.; Sanchez-Ferrer, C.F.; Peiro, C. Visfatin/Nampt: An Adipokine with Cardiovascular Impact. Mediators Inflamm. 2013, 2013, 946427. [Google Scholar] [CrossRef]
- Goldberg, E.L.; Dixit, V.D. Drivers of Age-Related Inflammation and Strategies for Healthspan Extension. Immunol. Rev. 2015, 265, 63–74. [Google Scholar] [CrossRef]
- Xia, S.; Zhang, X.; Zheng, S.; Khanabdali, R.; Kalionis, B.; Wu, J.; Wan, W.; Tai, X. An Update on Inflamm-Aging: Mechanisms, Prevention, and Treatment. J. Immunol. Res. 2016, 2016, 8426874. [Google Scholar] [CrossRef]
- Jadeja, R.N.; Powell, F.L.; Jones, M.A.; Fuller, J.; Joseph, E.; Thounaojam, M.C.; Bartoli, M.; Martin, P.M. Loss of NAMPT in Aging Retinal Pigment Epithelium Reduces NAD(+) Availability and Promotes Cellular Senescence. Aging 2018, 10, 1306–1323. [Google Scholar] [CrossRef]
- Ming, G.F.; Tang, Y.J.; Hu, K.; Chen, Y.; Huang, W.H.; Xiao, J. Visfatin Attenuates the Ox-LDL-Induced Senescence of Endothelial Progenitor Cells by Upregulating SIRT1 Expression through the PI3K/Akt/ERK Pathway. Int. J. Mol. Med. 2016, 38, 643–649. [Google Scholar] [CrossRef][Green Version]
- Villalobos, L.A.; Uryga, A.; Romacho, T.; Leivas, A.; Sanchez-Ferrer, C.F.; Erusalimsky, J.D.; Peiro, C. Visfatin/Nampt Induces Telomere Damage and Senescence in Human Endothelial Cells. Int. J. Cardiol. 2014, 175, 573–575. [Google Scholar] [CrossRef][Green Version]
- Kitagawa, M.; Ueda, H.; Iizuka, S.; Sakamoto, K.; Oka, H.; Kudo, Y.; Ogawa, I.; Miyauchi, M.; Tahara, H.; Takata, T. Immortalization and characterization of human dental pulp cells with odontoblastic differentiation. Arch. Oral Biol. 2007, 52, 727–731. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wei, D.; Xiao, H. Methods of cellular senescence induction using oxidative stress. Humana Press, Totowa, NJ Biological Aging. Methods Mol. Biol. 2013, 1048, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Bak, K.J.; Ok, C.Y.; Park, H.J.; Jang, H.O.; Bae, M.K.; Bae, S.K. Melatonin Rescues Human Dental Pulp Cells from Premature Senescence Induced by H2O2. Int J. Oral Biol. 2017, 42, 91–97. [Google Scholar] [CrossRef]
- Hasmann, M.; Schemainda, I. FK866, a Highly Specific Noncompetitive Inhibitor of Nicotinamide Phosphoribosyltransferase, Represents a Novel Mechanism for Induction of Tumor Cell Apoptosis. Cancer Res. 2003, 63, 7436–7442. [Google Scholar] [PubMed]
- Kay, J.; Thadhani, E.; Samson, L.; Engelward, B. Inflammation-Induced DNA Damage, Mutations and Cancer. DNA Repair 2019, 83, 102673. [Google Scholar] [CrossRef] [PubMed]
- Dodig, S.; Cepelak, I.; Pavic, I. Hallmarks of Senescence and Aging. Biochem. Med. 2019, 29, 030501. [Google Scholar] [CrossRef]
- Marazita, M.C.; Dugour, A.; Marquioni-Ramella, M.D.; Figueroa, J.M.; Suburo, A.M. Oxidative Stress-Induced Premature Senescence Dysregulates VEGF and CFH Expression in Retinal Pigment Epithelial Cells: Implications for Age-Related Macular Degeneration. Redox Biol. 2016, 7, 78–87. [Google Scholar] [CrossRef]
- Wang, Y.; Ji, L.; Jiang, R.; Zheng, L.; Liu, D. Oxidized High-Density Lipoprotein Induces the Proliferation and Migration of Vascular Smooth Muscle Cells by Promoting the Production of ROS. J. Atheroscler. Thromb. 2014, 21, 204–216. [Google Scholar] [CrossRef]
- Banath, J.P.; Klokov, D.; MacPhail, S.H.; Banuelos, C.A.; Olive, P.L. Residual gammaH2AX Foci as an Indication of Lethal DNA Lesions. BMC Cancer 2010, 10, 4. [Google Scholar] [CrossRef]
- Lee, M.S.; Yaar, M.; Eller, M.S.; Runger, T.M.; Gao, Y.; Gilchrest, B.A. Telomeric DNA Induces p53-Dependent Reactive Oxygen Species and Protects Against Oxidative Damage. J. Dermatol. Sci. 2009, 56, 154–162. [Google Scholar] [CrossRef][Green Version]
- Borodkina, A.V.; Deryabin, P.I.; Giukova, A.A.; Nikolsky, N.N. “Social Life” of Senescent Cells: What is SASP and Why Study it? Acta Nat. 2018, 10, 4–14. [Google Scholar] [CrossRef]
- Fernandez-Sanchez, A.; Madrigal-Santillan, E.; Bautista, M.; Esquivel-Soto, J.; Morales-Gonzalez, A.; Esquivel-Chirino, C.; Durante-Montiel, I.; Sanchez-Rivera, G.; Valadez-Vega, C.; Morales-Gonzalez, J.A. Inflammation, Oxidative Stress, and Obesity. Int. J. Mol. Sci. 2011, 12, 3117–3132. [Google Scholar] [CrossRef] [PubMed]
- Salvestrini, V.; Sell, C.; Lorenzini, A. Obesity may Accelerate the Aging Process. Front. Endocrinol. 2019, 10, 266. [Google Scholar] [CrossRef] [PubMed]
- Iwabu, M.; Okada-Iwabu, M.; Yamauchi, T.; Kadowaki, T. Adiponectin/adiponectin Receptor in Disease and Aging. NPJ Aging Mech. Dis. 2015, 1, 15013. [Google Scholar] [CrossRef]
- Yang, N.C.; Song, T.Y.; Chang, Y.Z.; Chen, M.Y.; Hu, M.L. Up-Regulation of Nicotinamide Phosphoribosyltransferase and Increase of NAD+ Levels by Glucose Restriction Extend Replicative Lifespan of Human Fibroblast Hs68 Cells. Biogerontology 2015, 16, 31–42. [Google Scholar] [CrossRef]
- Pi, C.; Yang, Y.; Sun, Y.; Wang, H.; Sun, H.; Ma, M.; Lin, L.; Shi, Y.; Li, Y.; Li, Y.; et al. Nicotinamide Phosphoribosyltransferase Postpones Rat Bone Marrow Mesenchymal Stem Cell Senescence by Mediating NAD(+)-Sirt1 Signaling. Aging 2019, 11, 3505–3522. [Google Scholar] [CrossRef]
- Ohtani, N. Deciphering the Mechanism for Induction of Senescence-Associated Secretory Phenotype (SASP) and its Role in Aging and Cancer Development. J. Biochem. 2019. [Google Scholar] [CrossRef]
- Rodier, F.; Campisi, J. Four Faces of Cellular Senescence. J. Cell Biol. 2011, 192, 547–556. [Google Scholar] [CrossRef]
- Nacarelli, T.; Lau, L.; Fukumoto, T.; Zundell, J.; Fatkhutdinov, N.; Wu, S.; Aird, K.M.; Iwasaki, O.; Kossenkov, A.V.; Schultz, D.; et al. NAD(+) Metabolism Governs the Proinflammatory Senescence-Associated Secretome. Nat. Cell Biol. 2019, 21, 397–407. [Google Scholar] [CrossRef]
- Xu, T.Y.; Zhang, S.L.; Dong, G.Q.; Liu, X.Z.; Wang, X.; Lv, X.Q.; Qian, Q.J.; Zhang, R.Y.; Sheng, C.Q.; Miao, C.Y. Discovery and Characterization of Novel Small-Molecule Inhibitors Targeting Nicotinamide Phosphoribosyltransferase. Sci. Rep. 2015, 5, 10043. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Age | No. of Patients | Gender | ||
|---|---|---|---|---|
| Male | Female | |||
| Young | ~19 | 4 | 4 | 0 |
| ~29 | 3 | 2 | 1 | |
| Old | ~39 | 3 | 0 | 3 |
| ~49 | 3 | 2 | 1 | |
| Total | 13 | 8 | 5 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ok, C.Y.; Park, S.; Jang, H.-O.; Takata, T.; Bae, M.-K.; Kim, Y.-D.; Ryu, M.H.; Bae, S.-K. Visfatin Induces Senescence of Human Dental Pulp Cells. Cells 2020, 9, 193. https://doi.org/10.3390/cells9010193
Ok CY, Park S, Jang H-O, Takata T, Bae M-K, Kim Y-D, Ryu MH, Bae S-K. Visfatin Induces Senescence of Human Dental Pulp Cells. Cells. 2020; 9(1):193. https://doi.org/10.3390/cells9010193
Chicago/Turabian StyleOk, Chang Youp, Sera Park, Hye-Ock Jang, Takashi Takata, Moon-Kyoung Bae, Yong-Deok Kim, Mi Heon Ryu, and Soo-Kyung Bae. 2020. "Visfatin Induces Senescence of Human Dental Pulp Cells" Cells 9, no. 1: 193. https://doi.org/10.3390/cells9010193
APA StyleOk, C. Y., Park, S., Jang, H.-O., Takata, T., Bae, M.-K., Kim, Y.-D., Ryu, M. H., & Bae, S.-K. (2020). Visfatin Induces Senescence of Human Dental Pulp Cells. Cells, 9(1), 193. https://doi.org/10.3390/cells9010193